Introduction
We have been interested in the coordination behaviour of a broad range of neutral or anionic chelating ligands containing N and/or O donor atoms, such as β-diketones [
1],
cis-diols [
2], oximes [
2], 2-pyridyl alcohols [
3], diazabutadienes [
4] and pyrazolylpyridines [
5,
6]. Most of this work has centred on either high-valent d
0 transition metal complexes, mainly Mo
VI, as catalysts for olefin epoxidation [
2,
3,
4,
5], or trivalent lanthanide ions for optical applications [
1,
6]. Ligands derived from 2-(3-pyrazolyl)pyridine (
Figure 1) have proven to be particularly interesting for both applications. For example, the replacement of the coordinated water molecules in the tris(β-diketonate) complex Eu(NTA)
3(H
2O)
2 (NTA = 1-(2-naphthoyl)-3,3,3-trifluoroacetonate) with the ligand ethyl-[3-(2-pyridyl)-1-pyrazolyl]acetate modifies the luminescence properties of the parent triscomplex in a favourable way [
6]. In these complexes the pyrazole moiety is in its neutral form and is bidentate, with the metal ion coordinated to the pyridyl nitrogen and the pyrazole nitrogen. Deprotonation of the pyrazole unit could, in principle, allow several other coordination modes. For example,
endo-bidentate (
η2) coordination of pyrazolate ligands has been established for the lanthanides [
7]. However, in attempts to prepare an anionic complex of the type [Eu(BTA)
3(pypz)]
– (BTA = 1-benzoyl-3,3,3-trifluoroacetonate) with chelating
η2-pyrazolate coordination, we have reproducibly obtained a europium-free compound that contains sodium ions ligated by BTA anions and neutral pypzH molecules. The structure of this compound is striking because of the presence of an unusual centrosymmetric Na
+ hybrid tetramer. The bonding mode of pyrazolylpyridines to transition metals is very well known; in contrast we know little of their bonding mode to alkali-metal cations. In fact, information on the structural chemistry of alkali metal ions with nitrogen chelating ligands is still relatively scarce, especially for sodium and potassium [
8]. Herein, the solid state structure of the title compound is described and the sodium ion coordination environments are compared with existing {Na
