Experimental
General
All required chemicals were purchased from the Merck or Fluka chemical companies. Dichloromethane and triethylamine were dried by distillation over CaH2 and then stored over 4Å molecular sieves. IR spectra were run on a Shimadzu FT-IR 8300 spectrophotometer. 1H- and 13C- NMR spectra were recorded in DMSO-d6 or CDCl3 using a Bruker Avance DPX instrument (1H-NMR at 250 MHz, 13C-NMR at 62.9 MHz, respectively). Chemical shifts are reported in ppm (δ) downfield from TMS. All the coupling constants (J) are given in Hertz. The mass spectra were recorded on a Shimadzu GC-MS QP 1000 EX instrument. Elemental analyses were run on a Thermo Finnigan Flash EA-1112 series. Melting points were determined in open capillaries with a Buchi 510 melting point apparatus and are not corrected. Thin-layer chromatography was carried out on silica gel 254 analytical sheets obtained from Fluka. Column chromatography was performed on Merck Kieselguhr (230-270 mesh).
General procedurefor synthesis of Schiff bases 7a-f.
A mixture of p-ethoxyaniline (20.0 mmol) and corresponding aldehyde (20.0 mmol) was refluxed in EtOH for 2-4 hours. After cooling the solutions, the precipitate formed was filtered off and washed with ethanol to give pure Schiff bases 7a-f as colored solid or crystals in excellent yields.
(4-Nitrobenzylidene)-(4-ethoxyphenyl)amine (7a). Brown solid (from p-phenetidine and 4-nitro-benzaldehyde); yield 97 %; m.p. 124-126 oC; IR (KBr) (cm-1) 1620.1 (C=N); 1H-NMR (CDCl3) δ 1.33 (Me, t, 3H), 3.88 (OCH2, q, 2H), 6.81-8.18 (ArH, m, 8H), 8.44 (HC=N, s, 1H); 13C-NMR (CDCl3) δ 14.82 (Me), 63.73 (OCH2), 115.05-154.52 (aromatic carbons), 158.66 (C=N); GC-MS m/z = 270 [M+]; Anal. calcd. for C15H14N2O3: C, 66.66; H, 5.22; N, 10.36. Found: C, 66.62; H, 5.39; N, 10.32.
(4-Chlorobenzylidene)-(4-ethoxyphenyl)amine (7b). Milky-coloured solid (from p-phenetidine and 4-chlorobenzaldehyde); yield 94 %; m.p. 92-94 °C; IR (KBr) (cm-1) 1620.1 (C=N);1H-NMR (CDCl3) δ 1.43 (Me, t, 3H), 4.00 (OCH2, q, 2H), 6.87-7.80 (ArH, m, 8H), 8.39 (HC=N, s, 1H); 13C-NMR (CDCl3) δ 14.84 (Me), 63.63 (OCH2), 114.94-156.43 (aromatic carbons), 157.85 (C=N); GC-MS m/z = 261 [M+, 37Cl], 259 [M+, 35Cl]; Anal. calcd. for C15H14ClNO: C, 69.36; H, 5.43; N, 5.39. Found: C, 69.29; H, 5.49; N, 5.44.
(4-Methoxybenzylidene)-(4-ethoxyphenyl)amine (7c). Milky-colour solid (from p-phenetidine and 4-methoxybenzaldehyde); yield 95 %; m.p. 128-130°C; IR (KBr) (cm-1) 1612.4 (C=N); 1H-NMR (CDCl3) δ 1.41 (Me, t, 3H), 3.86 (OMe, s, 3H), 4.03 (OCH2, q, 2H), 6.88-7.83 (ArH, m, 8H), 8.39 (HC=N, s, 1H); 13C-NMR (CDCl3) δ 14.88 (Me), 55.37 (OMe), 63.64 (OCH2), 114.12-157.73 (aromatic carbons), 161.95 (C=N); GC-MS m/z = 255 [M+]; Anal. calcd. for C16H17NO2: C, 75.27; H, 6.71; N, 5.49. Found: C, 75.17; H, 6.80; N, 5.45.
(4-Methylbenzylidene)-(4-ethoxyphenyl)amine (7d). Yellow solid (from p-phenetidine and 4-methyl-benzaldehyde); yield 93 %; m.p. 87-89 °C; IR (KBr) (cm-1) 1609.8 (C=N); 1H-NMR (CDCl3) δ 1.31 (Me, t, 3H), 2.36 (Me, s, 3H), 4.00 (OCH2, q, 2H), 6.85-7.76 (ArH, m, 8H), 8.39 (HC=N, s, 1H); 13C-NMR (CDCl3) δ 14.84, 21.54 (2Me), 63.57 (OCH2), 114.87-157.48 (aromatic carbons), 158.16 (C=N); GC-MS m/z = 239 [M+]; Anal. calcd. for C16H17NO: C, 80.30; H, 7.16; N, 5.85 Found: C, 80.35; H, 7.23; N, 5.89.
(4-Cinnamylidene)-(4-ethoxyphenyl)amine (7e). Light yellow solid (from p-phenetidine and cinnamaldehyde); yield 96 %; m.p. 76-78 oC; IR (KBr) (cm-1) 1622.5 (C=N); 1H-NMR (CDCl3) δ 1.40 (Me, t, 3H), 4.01 (OCH2, q, 2H), 6.87-7.52 (ArH and CH=CH, m, 11H), 8.26 (HC=N, d, 1H); 13C- NMR (CDCl3) δ 14.84 (Me), 63.61 (OCH2), 114.94-157.78 (C=C and aromatic carbons), 159.29 (C=N); GC-MS m/z = 251 [M+]; Anal. calcd. for C17H17NO: C, 81.24; H, 6.82; N, 5.57. Found: C, 81.19; H, 6.78; N, 5.52.
(3,4-Dimethoxybenzylidene)-(4-ethoxyphenyl)amine (7f). Green-yellow solid (from p-phenetidine and 3,4-dimethoxybenzaldehyde); yield 94 %; m.p. 82-84 °C. IR (KBr) (cm-1) 1619.7 (C=N); 1H-NMR (CDCl3) δ 1.39 (Me, t, 3H), 3.89, 3.95 (2OMe, 2s, 6H), 4.00 (OCH2, q, 2H), 6.85-7.59 (ArH, m, 7H), 8.34 (HC=N, s, 1H); 13C-NMR (CDCl3) δ 14.86 (Me), 55.91 (OMe), 63.57 (OCH2), 108.73-157.35 (aromatic carbons), 157.78 (C=N); GC-MS m/z = 285 [M+]; Anal. calcd. for C17H19NO3: C, 71.56; H, 6.71; N, 4.91. Found: C, 71.60; H, 6.67; N, 4.94.
