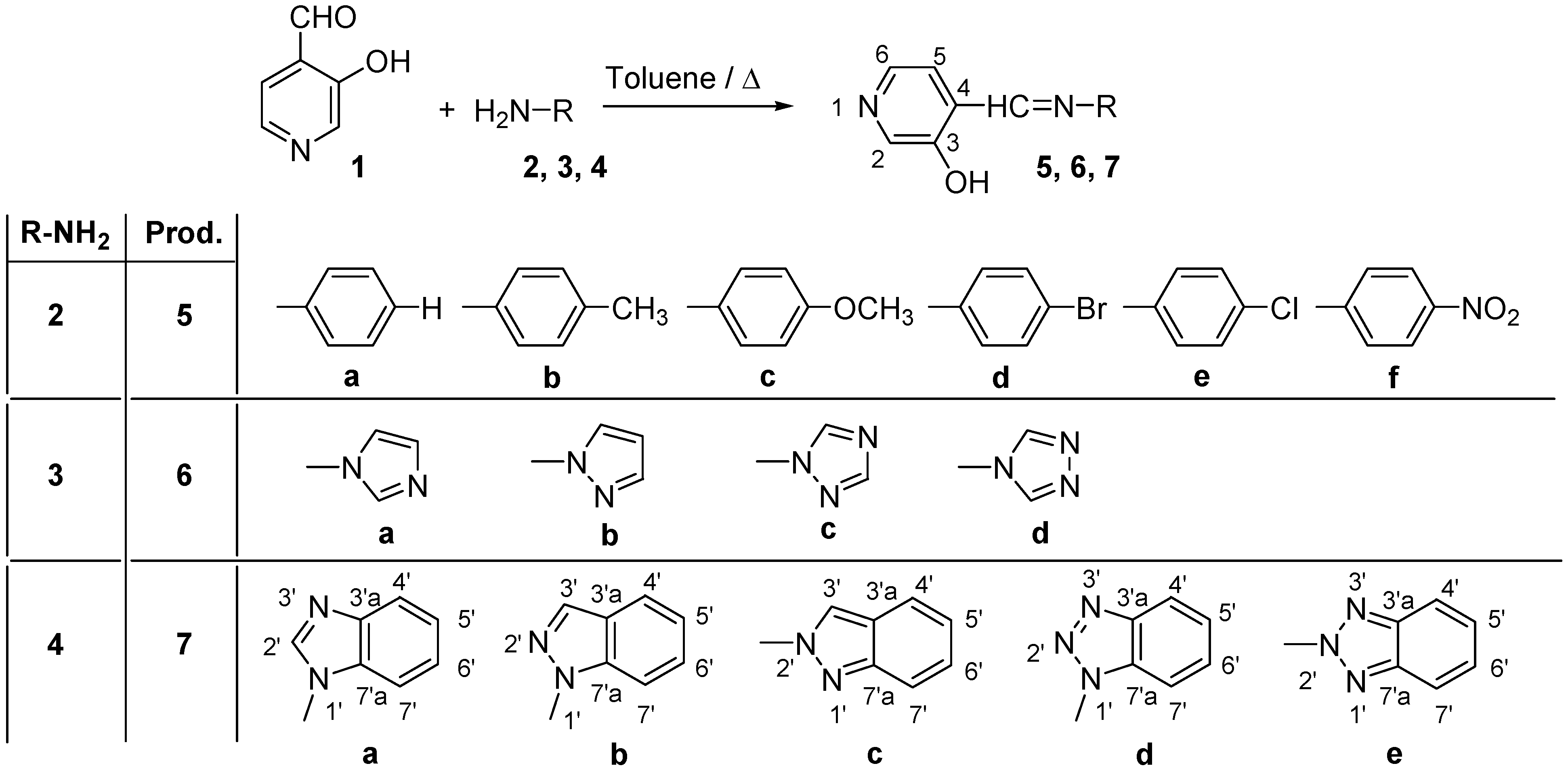
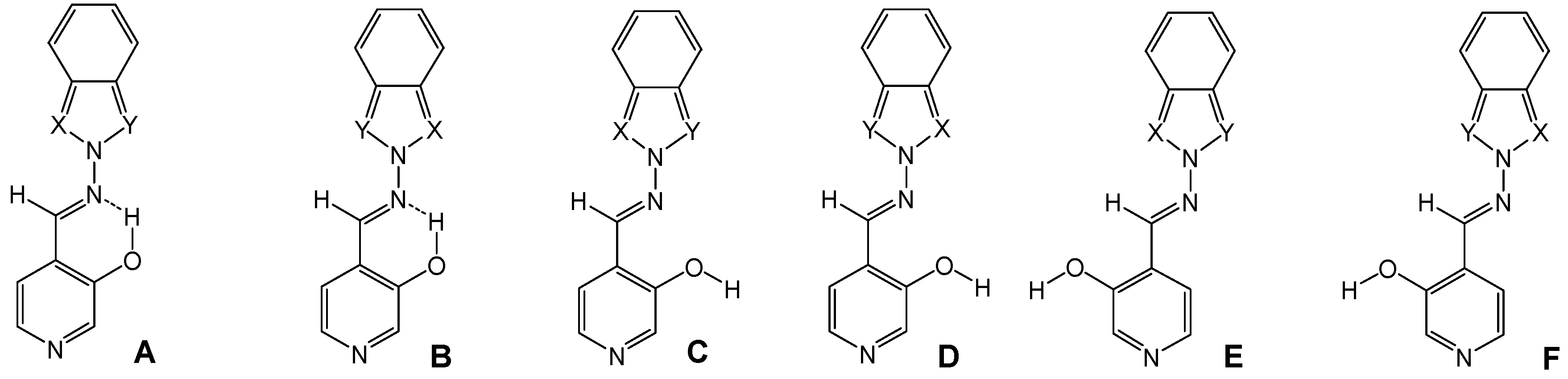
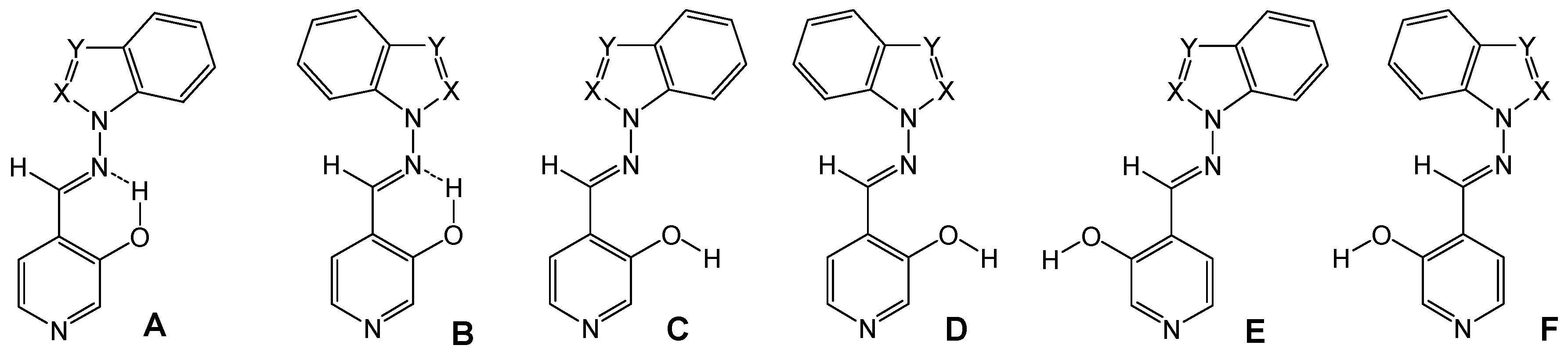
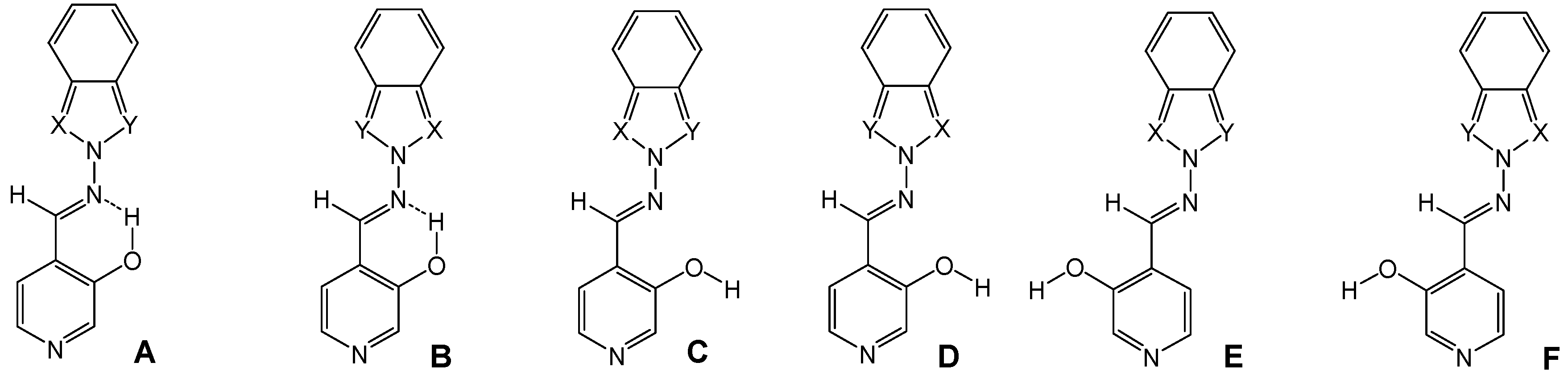
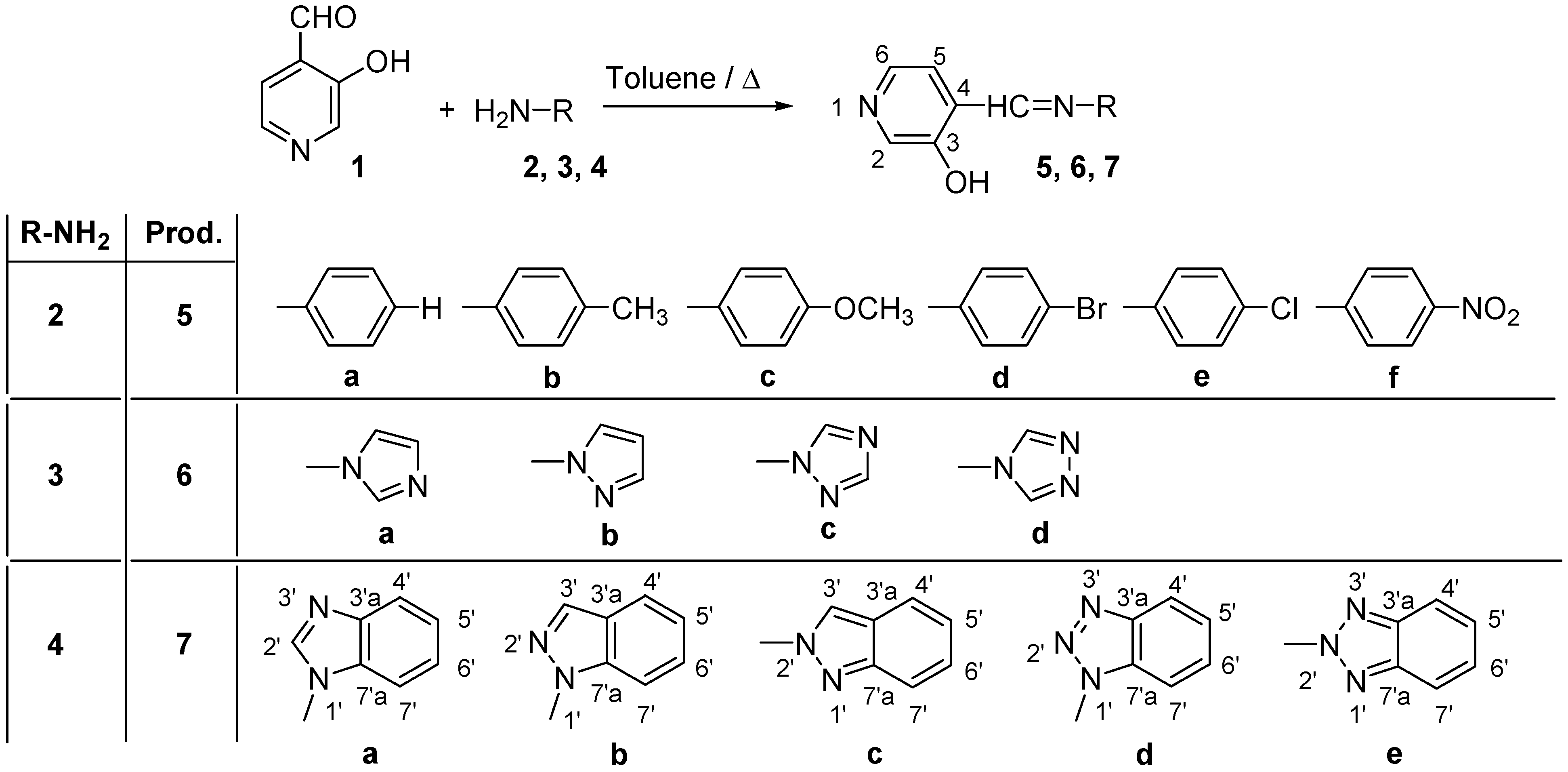