xO
y} aggregates found in a search of the Cambridge Structural Database (CSD).
Figure 1.
The pyrazolylpyridine ligand used in this work [2-(3-pyrazolyl)pyridine, abbreviated pypzH].
Figure 1.
The pyrazolylpyridine ligand used in this work [2-(3-pyrazolyl)pyridine, abbreviated pypzH].
Results and Discussion
NaH was reacted with a THF solution of Eu(BTA)3(pypzH) in a 2:1 stoichiometry. In all instances, recrystallisation of the crude product from chloroform/n-hexane gave a colourless microcrystalline solid with the composition [Na4(pypzH)2(BTA)4]·3H2O (1). The number of water molecules was confirmed by carrying out thermogravimetric analysis under air (not shown). Thus, compound 1 undergoes a gradual weight loss of 4% from room temperature up to 150 ºC, which is attributed to the removal of three water molecules per formula unit. The next step takes place between 150 and 280 ºC (37.6%) and is attributed to the partial decomposition of the organic ligands. A final weight loss of 11.3% takes place between 375 and 485 ºC, leaving a residual mass of 39.7%. The TGA curve for compound 1 showed no steps characteristic of either “free” 1-benzoyl-3,3,3-trifluoroacetone or 2-(3-pyrazolyl)pyridine. Compound 1 was further characterised by IR, Raman, 1H and 13C-NMR spectroscopy (see experimental section for details).
Recrystallisation of
1 by slow diffusion of n-hexane into a chloroform solution of the crude product led to the formation of small single-crystals, which were ultimately formulated as [Na
4(pypzH)
2(μ
4-BTA)
2(μ
2-BTA)
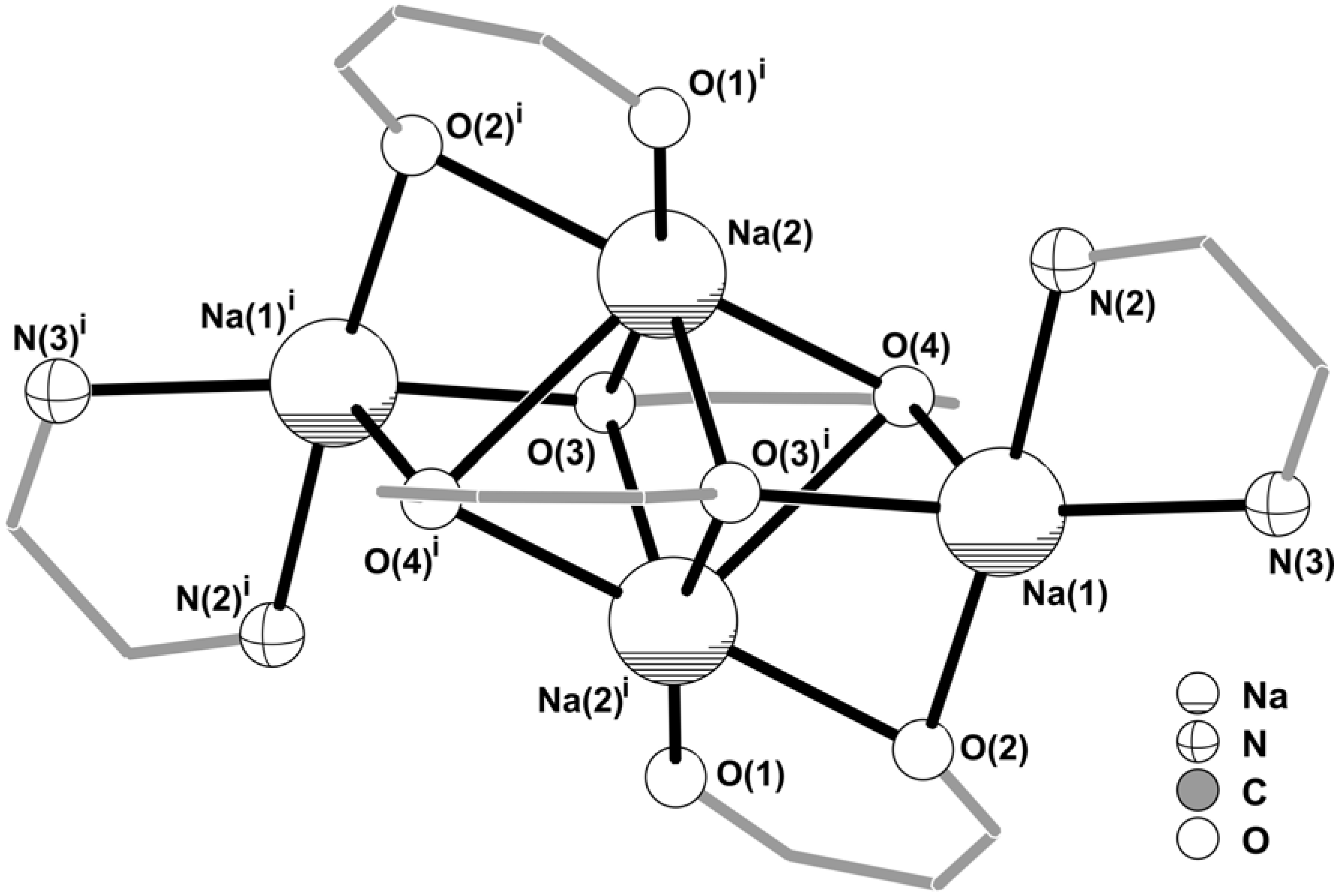
2] on the basis of single-crystal X-ray diffraction studies. The striking feature of this material resides in the presence of a centrosymmetric Na
+ hybrid tetramer, which fully occupies the contents of the triclinic unit cell. The crystal structure contains only two individual Na
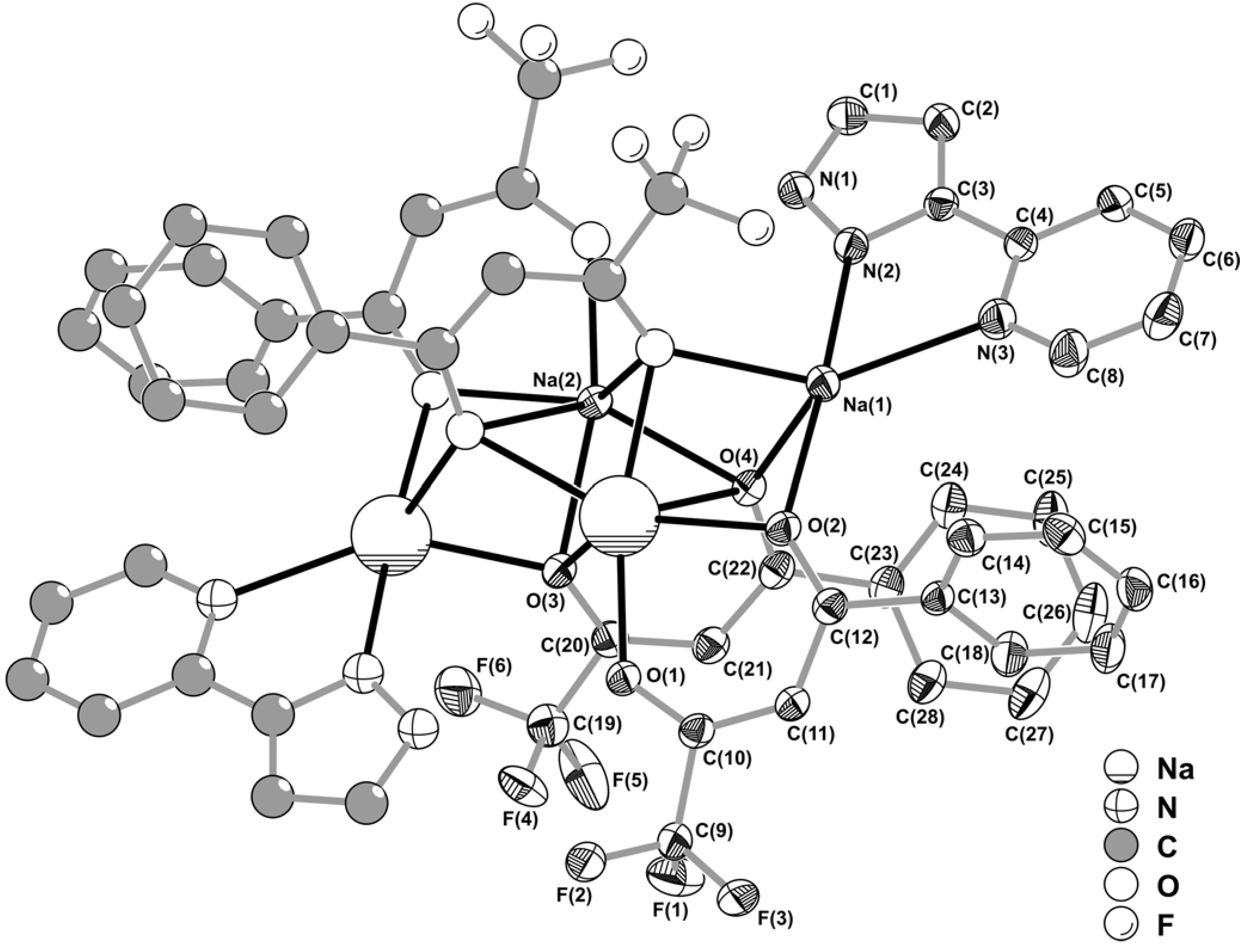
+ cations, Na(1) and Na(2), bound to the above mentioned organic residues, of which only two BTA and one pypzH are crystallographically independent (
Figure 2).