Typical experimental procedure for the synthesis of 2-azetidinones 8a-n
Method A. A solution of the corresponding acyl chlorides (1.50 mmol) in dry CH2Cl2 (10 mL) was slowly added to a solution of Schiff bases 7a-f (1.00 mmol) and triethylamine (3.00 mmol) in CH2Cl2 (15 mL) at –10 oC. The reaction mixture was then allowed to warm to room temperature, stirred overnight and then it was washed successively with saturated sodium bicarbonate solution (20 mL) and brine (20 mL), dried (Na2SO4) and the solvent was evaporated to give the crude product which was then purified by column chromatography or recrystalization from EtOAc.
Method B. A solution of Schiff base 7a-b (1.0 eq.) was stirred with the corresponding substituted acetic acid (1.5 eq.), p-toluenesulfonyl chloride (1.5 eq.) and triethylamine (4-5 eq.) in dry CH2Cl2 at room temperature. After 8 to 10 h, the mixture was washed with saturated sodium bicarbonate solution and brine, dried over sodium sulfate and the solvent was evaporated to give the crude product which was then purified by recrystalization from EtOAc, unless stated otherwise.
2-(1-(4-Ethoxyphenyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (8a). Yield: 81%; mp: 190-192 °C; IR (CHCl3) cm-1: 1738.0, 1776.2 (CO, phth), 1788.8 (CO, β-lactam); 1H-NMR (DMSO-d6) δ 1.29 (Me, t, 3H), 3.92 (OCH2, q, 2H), 5.32 (H-4, d, 1H, J=2.5), 5.70 (H-3, d, 1H, J=2.5), 6.92-8.27 (ArH, m, 12H); 13C-NMR (DMSO-d6) δ 14.40 (Me), 58.33 (OCH2), 61.52 (C-4), 63.26 (C-3), 115.00-155.38 (aromatic carbons), 161.11 (CO, phth), 166.63 (CO, β-lactam); GC-MS m/z = 457 [M+]; Anal. calcd. for C25H19N3O6: C, 65.64; H, 4.19; N, 9.19. Found: C, 65.69; H, 4.13; N, 9.22.
2-(2-(4-Chlorophenyl)-1-(4-ethoxyphenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (8b). Yield: 87 %; mp: 211-213 °C IR (CHCl3) cm-1: 1720.4, 1758.9 (CO, phth), 1786.6 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.37 (Me, t, 3H), 3.97 (OCH2, q, 2H), 5.22 (H-4, d, 1H, J=2.5), 5.32 (H-3, d, 1H, J=2.5), 6.78-7.76 (ArH, m, 12H); 13C-NMR (CDCl3) δ 14.78 (Me), 60.68 (OCH2), 62.68 (C-4), 63.68 (C-3), 115.02-156.01 (aromatic carbons), 161.23 (CO, phth), 166.78 (CO, β-lactam); GC-MS m/z = 448 [M+, 37Cl], 446 [M+, 35Cl]; Anal. Calcd for C25H19ClN2O4: C, 67.19; H, 4.29; N, 6.27. Found: C, 67.16; H, 4.27; N, 6.24.
2-(1-(4-Ethoxyphenyl)-2-(4-methoxyphenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (8c). Yield: 80 %; mp: 199-201 oC IR (CHCl3) cm-1: 1724.2, 1758.9 (CO, phth), 1778.0 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.09 (Me, t, 3H), 3.74 (OCH2, q, 2H), 4.19 (OMe, s, 3H), 4.96 (H-4, d, 1H, J=2.5), 5.20 (H-3, d, 1H, J=2.5), 6.62-7.73 (ArH, m, 12H); 13C-NMR (CDCl3) δ 14.50 (Me), 55.04 (OCH2), 59.65 (OMe), 61.90 (C-4), 63.05 (C-3), 114.27-161.43 (aromatic carbons), 164.24 (CO, phth), 167.57 (CO, β-lactam); GC-MS m/z = 442 [M+]; Anal. calcd. for C26H22N2O5: C, 70.58; H, 5.01; N, 6.33. Found: C, 70.63; H, 5.05; N, 6.28.
2-(1-(4-Ethoxyphenyl)-2-oxo-4-p-tolylazetidin-3-yl)isoindoline-1,3-dione (8d). Yield: 84 % mp: 202-204 °C; IR (KBr) cm-1: 174.2, 1776.2 (CO, phth), 1788.7 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.35 (Me, t, 3H), 2.33 (Me, s, 3H), 3.94 (OCH2, q, 2H), 5.25 (H-4, d, 1H, J=2.5), 5.32 (H-3, d, 1H, J=2.5), 6.68-7.85 (ArH, m, 12H); 13C-NMR (CDCl3) δ 14.30, 20.73 (2Me), 60.70 (OCH2), 62.27 (C-4), 63.13 (C-3), 114.41-155.32 (aromatic carbons), 161.11 (CO, phth), 166.35 (CO, β-lactam); GC-MS m/z = 426 [M+]; Anal. calcd. for C26H22N2O4: C, 70.58; H, 5.01; N, 6.33. Found: C, 70.64; H, 5.05; N, 6.37.
2-(1-(4-Ethoxyphenyl)-2-oxo-4-styrylazetidin-3-yl)isoindoline-1,3-dione (8e). Yield: 88 %; mp: 161-163 °C; IR (CHCl3) cm-1: 1724.2, 1758.5 (CO, phth), 1774.7 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.37 (Me, t, 3H), 2.33 (Me, s, 3H), 3.97 (OCH2, q, 2H), 5.03 (H-4, dd, 1H, J=5.5, 8.5), 5.68 (H-3, d, 1H, J=5.5), 6.32 (H-5, dd, J=8.5, 16.0), 6.85 (H-6, d, 1H, J=9.0), 7.19-7.82 (ArH, m, 13H); 13C-NMR (CDCl3) δ 14.78 (Me), 57.69 (OCH2), 61.04 (C-4), 63.67 (C-3), 114.99-155.82 (C=C, aromatic carbons), 160.56 (CO, phth), 167.28 (CO, β-lactam); GC-MS m/z = 438 [M+]; Anal. calcd. for C27H22N2O4: C, 73.96; H, 5.06; N, 6.39. Found: C, 74.02; H, 5.09; N, 6.33.
1-(4-Ethoxyphenyl)-4-(4-nitrophenyl)-3-phenoxyazetidin-2-one (8f). Purified by column chromatography (eluent: 6:4 hexane-EtOAc); yield: 91 %; mp: 180-182 °C; IR (KBr) cm-1: 1743.5 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.30 (Me, t, 3H), 3.89 (OCH2, q, 2H), 5.39 (H-4, d, 1H, J=4.8), 5.55 (H-3, d, 1H, J=4.8), 6.68-8.08 (ArH, m, 13H); 13C-NMR (CDCl3) δ 14.74 (Me), 61.11 (OCH2), 63.72 (C-4), 81.24 (C-3), 115.17-156.49 (aromatic carbons), 161.82 (CO, β-lactam); GC-MS m/z = 404[M+]; Anal. calcd. for C23H20N2O5: C, 68.31; H, 4.98; N, 6.93. Found: C, 68.28; H, 5.05; N, 6.88.