Synthesis of compounds 7a-7e
The compounds were prepared in nearly quantitative yields (85-90%) by refluxing equimolar amounts of 1 [
6] and the corresponding amines
4a-
4e [
23] in toluene during 7 h and then stirring overnight.
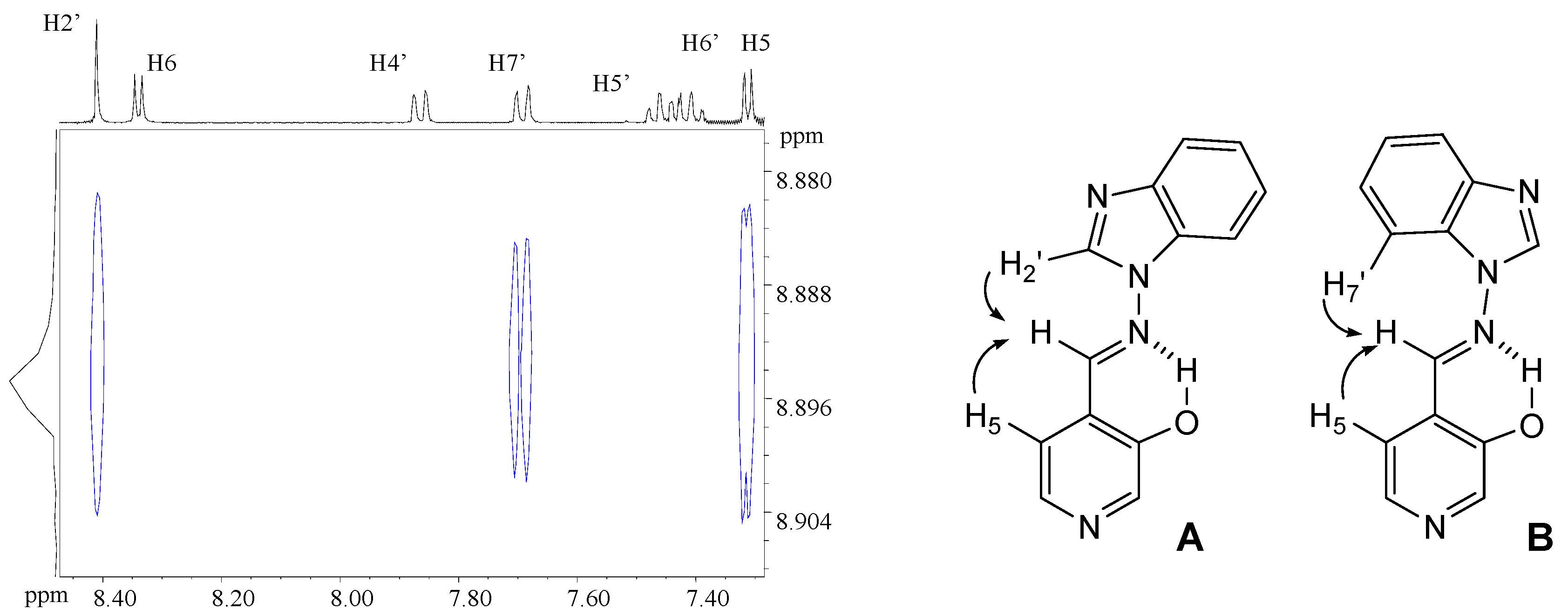
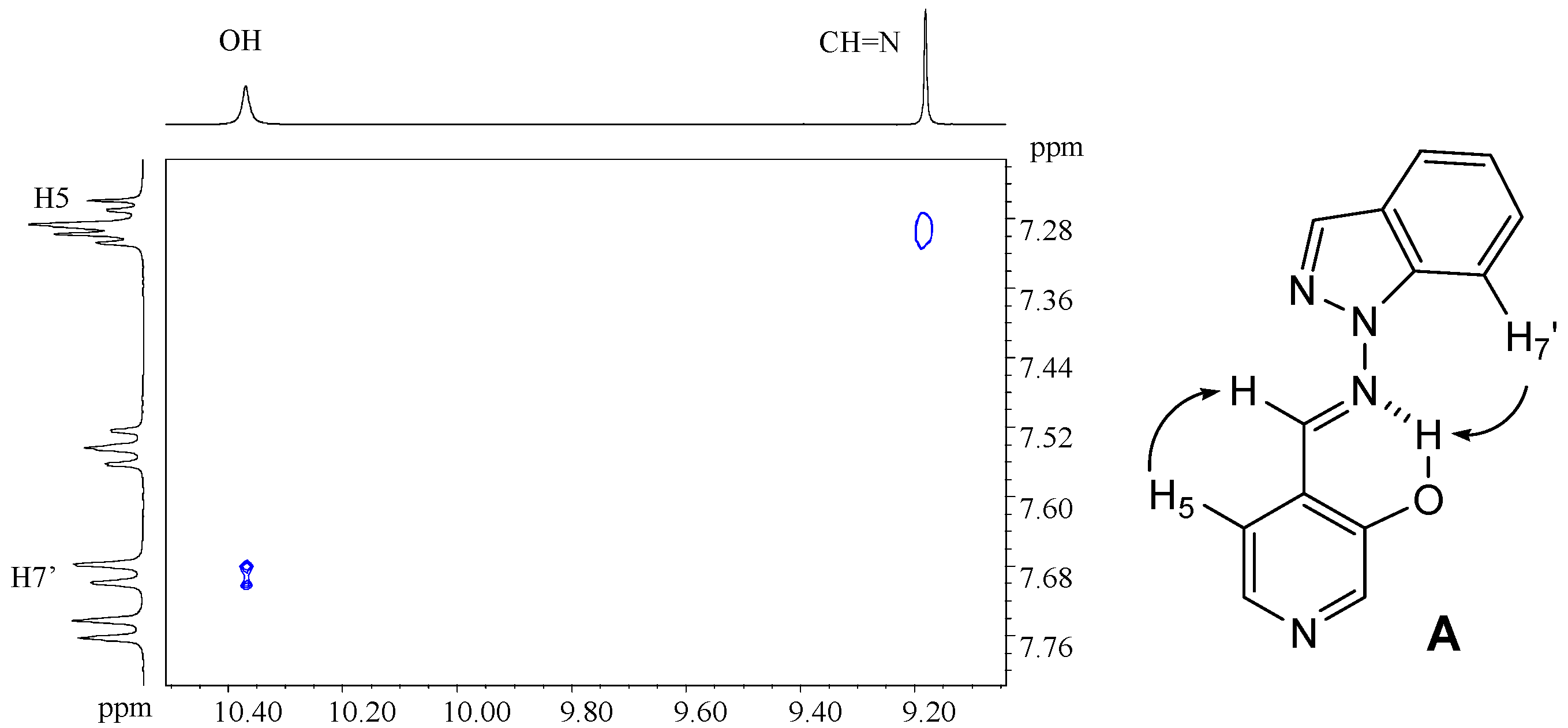
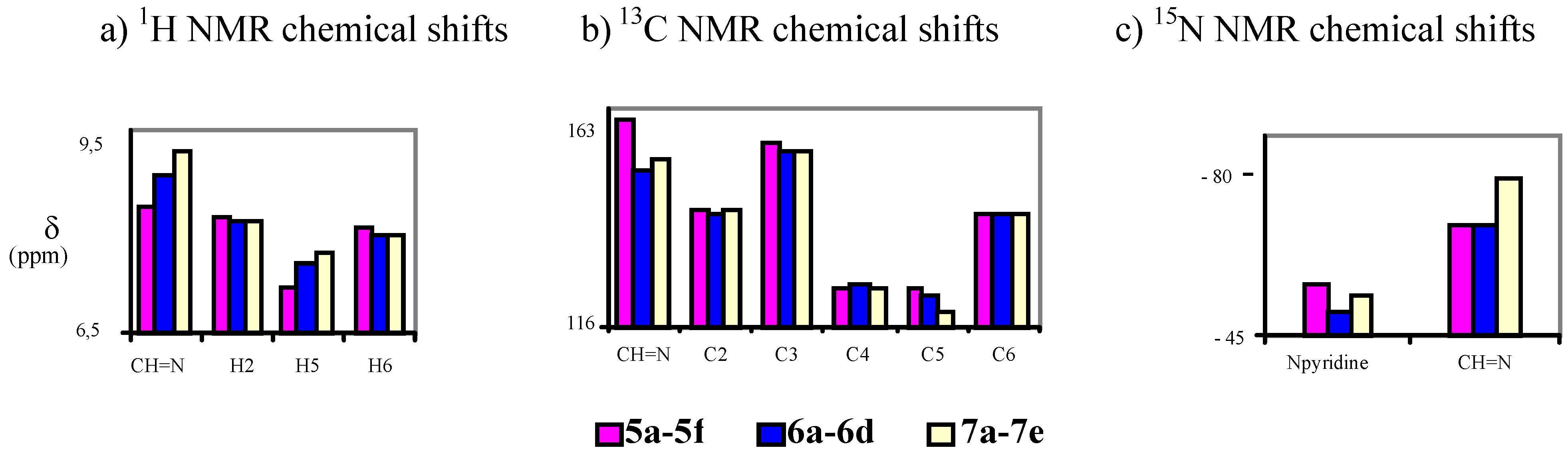