Figure 2.
Schematic representation of the neutral centrosymmetric tetrasodium [Na
4(pypzH)
2(μ
4-BTA)
2(μ
2-BTA)
2] molecular aggregate present in the crystal structure of
1, showing the labelling scheme for all non-hydrogen crystallographically independent atoms. Thermal ellipsoids are represented at the 30% probability level. Hydrogen atoms and part of the disordered –CF
3 terminal group [C(19)-F(4')-F(5')-F(6') – 29.2(2)% rate of occupancy] have been omitted for clarity. See
Table 1 for selected bond lengths and angles.
Figure 2.
Schematic representation of the neutral centrosymmetric tetrasodium [Na
4(pypzH)
2(μ
4-BTA)
2(μ
2-BTA)
2] molecular aggregate present in the crystal structure of
1, showing the labelling scheme for all non-hydrogen crystallographically independent atoms. Thermal ellipsoids are represented at the 30% probability level. Hydrogen atoms and part of the disordered –CF
3 terminal group [C(19)-F(4')-F(5')-F(6') – 29.2(2)% rate of occupancy] have been omitted for clarity. See
Table 1 for selected bond lengths and angles.
Table 1.
Selected bond lengths (Å) and angles (º) for the two distinct Na+ coordination environments forming the neutral tetrasodium [Na4(pypzH)2(μ4-BTA)2(μ2-BTA)2] molecular aggregate present in the crystal structure of 1.a
Table 1.
Selected bond lengths (Å) and angles (º) for the two distinct Na+ coordination environments forming the neutral tetrasodium [Na4(pypzH)2(μ4-BTA)2(μ2-BTA)2] molecular aggregate present in the crystal structure of 1.a
| Na(1)–O(2) | 2.453(3) | Na(2)–O(1)i | 2.234(3) |
| Na(1)–O(3)i | 2.333(3) | Na(2)–O(2)i | 2.395(3) |
| Na(1)–O(4) | 2.370(3) | Na(2)–O(3) | 2.332(3) |
| Na(1)–N(2) | 2.444(3) | Na(2)–O(3)i | 2.534(3) |
| Na(1)–N(3) | 2.511(4) | Na(2)–O(4) | 2.420(3) |
| | | Na(2)–O(4)i | 2.747(3) |
| | | | |
| O(2)–Na(1)–N(3) | 117.55(11) | O(1)i –Na(2)–O(2)i | 80.43(10) |
| O(3)i –Na(1)–O(2) | 83.60(9) | O(1)i –Na(2)–O(3) | 165.29(11) |
| O(3)i –Na(1)–O(4) | 77.65(10) | O(1)i –Na(2)–O(3)i | 84.81(11) |
| O(3)i –Na(1)–N(2) | 93.66(10) | O(1)i –Na(2)–O(4) | 121.57(11) |
| O(3)i –Na(1)–N(3) | 146.82(12) | O(1)i –Na(2)–O(4)i | 106.04(10) |
| O(4)–Na(1)–O(2) | 79.32(10) | O(2)i –Na(2)–O(3)i | 128.33(10) |
| O(4)–Na(1)–N(2) | 95.26(12) | O(2)i –Na(2)–O(4) | 153.09(11) |
| O(4)–Na(1)–N(3) | 128.79(12) | O(2)i –Na(2)–O(4)i | 73.21(9) |
| N(2)–Na(1)–O(2) | 174.33(12) | O(3)–Na(2)–O(2)i | 84.90(10) |
| N(2)–Na(1)–N(3) | 67.18(11) | O(3)–Na(2)–O(3)i | 105.43(10) |
| | | O(3)–Na(2)–O(4) | 72.25(9) |
| | | O(3)–Na(2)–O(4)i | 70.44(10) |
| | | O(3)i –Na(2)–O(4)i | 63.95(9) |
| | | O(4)–Na(2)–O(3)i | 73.01(9) |
| | | O(4)–Na(2)–O(4)i | 110.64(8) |
Na(2) is only coordinated to BTA residues which, despite appearing as typical O,O-chelating ligands via the ketone functional groups, exhibit remarkably different coordination geometries. One BTA moiety, chelated via O(1)/O(2) in a typical
syn,syn bidentate fashion (see
Figure 2 and
Figure 3), exhibits a bite angle with Na(2) of 80.43(10)º, which is significantly higher than expected, even though not chemically unrealistic: a search in the CSD (Version 5.27, November 2005) [
9,
10] reveals that the bite angle of chelating β-diketonates with Na
+ are normally found in the 62.2-86.7º range with a median value of 73.9º (from 63 entries). The rather large value of 80.43(10)º might be structurally explained by the
anti-interaction of O(2) with Na(1), as depicted in
Figure 2 and
Figure 3, which seems to dislocate the former atom, thus “opening” the O,O-chelate ring. The second BTA moiety not only chelates to Na(2) via O(3)/O(4) but also establishes a physical bridge between symmetry-related Na
+ centres (
Figure 2 and
Figure 3), imposing a Na(2)∙∙∙Na(2)
i separation of 2.952(3) Å [symmetry code: (i)
1‑x,
1‑y,
1‑z]. Therefore, with the organic molecule being structurally located between the two Na
+ cations, the coordination is best described as typical
skew,
skew-chelating fashion with two distinct bite angles, 72.25(9)º and 63.95(9)º, still well within the expected range for such types of interactions (see above). This
skew,
skew-O,O-chelating fashion with Na
+ cations, which leads to the formation of {Na
2O
4} clusters, has been rarely observed among crystal structures containing organic moieties. In fact, only a search in the CSD allowed a reliable and unambiguous survey for crystal structures containing this structural motif since the coordination environment of Na
+ cations, which are usually employed as counter-ions, is rarely described in detail. Hence, this type of {Na
2O
4} cluster has only been reported in a couple of alkali metal salt complexes of metallacrowns [
11,
12,
13], palladium [
14] and gallium [
15] complexes, and in very few organic salts [
16,
17].