4-(4-Chlorophenyl)-1-(4-ethoxyphenyl)-3-phenoxyazetidin-2-one (8g). Yield: 88 %; mp: 164-166 °C; IR (KBr) cm-1:1746.5 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.31 (Me, t, 3H), 3.87 (OCH2, q, 2H), 5.24 (H-4, d, 1H, J=4.8), 5.45 (H-3, d, 1H, J=4.8), 6.68-7.23 (ArH, m, 13H); 13C-NMR (CDCl3) δ 14.77 (Me), 61.41 (OCH2), 63.68 (C-4), 81.09 (C-3), 115.03-156.78 (aromatic carbons), 162.26 (CO, β-lactam); GC-MS m/z = 395[M+, 37Cl], 393 [M+, 35Cl]; Anal. calcd. for C23H20ClNO3: C, 70.14; H, 5.12; N, 3.56. Found: C, 70.24; H, 5.17; N, 3.50.
1-(4-Ethoxyphenyl)-4-(4-methoxyphenyl)-3-phenoxyazetidin-2-one (8h). Purified by column chromatography (eluent: 7:3 hexane-EtOAc); yield: 90 %; mp: 168-170 °C; IR (KBr) cm-1:1753.5 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.30 (Me, t, 3H), 3.64 (OMe, s, 3H), 3.88 (OCH2, q, 2H), 5.21 (H-4, d, 1H, J=4.7), 5.41 (H-3, d, 1H, J=4.7), 6.69-7.23 (ArH, m, 13H); 13C-NMR (CDCl3) δ 14.79 (Me), 55.17 (OMe), 61.79 (OCH2), 63.65 (C-4), 81.23 (C-3), 113.84-159.84 (aromatic carbons), 162.56 (CO, β-lactam); GC-MS m/z = 389 [M+]; Anal. calcd. for C24H23NO4: C, 74.02; H, 5.95; N, 3.60. Found: C, 73.97; H, 5.90; N, 3.64.
1-(4-Ethoxyphenyl)-3-phenoxy-4-p-tolylazetidin-2-one (8i). Yield: 94 %; mp: 165-167 °C; IR (CHCl3) cm-1: 1751.2 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.31 (Me, t, 3H), 2.25 (Me, s, 3H), 3.91 (OCH2, q, 2H), 5.28 (H-4, d, 1H, J=4.8), 5.47 (H-3, d, 1H, J=4.8), 6.74-7.30 (ArH, m, 13H); 13C-NMR (CDCl3) δ 14.76, 21.17 (2Me), 61.98 (OCH2), 63.60 (C-4), 81.24 (C-3), 114.91-157.08 (aromatic carbons), 162.55 (CO, β-lactam); GC-MS m/z = 373 [M+]; Anal. calcd. for C24H23NO3: C, 77.19; H, 6.21; N, 3.75. Found: C, 77.25; H, 6.26; N, 3.73.
1-(4-Ethoxyphenyl)-3-phenoxy-4-styrylazetidin-2-one (8j). Yield: 91 %; mp: 171-173 °C; IR (CHCl3) cm-1: 1749.3 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.30 (Me, t, 3H), 3.90 (OCH2, q, 2H), 4.90 (H-4, dd, 1H, J=4.9, 8.5), 5.37 (H-3, d, 1H, J=4.9), 6.23 (H-5, dd, J=8.5, 16.0), 6.75 (H-6, d, 1H, J=16.0), 6.87-7.38 (ArH, m, 14H); 13C-NMR (CDCl3) δ 14.80 (Me), 61.12 (OCH2), 63.70 (C-4), 81.48 (C-3), 115.01-157.42 (C=C, aromatic carbons), 162.23 (CO, β-lactam); GC-MS m/z = 385 [M+]; Anal. calcd. for C25H23NO3: C, 77.90; H, 6.01; N, 3.63. Found: C, 77.97; H, 6.06; N, 3.60.
4-(3,4-Dimethoxyphenyl)-1-(4-ethoxyphenyl)-3-phenoxyazetidin-2-one (8k). Yield: 95 %; mp: 186-188 °C; IR (KBr) cm-1: 1758.2 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.36 (Me, t, 3H), 3.75, 3.81 (2OMe, 2s, 6H), 3.95 (OCH2, q, 2H), 5.28 (H-4, d, 1H, J=4.2), 5.52 (H-3, d, 1H, J=4.2), 6.74-7.33 (ArH, m, 12H); 13C-NMR (CDCl3) δ 14.77 (Me), 55.76, 55.94 (2OMe), 62.05 (OCH2), 63.64 (C-4), 81.11 (C-3), 110.78-156.96 (aromatic carbons), 162.50 (CO, β-lactam); GC-MS m/z = 419 [M+]; Anal. calcd. for C25H25NO5: C, 71.58; H, 6.01; N, 3.34. Found: C, 71.63; H, 5,98; N, 3.38.
1-(4-Ethoxyphenyl)-3-(naphthalen-2-yloxy)-4-(4-nitrophenyl)azetidin-2-one (8l). Yield: 84 %; mp: 174-176 °C; IR (KBr) cm-1: 1750.6 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.39 (Me, t, 3H), 3.95 (OCH2, q, 2H), 5.51 (H-4, d, 1H, J=4.8), 5.74 (H-3, d, 1H, J=4.8), 6.79-8.11 (ArH, m, 15H); 13C-NMR (CDCl3) δ 14.75 (Me), 61.08 (OCH2), 63.73 (C-4), 81.16 (C-3), 108.98-156.28 (aromatic carbons), 161.70 (CO, β-lactam); GC-MS m/z = 454 [M+]; Anal. calcd. for C27H22N2O5: C, 71.35; H, 4.88; N, 6.16. Found: C, 71.41; H, 4.92; N, 6.20.
3-(2,4-Dichlorophenoxy)-1-(4-ethoxyphenyl)-4-(4-nitrophenyl)azetidin-2-one (8m). Purified by column chromatography (eluent: 6:4 hexane-EtOAc); yield: 89 %; mp: 160-162 °C; IR (KBr) cm-1: 1747.8 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.37 (Me, t, 3H), 3.96 (OCH2, q, 2H), 5.52 (H-4, d, 1H, J=5.1), 5.56 (H-3, d, 1H, J=5.1), 6.78-8.22 (ArH, m, 11H); 13C-NMR (CDCl3) δ 14.74 (Me), 60.44 (OCH2), 63.73 (C-4), 81.84 (C-3), 115.19-156.38 (aromatic carbons), 161.26 (CO, β-lactam); GC-MS m/z = 476 [M+, 37Cl], 474, 472 [M+, 35Cl]; Anal. calcd. for C23H18Cl2N2O5: C, 58.37; H, 3.83; N, 5.92. Found: C, 58.32; H, 3.88; N, 5.89.