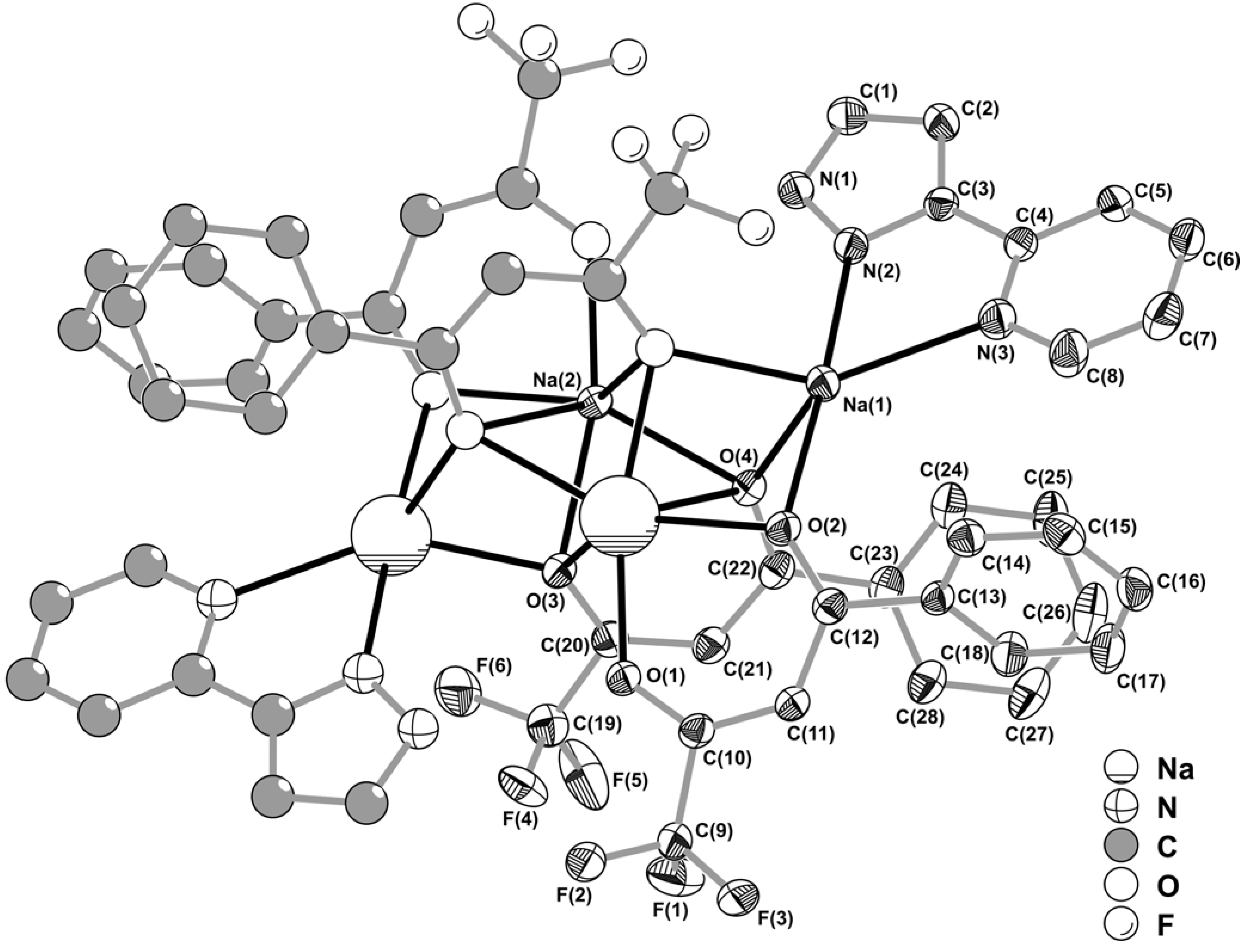
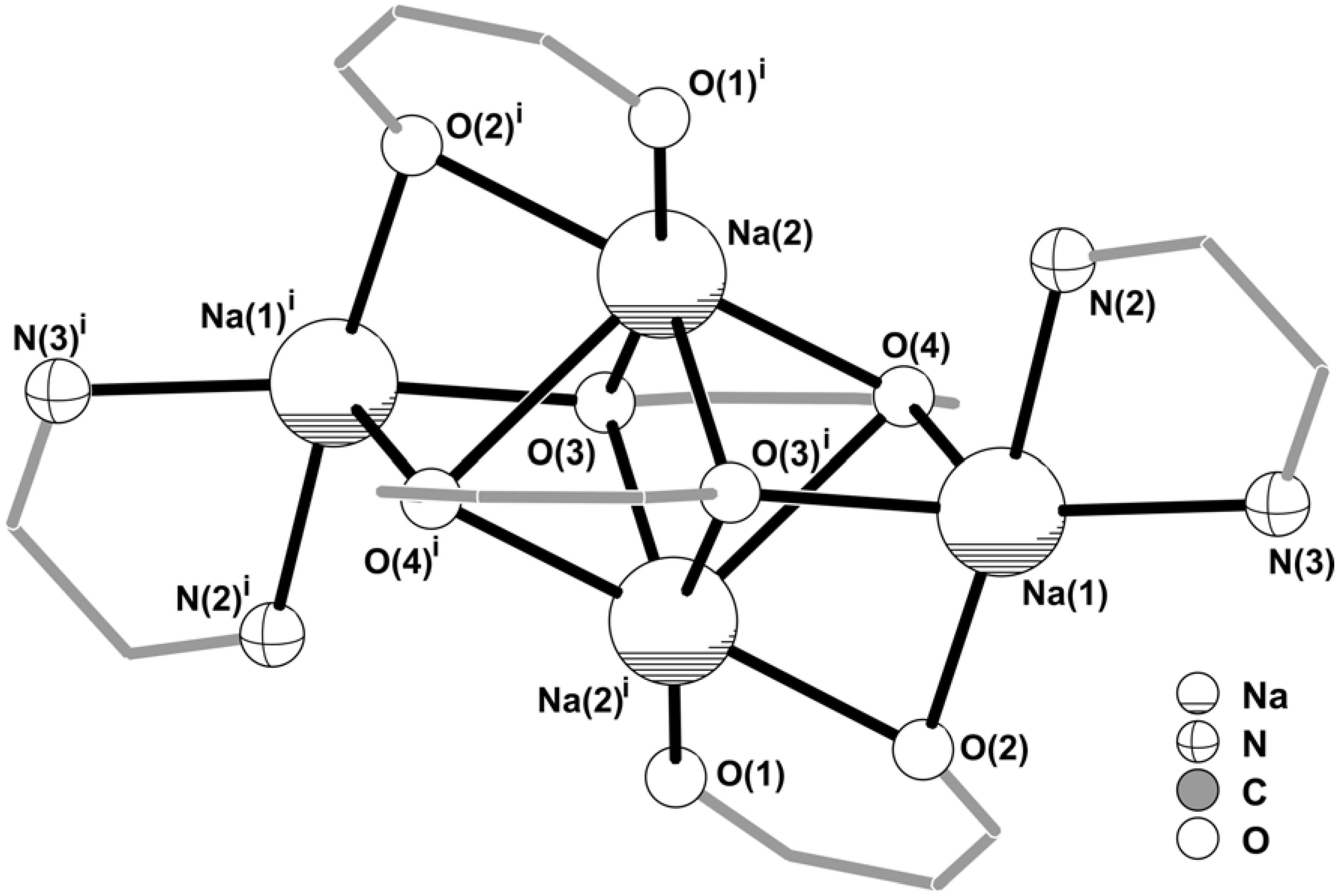
Figure 3.
Core of the centrosymmetric tetrasodium [Na4(pypzH)2(μ4-BTA)2(μ2-BTA)2] molecular aggregate emphasising the two highly distorted {NaN2O3} and {NaO6} coordination environments for the Na+ cations. Intermetallic distances for the tetrasodium core: Na(1)∙∙∙Na(2)i 3.315(2) Å; Na(1)∙∙∙Na(2) 3.702(2) Å; Na(2)∙∙∙Na(2)i 2.952(3) Å. For clarity, the coordinated pyrazolylpyridine and BTA organic ligands have been replaced by 5- and 6- membered rings. Symmetry transformation used to generate equivalent atoms: (i) 1‑x, 1‑y, 1‑z.
Figure 3.
Core of the centrosymmetric tetrasodium [Na4(pypzH)2(μ4-BTA)2(μ2-BTA)2] molecular aggregate emphasising the two highly distorted {NaN2O3} and {NaO6} coordination environments for the Na+ cations. Intermetallic distances for the tetrasodium core: Na(1)∙∙∙Na(2)i 3.315(2) Å; Na(1)∙∙∙Na(2) 3.702(2) Å; Na(2)∙∙∙Na(2)i 2.952(3) Å. For clarity, the coordinated pyrazolylpyridine and BTA organic ligands have been replaced by 5- and 6- membered rings. Symmetry transformation used to generate equivalent atoms: (i) 1‑x, 1‑y, 1‑z.
The structural arrangement described above leads to the formation of a centrosymmetric [Na
2(μ
4-BTA)
2(μ
2-BTA)
2]
2– anion (
Figure 3) in which the core can be formally described by a {Na
2O
6} cluster, only found in three organic-inorganic structures reported to date containing Na
+ cations [
11,
12,
13]. The spatial distribution of the four BTA ligands creates three coordinatively vacant positions formed by O(2), O(3) and O(4) which are instead interacting with Na(1) (
Figure 2), thus forming a {Na
4O
6} cluster (
Figure 3) which, to the best of our knowledge, is unprecedented for Na
+. Moreover, this topological feature is also completely new for group 1 metal centres, and only finds a structural parallel for a strontium complex very recently reported by Davies
et al. [
18]. Externally to this {Na
4O
6} cluster pyrazolylpyridine organic molecules are N,N-chelated to Na(1) with a bite angle of 67.18(11)º which is in good agreement with statistical data retrieved from the CSD for a similar structural arrangement (values in the 57.7-89.0º range, from 160 entries with a median of 71.6º).
As usually happens for Na
+ cations, the local coordination environments of Na(1) and Na(2) in
1, {NaN
2O
3} and {NaO
6} respectively, are highly irregular and no polyhedral resemblance can be assigned for either centre. As summarised in
Table 1, the Na–O distances are found in the 2.333(3)-2.453(3) Å and 2.234(3)-2.747(3) Å ranges for Na(1) and Na(2). A search in the CSD for typical Na–O interactions with β-diketonates residues reveals that the values are usually found in the 2.27-2.84 Å range, with a median of 2.52 Å (from 63 entries). Thus, even though the majority of the registered Na–O distances for
1 are in good agreement with the expected values, the Na(2)–O(1) distance is shorter than expected. Taking into account the above discussion about the rather large O(1)–Na(2)–O(2) bite angle, we may conclude that the
syn,
anti-bridge of O(2) between neighbouring Na(1) and Na(2) (
Figure 2) imposes significant strain in the β-diketonate group of the BTA residue, thus forcing O(1) to move towards Na(2). The Na(1)–N distances registered for
1 (average value of 2.48 Å – see
Table 1) are also typical of coordinated aromatic ligands with N-donor atoms (90 entries in the CSD, median value of 2.49 Å for values found in the 2.31-3.12 Å range).