1-(4-Ethoxyphenyl)-3-methoxy-4-p-tolylazetidin-2-one (8n). Yield: 92 %; mp: 133-135 °C; IR (KBr) cm-1: 1744.5 (CO, β-lactam); 1H-NMR (CDCl3) δ 1.34 (Me, t, 3H), 2.34 (Me, s, 3H), 3.94 (OCH2, q, 2H), 4.76 (H-4, d, 1H, J=4.7), 5.12 (H-3, d, 1H, J=4.7), 6.73-7.28 (ArH, m, 15H); 13C-NMR (CDCl3) δ 14.77, 21.24 (2Me), 61.61 (OCH2), 63.59 (C-4), 84.74 (C-3), 114.85-155.64 (aromatic carbons), 163.78 (CO, β-lactam); GC-MS m/z = 311 [M+]; Anal. calcd. for C19H21NO3: C, 73.29; H, 6.80; N, 4.50. Found: C, 73.34; H, 6.85; N, 4.47.
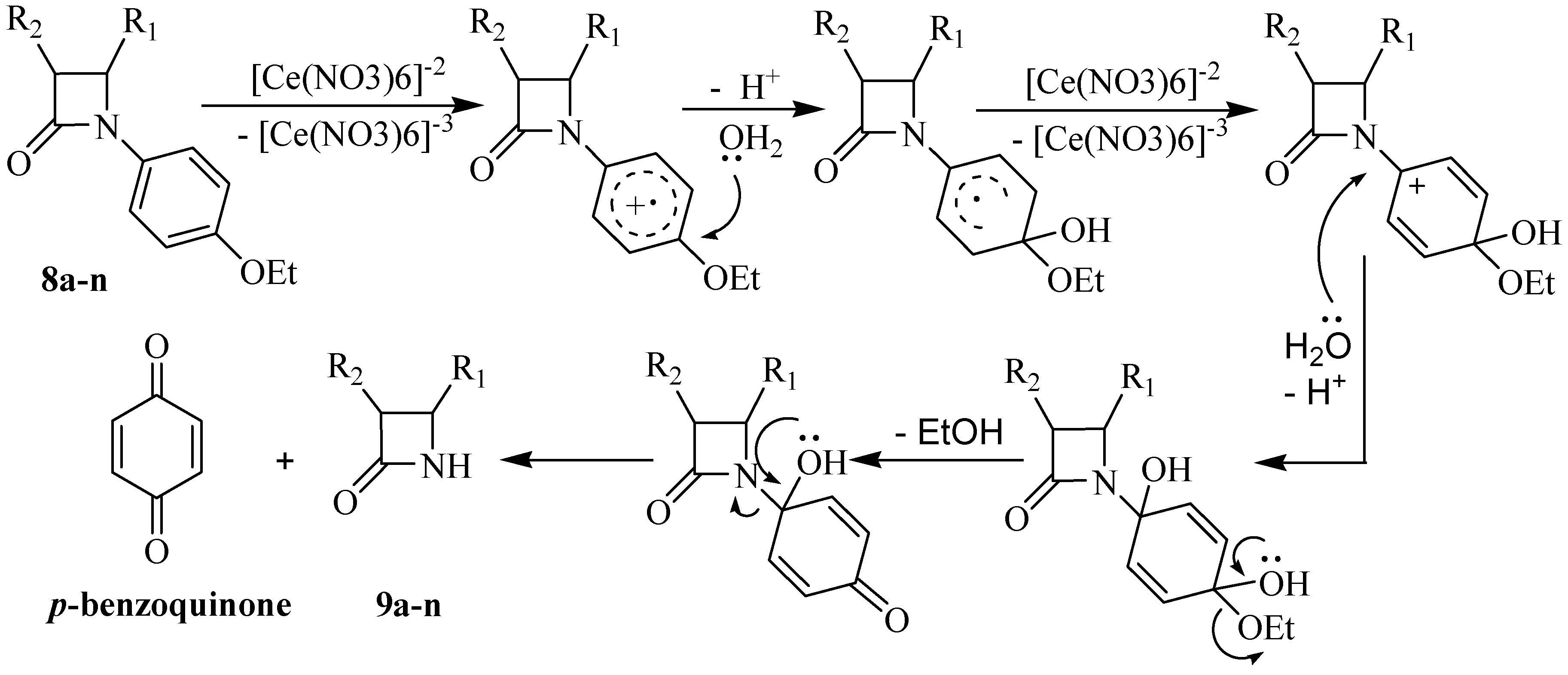
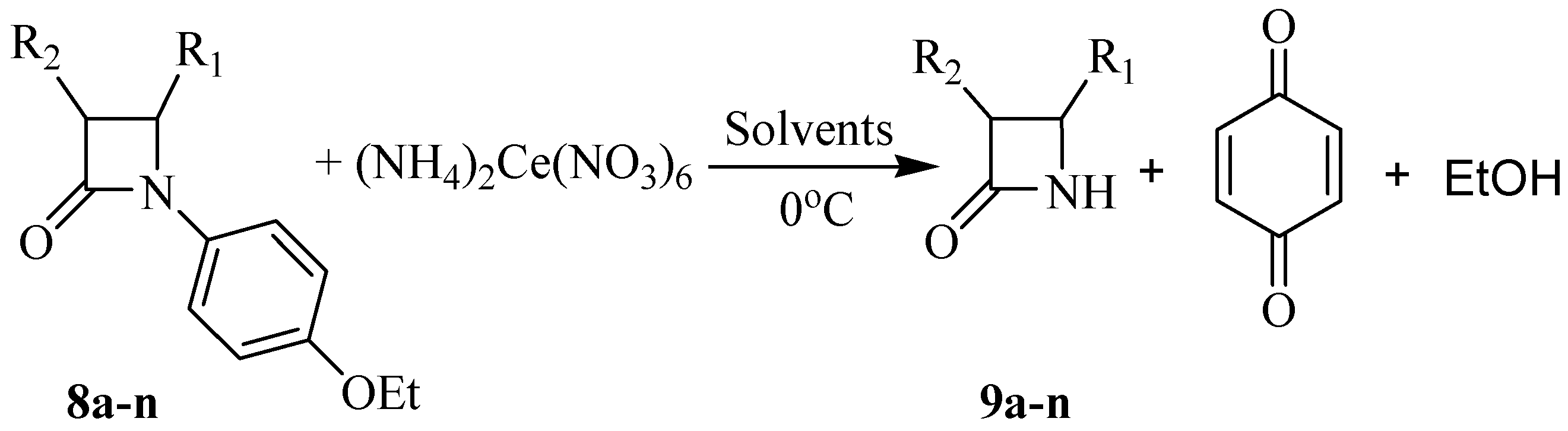
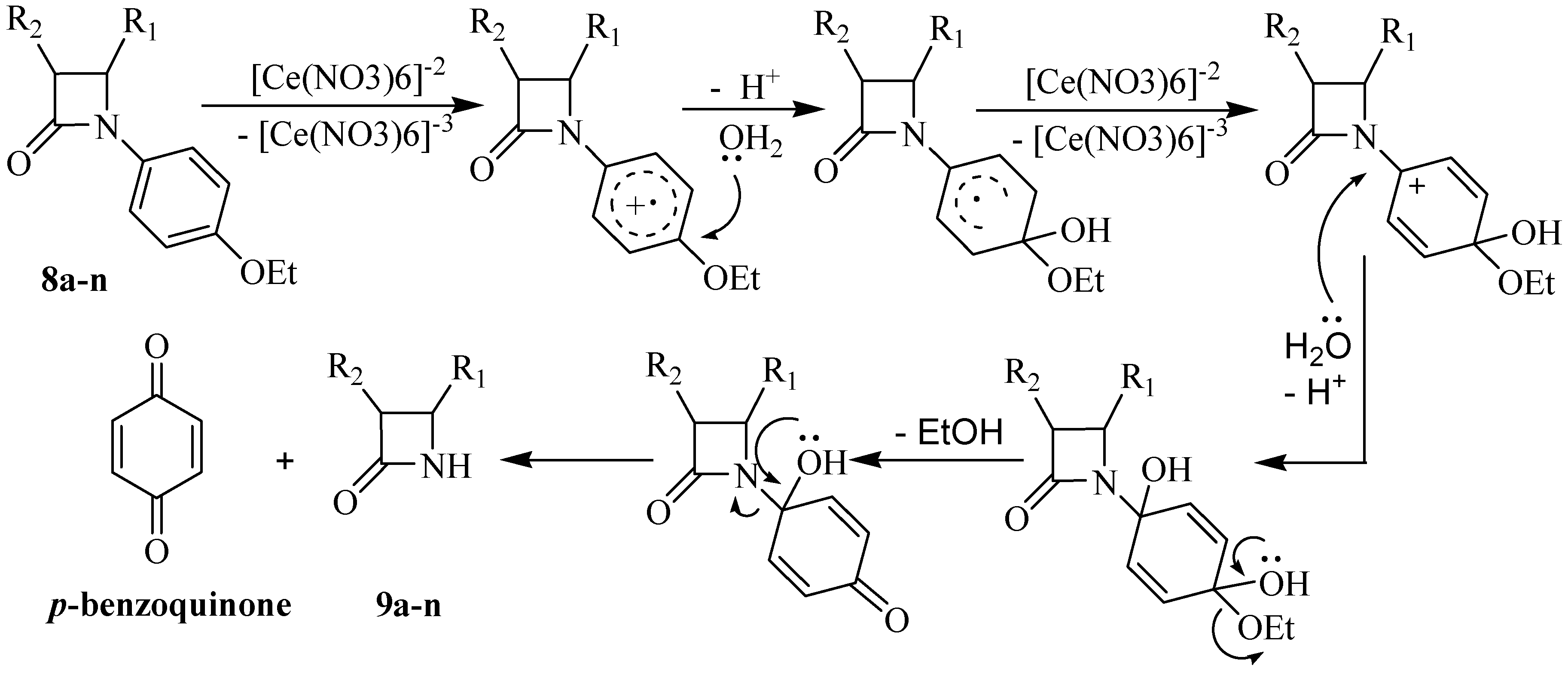
Typical experimental procedure for the synthesis of N-unsubstituted β-lactams 9a-n
A solution of (NH
4)
2Ce(NO
3)
6 (CAN, 2.0-3.5 mmol) in water (15 mL) was added dropwise to a solution of the β-lactam
8a-n (1.00 mmol) in CH
3CN or THF (30 mL) at the temperature mentioned in
Table 3. The mixture was stirred at corresponding temperature for the mentioned time, then water (30 mL) was added and the mixture was extracted with EtOAc (3×20 mL) and washed with 10 % aqueous NaHCO
3 (40 mL). The aqueous layer of NaHCO
3 was extracted again with EtOAc (15 mL) and all organic layers were combined and washed successively with 10 % NaHSO
3 (2×30 mL), 10 % NaHCO
3 (20 mL) and brine (20 mL) and then dried over sodium sulfate. After filtration and evaporation of the solvent
in vacuo, the crude product was purified by column chromatography or recrystalization from diethyl ether, as indicated.