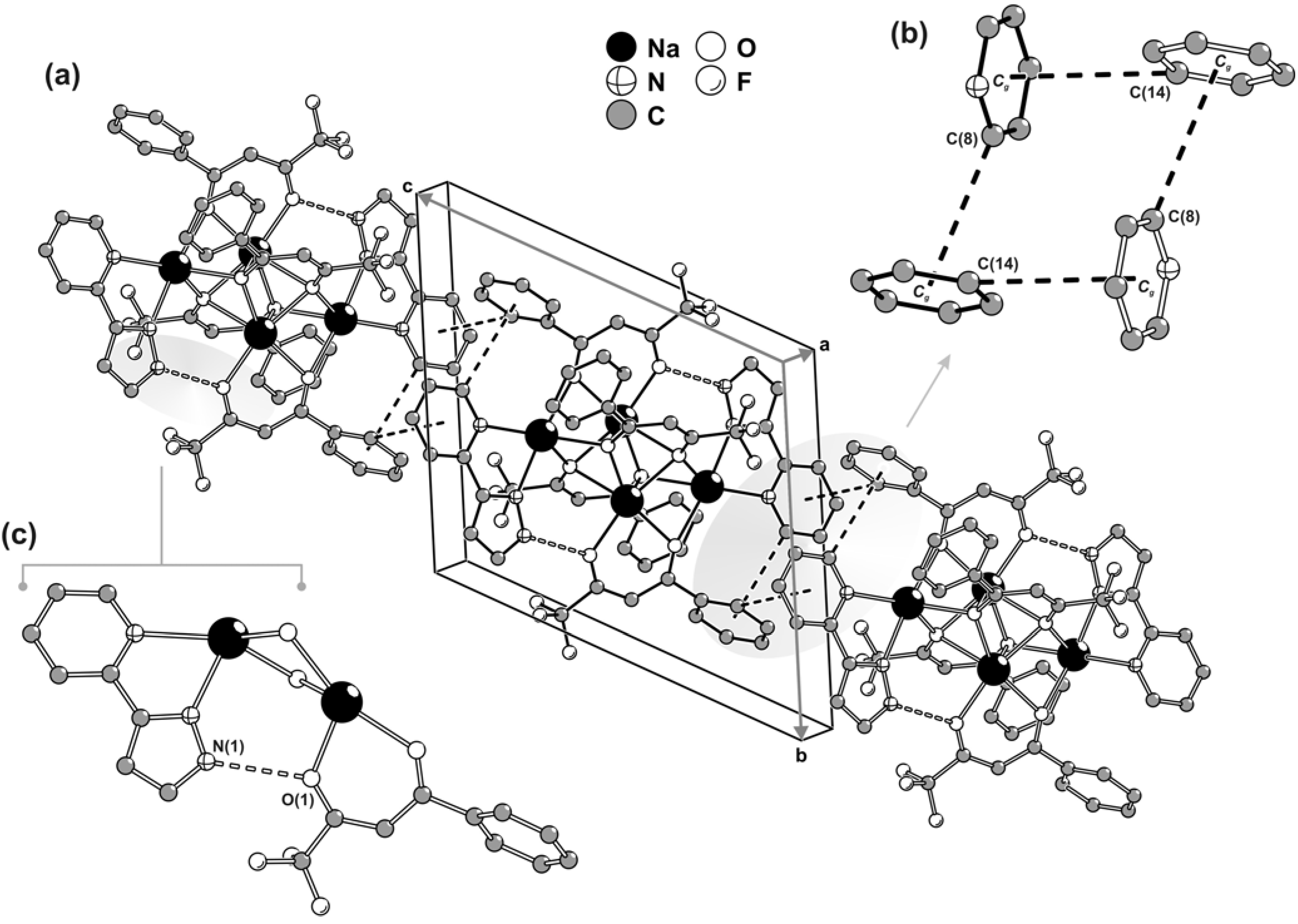
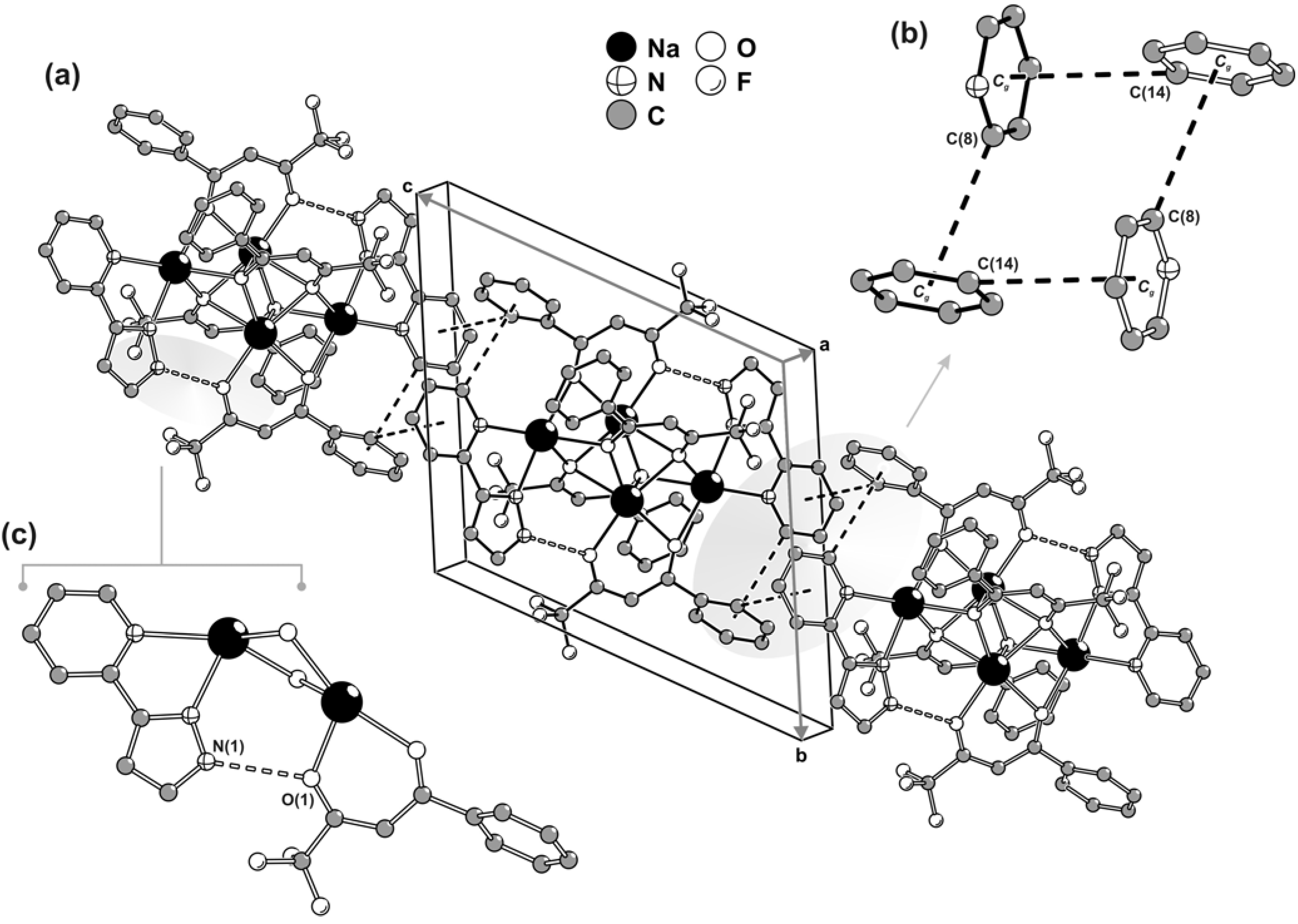
Surprisingly, even though all of the organic residues contain aromatic rings, the crystal packing of individual centrosymmetric tetrasodium [Na
4(pypzH)
2(μ
4-BTA)
2(μ
2-BTA)
2] molecular moieties is not mediated by the expected
a priori π-π interactions. Close packing is instead essentially driven through geometrical aspects (
i.e., the need to efficiently fill in the space) combined with weak C–H∙∙∙π interactions. Along the [001] direction of the unit cell, aromatic rings of one pypzH molecule and one BTA residue belonging to neighbouring tetrasodium complexes are engaged in a cooperative, and rather intriguing, rectangular arrangement of C–H∙∙∙π interactions as depicted in
Figure 4a,b. Even though the C(8)–H(8)∙∙∙
Cg [D∙∙∙A of 3.615(2) Å] interaction approaches linearity with the angle being
ca. 160º, the C(14)–H(14)∙∙∙
Cg interaction [D∙∙∙A of 3.737(2) Å] is significantly weaker with the DHA angle being only
ca. 134º. This occurs because, structurally, the interaction with the neighbouring aromatic ring is divided between C(14) and C(15), but it is slightly more favourable for the former atom [as C(15)–H(15)∙∙∙
Cg is 3.858(2) Å with <DHA being
ca. 122º], with this being the only interaction represented in
Figure 4a,b.
In addition to the aforementioned C–H∙∙∙π interactions, the material also contains classical strong hydrogen bonds, even though these do not directly contribute to the packing driving forces. In fact, within each tetranuclear [Na
4(pypzH)
2(μ
4-BTA)
2(μ
2-BTA)
2] complex, the N–H groups of pypzH are engaged in strong hydrogen bonds with the neighbouring β-diketonate groups as depicted in
Figure 4c.
Experimental
General
Elemental analysis was performed at the University of Aveiro. Thermogravimetric analysis (TGA) was carried out using a Shimadzu TGA-50 system at a heating rate of 5 ºC min–1 under a static atmosphere of air. IR spectra were obtained as KBr pellets using a FTIR Mattson-7000 infrared spectrophotometer. Raman spectra were collected on a Bruker RFS100/S FT instrument (Nd:YAG laser, 1064 nm excitation, InGaAs detector). 1H and 13C-NMR spectra were measured in solution using a Bruker CXP 300 spectrometer. Chemical shifts are quoted in parts per million from tetramethylsilane.
Anhydrous tetrahydrofuran (THF) and 60% sodium hydride dispersed in mineral oil were purchased from Aldrich and used as received. 2-(3-pyrazolyl)pyridine (pypzH) [
19] and Eu(BTA)
3(pypzH) [
1,
20] were prepared using literature procedures, and characterised by elemental analysis, FTIR,
1H and
13C-NMR spectroscopies.
[Na4(pypzH)2(μ4-BTA)2(μ2-BTA)2]·3H2O (1)
A suspension of NaH (18 mg, 0.75 mmol) in THF (5 mL) was added to a solution of Eu(BTA)3(pypzH) (350 mg, 0.37 mmol) in THF (10 mL). The mixture was stirred under nitrogen, at room temperature, for 7 h, and the liberation of hydrogen was observed. The solution was then filtered off and evaporated to dryness. The resultant solid was washed with n-hexane (10 mL) and diethyl ether (10 mL), and finally dried under reduced pressure. The white solid was recrystallised from CHCl3/n-hexane to give compound 1 (121 mg, 50%). Anal. Calcd for C56H38F12N6Na4O8·3H2O (1296.9): C, 51.86; H, 3.42; N, 6.48. Found: C, 52.17; H, 3.16; N, 6.42%. Selected IR (KBr): ν = 3434m, 3194m, 3066w, 2994w, 2935w, 1631vs, 1599m, 1576s, 1532vs, 1430m, 1361m, 1286vs, 1240m, 1181vs, 1151s, 1124s, 1088w, 1075w, 1058w, 1022w, 957w, 942w, 807w, 793w, 763s, 711s, 632m, 576m cm–1. Selected Raman: 3154w, 3056m, 1644m, 1600vs, 1573m, 1518s, 1475m, 1450m, 1442m, 1362m, 1317m, 1303m, 1282s, 1242m, 1157m, 1058m, 1001vs, 960m, 946m, 939m, 717m, 633m, 618m, 406w, 369m, 284m, 192m, 168m cm–1. 1H-NMR (300.13 MHz, 25 ºC, acetone-d6): δ = 12.37 (br, N–H), 8.60 (d, J = 4.58 Hz, pypzH), 8.04-7.92 (br, pypzH), 7.88-7.80 [m, phenyl-H (BTA)], 7.73 [br, pypzH and phenyl-H (BTA)], 7.45-7.36 [m, phenyl-H (BTA)], 7.31-7.27 (m, pypzH), 6.92 (d, J = 1.79 Hz, pypzH), 6.05 [s, CH (BTA)] ppm. 13C-NMR (75.47 MHz, 25 ºC, acetone-d6): δ = 187.5, 150.0, 143.0, 137.1, 130.8, 128.7, 127.6, 120.2, 104.0, 88.1 ppm.