2-(2-(4-Nitrophenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (9a). Purified by recrystalization; yield: 77 %; mp: 210-212 °C; IR (CHCl3) cm-1: 1736.3, 1776.7 (CO, phth), 1785.5 (CO, β-lactam), 3373.5 (NH); 1H-NMR (DMSO-d6) δ 4.99 (H-3, d, 1H, J=2.5), 5.70 (H-4, dd, 1H, J=1.3, 2.5), 7.38-8.27 (ArH, m, 8H), 9.11 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 59.51 (C-4), 62.49 (C-3), 123.19-147.12 (aromatic carbons), 164.33 (CO, phth), 166.24 (CO, β-lactam); GC-MS m/z = 337 [M+]; Anal. calcd. for C17H11N3O5: C, 60.54; H, 3.29; N, 12.46. Found: C, 60.60; H, 3.32; N, 12.51.
2-(2-(4-Chlorophenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (9b). Purified by recrystalization; yield: 74 %; mp: 196-198 °C; IR (CHCl3) cm-1: 1733.9, 1777.0 (CO, phth), 1785.0 (CO, β-lactam), 3373.5 (NH); 1H-NMR (DMSO-d6) δ 4.92 (H-3, d, 1H, J=2.5), 5.04 (H-4, dd, 1H, J=2.5, 3.2), 7.41-7.92 (ArH, m, 8H), 9.01 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 54.96 (C-4), 62.53 (C-3), 123.37-138.08 (aromatic carbons), 164.48 (CO, phth), 166.70 (CO, β-lactam); GC-MS m/z = 328 [M+, 37Cl], 326 [M+, 35Cl]; Anal. calcd. for C17H11ClN2O3: C, 62.49; H, 3.39; N, 8.57. Found: C, 62.55; H, 3.43; N, 8.54.
2-(2-(4-Methoxyphenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (9c). Purified by recrystalization; yield: 81 %; mp: 190-192 °C; IR (KBr) cm-1: 1735.7, 1775.6 (CO, phth), 1790.2 (CO, β-lactam), 3354.4 (NH); 1H-NMR (DMSO-d6) δ 3.81 (OMe, s, 3H), 4.94 (H-3, d, 1H, J=2.5), 5.03 (H-4, dd, 1H, J=2.5, 3.1), 7.43-8.01 (ArH, m, 8H), 8.98 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 55.08 (OMe), 55.19 (C-4), 62.69 (C-3), 113.92-159.09 (aromatic carbons), 164.60 (CO, phth), 166.73 (CO, β-lactam); GC-MS m/z = 322 [M+]; Anal. calcd. for C18H14N2O4: C, 67.07; H, 4.38; N, 8.69. Found: C, 67.11; H, 4.34; N, 8.65.
2-(2-Oxo-4-p-tolylazetidin-3-yl)isoindoline-1,3-dione (9d). Purified by recrystalization; yield: 78 %; mp: 197-199 °C; IR (CHCl3) cm-1: 1740.0, 1775.0 (CO, phth), 1785.0 (CO, β-lactam), 3480.5 (NH); 1H-NMR (DMSO-d6) δ 2.35 (Me, s, 3H), 4.94 (H-4, dd, 1H, J=2.5, 3.5), 5.04 (H-3, d, 1H, J=2.5), 7.23-8.03 (ArH, m, 8H), 9.02 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 20.68 (Me), 55.43 (C-4), 62.63 (C-3), 123.39-137.27 (aromatic carbons), 164.56 (CO, phth), 166.71 (CO, β-lactam); GC-MS m/z = 306 [M+]; Anal. calcd. for C18H14N2O3: C, 70.58; H, 4.61; N, 9.15. Found: C, 70.62; H, 4.58; N, 9.21.
2-(2-Oxo-4-styrylazetidin-3-yl)isoindoline-1,3-dione (9e). Purified by recrystalization; yield: 82 %; mp: 168-170 °C; IR (CHCl3) cm-1: 1726.2, 1768.6 (CO, phth), 1784.0 (CO, β-lactam), 3417.0 (NH); 1H-NMR (DMSO-d6) δ 4.72 (H-4, m, 1H), 5.60 (H-3, d, 1H, J=5.2), 6.25 (H-5, dd, 1H, J=7.6, 16.0), 6.70 (H-6, d, 1H, J=16.0), 7.22-7.92 (ArH, m, 9H), 8.85 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 55.38 (C-4), 58.69 (C-3), 123.42-135.69 (C=C, aromatic carbons), 164.06 (CO, phth), 166.93 (CO, β-lactam); GC-MS m/z = 318 [M+]; Anal. calcd. for C19H14N2O3: C, 71.69; H, 4.43; N, 8.80. Found: C, 71.74; H, 4.49; N, 8.78.
4-(4-Nitrophenyl)-3-phenoxyazetidin-2-one (9f). Purified by column chromatography (eluent: 4:6 hexane-EtOAc); yield: 80 %; mp: 160-162 °C; IR (KBr) cm-1: 1774.4 (CO), 3247.9 (NH); 1H-NMR (DMSO-d6) δ 5.33 (H-3, d, 1H, J=4.8), 5.77 (H-4, dd, 1H, J=2.2, 4.8), 6.77-8.20 (ArH, m, 9H), 9.10 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 56.01 (C-4), 82.71 (C-3), 115.04-156.24 (aromatic carbons), 165.78 (CO, β-lactam); GC-MS m/z = 284 [M+]; Anal. calcd. for C15H12N2O4: C, 63.38; H, 4.25; N, 9.85. Found: C, 63.44; H, 4.30; N, 9.87.
4-(4-Chlorophenyl)-3-phenoxyazetidin-2-one (9g). Purified by recrystalization; yield: 82 %; mp: 188-190 °C; IR (KBr) cm-1: 1773.5 (CO), 3420.0 (NH); 1H-NMR (DMSO-d6) δ 5.09 (H-3, d, 1H, J=4.5), 5.61 (H-4, dd, 1H, J=2.1, 4.5), 6.77-7.32 (ArH, m, 9H), 8.89 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 56.91 (C-4), 82.26 (C-3), 114.98-156.34 (aromatic carbons), 165.90 (CO, β-lactam); GC-MS m/z = 275 [M+, 37Cl], 273 [M+, 35Cl]; Anal. calcd. for C15H12ClNO2: C, 65.82; H, 4.42; N, 5.12. Found: C, 65.78; H, 4.46; N, 5.08.
4-(4-Methoxyphenyl)-3-phenoxyazetidin-2-one (9h). Purified by recrystalization; yield: 86 %; mp: 157-159 °C; IR (CHCl3) cm-1: 1776.3 (CO), 3409.9 (NH); 1H-NMR (DMSO-d6) δ 3.66 (MeO, s, 3H), 5.02 (H-3, d, 1H, J=4.3), 5.52 (H-4, dd, 1H, J=1.8, 4.3), 6.55-7.35 (ArH, m, 9H), 9.08 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 54.83 (OMe), 56.45 (C-4), 81.76 (C-3), 113.19-158.68 (aromatic carbons), 166.84 (CO, β-lactam); GC-MS m/z = 269 [M+]; Anal. calcd. for C16H15NO3: C, 71.36; H, 5.61; N, 5.20. Found: C, 71.42; H, 5.66; N, 5.24.
3-Phenoxy-4-p-tolylazetidin-2-one (9i). Purified by recrystalization; yield: 84 %; mp: 180-182 °C; IR (KBr) cm-1: 1773.9 (CO), 3300.0 (NH); 1H-NMR (DMSO-d6) δ 1.95 (Me, s, 3H), 4.81 (H-3, d, 1H, J=4.4), 5.32 (H-4, dd, 1H, J=2.2, 4.4), 6.54-6.98 (ArH, m, 9H), 8.60 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 20.62 (Me), 56.52 (C-4), 82.34 (C-3), 115.07-156.66 (aromatic carbons), 166.14 (CO, β-lactam); GC-MS m/z = 253 [M+]; Anal. calcd. for C16H15NO2: C, 75.87; H, 5.97; N, 5.53. Found: C, 75.84; H, 6.03; N, 5.48.
3-Phenoxy-4-styrylazetidin-2-one (9j). Purified by column chromatography (eluent: 5:5 hexane-EtOAc); yield: 76 %; mp: 190-192 °C; IR (KBr) cm-1: 1775.7 (CO), 3310.0 (NH); 1H-NMR (DMSO-d6) δ 4.64 (H-4, m, 1H,), 5.51 (H-3, d, 1H, J= 4.3), 6.18 (H-5, dd, 1H, J= 7.4, 15.9), 6.65 (H-6, d, 1H, J= 15.9), 6.90-7.33 (ArH, m, 9H), 8.96 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 60.67 (C-4), 87.47 (C-3), 120.30-162.15 (C=C, aromatic carbons), 170.78 (CO, β-lactam); GC-MS m/z = 265 [M+]; Anal. calcd. for C17H15NO2: C, 76.96; H, 5.70; N, 5.28. Found: C, 76.92; H, 5.77; N, 5.24.
4-(3,4-Dimethoxyphenyl)-3-phenoxyazetidin-2-one (9k). Purified by recrystalization; yield: 86 %; mp: 140-142 °C; IR (KBr) cm-1: 1777.7 (CO), 3418.9 (NH); 1H-NMR (DMSO-d6) δ 3.58, 3.66 (2MeO, 2s, 6H), 5.03 (H-3, d, 1H, J=4.0), 5.58 (H-4, dd, 1H, J=2.2, 4.0), 6.64-7.26 (ArH, m, 8H), 8.83 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 55.23, 55.25 (2OMe), 56.40 (C-4), 81.13 (C-3), 111.02-156.59 (aromatic carbons), 165.98 (CO, β-lactam); GC-MS m/z = 299 [M+]; Anal. calcd. for C17H17NO4: C, 68.21; H, 5.72; N, 4.68. Found: C, 68.31; H, 5.79; N, 4.73.