Single-crystal X-ray diffraction
A suitable single-crystal of [Na
4(pypzH)
2(BTA)
4] (
1) was mounted on a glass fibre using perfluoropolyether oil [
21]. Data were collected at 180(2) K on a Nonius Kappa charge-coupled device (CCD) area-detector diffractometer (Mo K
α graphite-monochromated radiation, λ = 0.7107 Å), equipped with an Oxford Cryosystems cryostream and controlled by the Collect software package [
22]. Images were processed using the software packages of Denzo and Scalepack [
23], and data were corrected for absorption by the empirical method employed in Sortav [
24,
25]. The structure was solved by the direct methods of SHELXS-97 [
26] and refined by full-matrix least squares on
F2 using SHELXL-97 [
27]. All non-hydrogen atoms were directly located from difference Fourier maps and refined with anisotropic displacement parameters. Information concerning crystallographic data collection and structure refinement details are summarised in
Table 2.
Hydrogen atoms attached to carbon were located at their idealised positions using the
HFIX 43 instruction in SHELXL [
27], and included in subsequent refinement cycles in riding-motion approximation with isotropic thermal displacement parameters (
Uiso) fixed at 1.2 times
Ueq of the carbon atom to which they were bonded. Even though the hydrogen atom bound to the uncoordinated nitrogen of the crystallographically independent pyrazolylpyridine molecule was visible in difference Fourier maps during the last stages of the structural refinement, it was included in the final model using a similar procedure to that applied for the remaining hydrogen atoms [
HFIX 43 instruction with
Uiso = 1.2 ×
Ueq(N)].
Table 2.
Crystal and structure refinement data for [Na4(pypzH)2(μ4-BTA)2(μ2-BTA)2].
Table 2.
Crystal and structure refinement data for [Na4(pypzH)2(μ4-BTA)2(μ2-BTA)2].
| Formula | C56H38F12N6Na4O8 |
| Formula weight | 1242.88 |
| Crystal system | Triclinic |
| Space group | Pī |
| a/Å | 9.3348(19) |
| b/Å | 12.907(3) |
| c/Å | 13.694(3) |
| α/º | 115.96(3) |
| β/º | 101.85(3) |
| γ/º | 99.46(3) |
| Volume/Å3 | 1389.9(5) |
| Z | 1 |
| Dc/g cm–3 | 1.485 |
| μ(Mo-Kα)/mm–1 | 0.153 |
| F(000) | 632 |
| Crystal size/mm | 0.10×0.10×0.05 |
| Crystal type | Colourless blocks |
| θ range | 3.53 to 24.71 |
| Index ranges | -10 ≤ h ≤ 10
-15 ≤ k ≤ 15
-16 ≤ l ≤ 12 |
| Data completeness to θ = 24.71º | 98.9% |
| Reflections collected | 12050 |
| Independent reflections | 4681 (Rint = 0.0661) |
| Final R indices [I>2σ(I)]a,b | R1 = 0.0618, wR2 = 0.1543 |
| Final R indices (all data)a,b | R1 = 0.0995, wR2 = 0.1812 |
| Weighting schemec | m = 0.0330, n = 6.4200 |
| Largest diff. peak and hole | 0.460 and –0.325 eÅ–3 |
One –CF3 terminal group of a BTA residue was found to be severely affected by thermal disorder, with the corresponding fluorine atoms giving rise to prolates when treated anisotropically (i.e., large max/min ratio for the anisotropic displacement parameters). Instead, this group was modelled over two distinct crystallographic positions with variable rates of occupancy, which ultimately refined to 70.8(2)% and 29.2(2)%. Moreover, in order to ensure a chemically reasonable geometry for the disordered terminal –CF3 group the C–F and F∙∙∙F distances were restrained to common (but refineable) 1.31(1) and 2.01(1) Å values, respectively.
Even though crystal data was collected up to a resolution of 0.77 Å (i.e., θ = 27.48º), the crystal was a very small block and diffracted rather weakly at high angles. Applying a cut-off at 0.85 Å resolution (θ = 24.71º) resulted in a good number of reflections observed at the 2σ level (see
Table 1 – the data-to-parameter ratio is greater than 11) and a good merging reliability factor. Collection at higher angle is likely only to be possible using a rotating-anode source or synchrotron radiation. The last difference Fourier map synthesis showed the highest peak (0.460 eÅ
–3) located at 0.97 Å from F(1), and the deepest hole (–0.325 eÅ
–3) at 0.91 Å from N(2).
Crystallographic data (excluding structure factors) for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-606059. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 2EZ, U.K. (FAX: (+44) 1223 336033; e-mail:
deposit@ccdc.cam.ac.uk).



{kind=link}
{kind=link}
{kind=link}
{kind=link}