3-(Naphthalen-2-yloxy)-4-(4-nitrophenyl)azetidin-2-one (9l). Purified by column chromatography (eluent: 5:5 hexane-EtOAc); yield: 83 %; mp: 172-174 °C; IR IR (KBr) cm-1: 1769.6 (CO), 3354.4 (NH); 1H-NMR (DMSO-d6) δ 5.38 (H-3, d, 1H, J=4.5), 5.86 (H-4, dd, 1H, J=2.3, 4.5), 7.31-7.78 (ArH, m, 11H), 9.10 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 56.05 (C-4), 82.71 (C-3), 117.51-147.53 (aromatic carbons), 165.66 (CO, β-lactam); GC-MS m/z = 334 [M+]; Anal. calcd. for C19H14N2O4: C, 68.26; H, 4.22; N, 8.38. Found: C, 68.23; H, 4.26; N, 8.42.
3-(2,4-Dichlorophenoxy)-4-(4-nitrophenyl)azetidin-2-one (9m). Purified by column chromatography (eluent: 5:5 hexane-EtOAc); yield: 78 %; mp: 160-162 °C; IR (KBr) cm-1: 1775.5 (CO), 3320.5 (NH); 1H-NMR (DMSO-d6) δ 5.34 (H-3, d, 1H, J=3.5), 5.77 (H-4, dd, 1H, J=2.4, 3.5), 7.32-8.22 (ArH, m, 7H), 9.20 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 55.49 (C-4), 82.99 (C-3), 116.31-150.69 (aromatic carbons), 165.00 (CO, β-lactam); GC-MS m/z = 356 [M+, 37Cl], 354, 352 [M+, 35Cl]; Anal. calcd. for C15H10Cl2N2O4: C, 51.01; H, 2.85; N, 7.93. Found: C, 51.05; H, 2.92; N, 7.97.
3-Methoxy-4-p-tolylazetidin-2-one (9n). Purified by column chromatography (eluent: 4:6 hexane-EtOAc); yield: 77 %; mp: 92-94 °C; IR (KBr) cm-1: 1765.8 (CO), 3414.0 (NH); 1H-NMR (DMSO-d6) δ 2.11 (Me, s, 3H), 2.82 (OMe, s, 3H), 4.51 (H-4, dd, 1H, J=2.2, 4.4), 4.59 (H-3, d, 1H, J=4.4), 6.69-7.07 (ArH, m, 4H), 8.41 (NH, brs, 1H); 13C-NMR (DMSO-d6) δ 20.65 (Me), 56.23 (OMe), 57.12 (C-4), 86.26 (C-3), 127.35-136.75 (aromatic carbons), 167.48 (CO, β-lactam); GC-MS m/z = 191 [M+]; Anal. calcd. for C11H13NO2: C, 69.09; H, 6.85; N, 7.32. Found: C, 69.14; H, 6.92; N, 7.28.

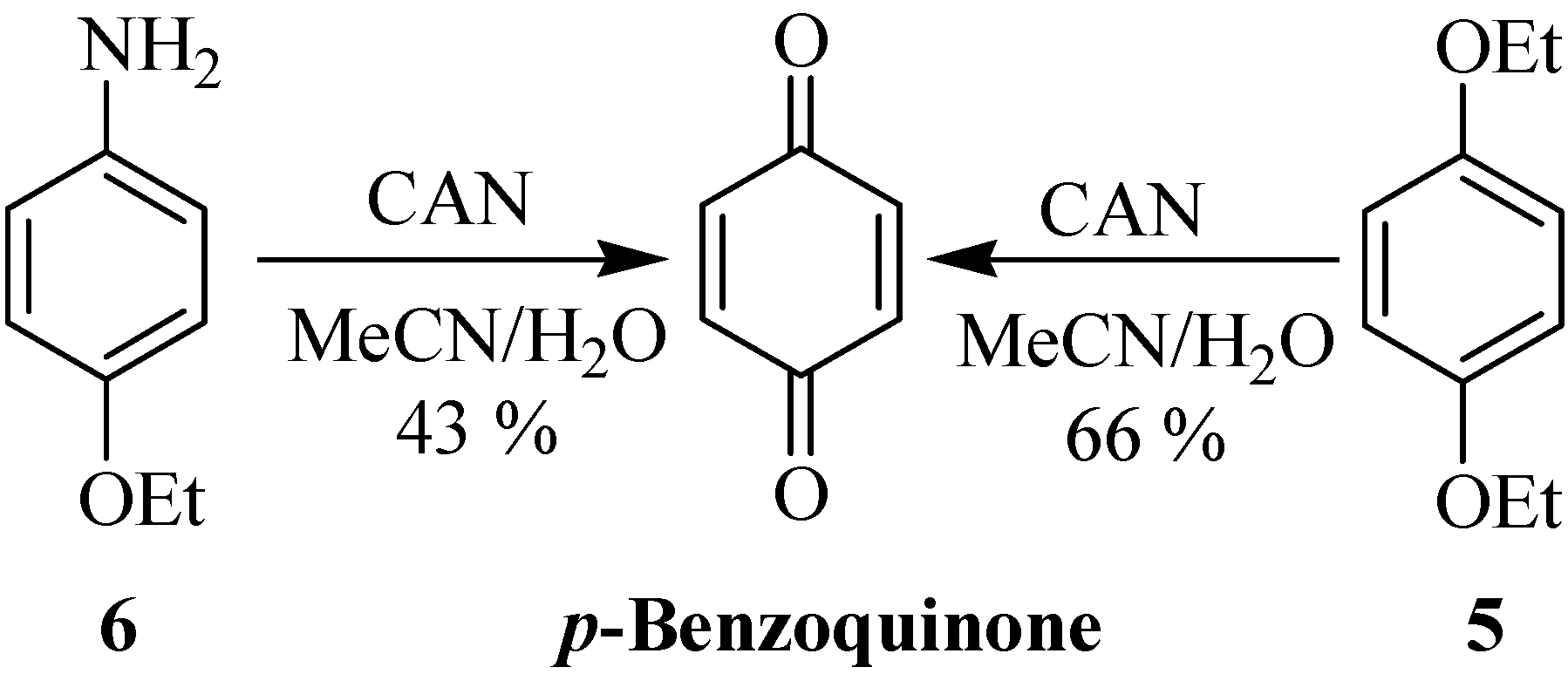
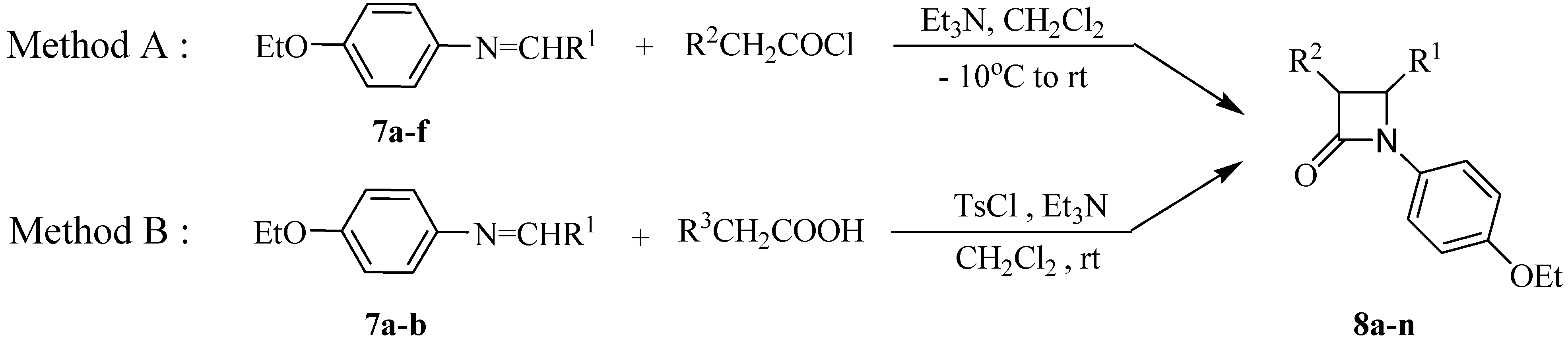

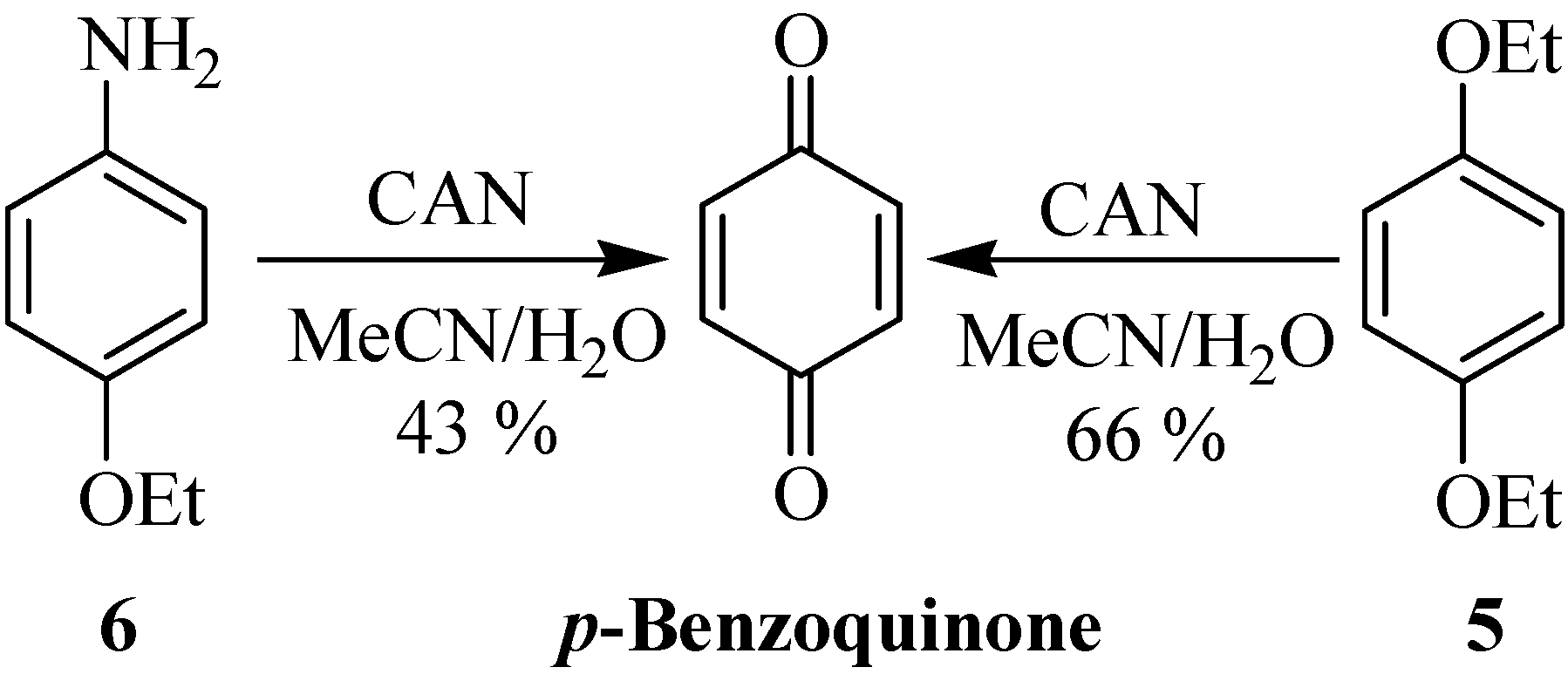{kind=link}
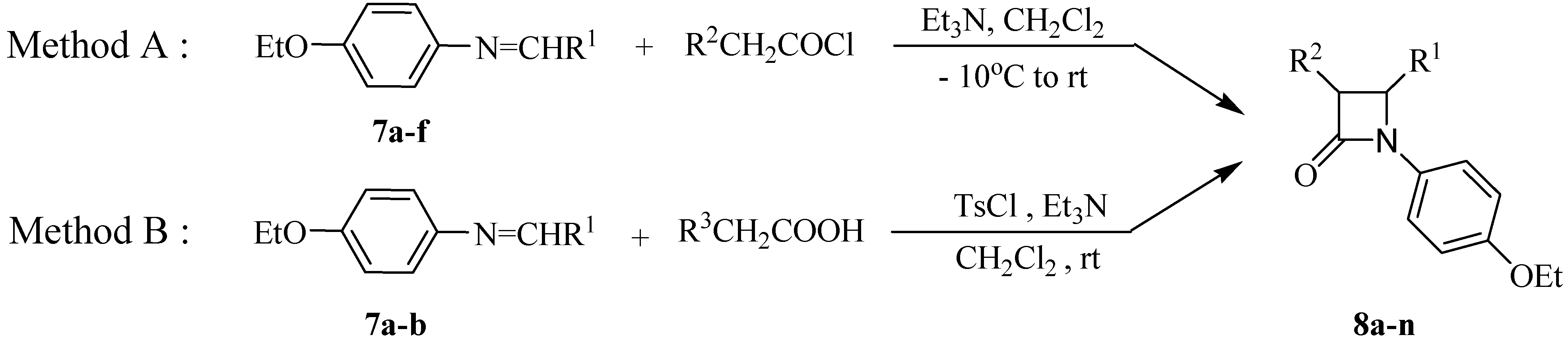{kind=link}
{kind=link}
{kind=link}