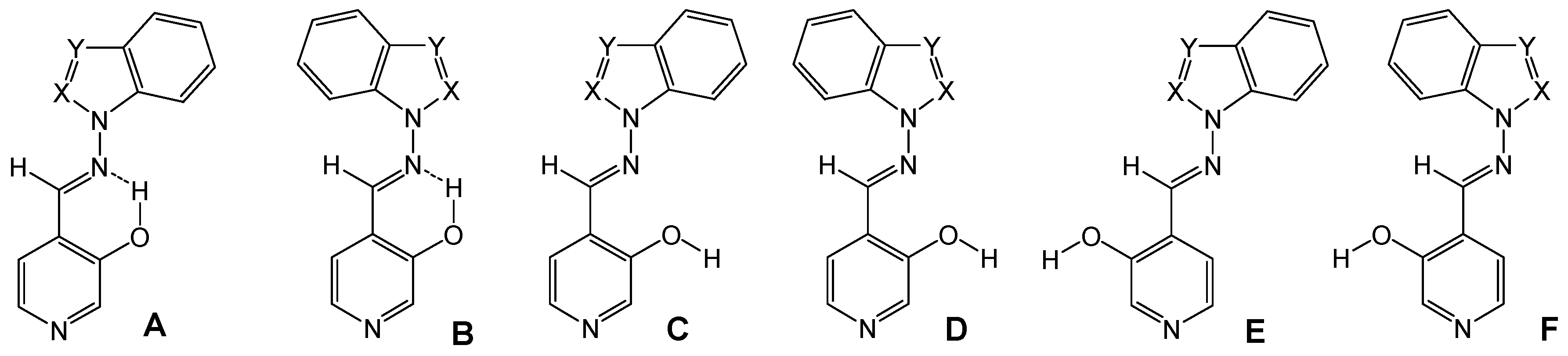
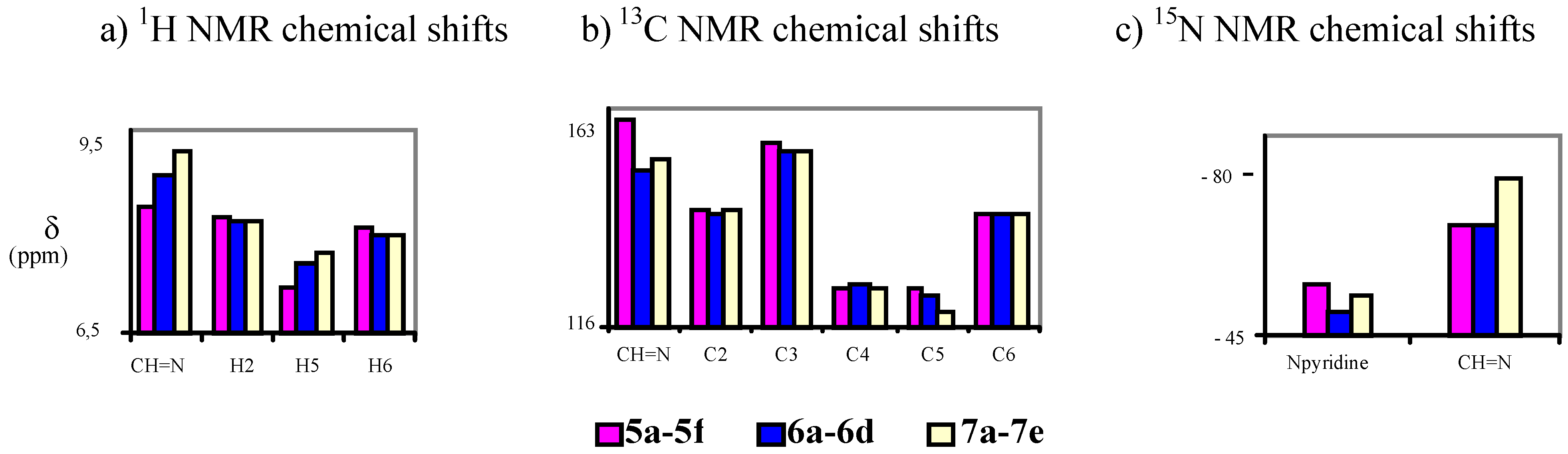
4-[(E)-(1H-benzimidazol-1-ylimino)methyl]pyridin-3-ol (7a). TLC [Rf 0.44 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (from C2H5OH), mp 239 ºC (microscope) and 239.1 ºC with decomposition at 255.5 (DSC); Anal. Calcd for C13H10N4O: C, 65.54; H, 4.23; N, 23.52. Found: C, 64.48; H, 4.33; N, 23.42; 1H-NMR (DMSO-d6) δ: 10.80 (s vbr, 1H, OH), 9.29 (s, 1H, CH=N), 9.07 (s, 1H, C2’-H), 8.38 (s br, 1H, C2-H), 8.18 (d br, 1H, 3J5-6= 4.8, C6-H), 7.84 (d, 1H, C5-H), 7.79 (d, 1H, 3J6’-7’= 8.0, C7’-H), 7.72 (d, 1H, 3J5’-4’= 7.9, C4’-H4), 7.40 (t, 1H, C6’-H), 7.31 (t, 1H, C5’-H); 13C-NMR (DMSO-d6) δ: 152.6 (C3), 146.9 (CH=N, 1J=169.6), 141.8 (C3’a), 140.5 (C6, 1J=181.2, 3J=11.9), 139.9 (C2, 1J=179.2, 3J=11.3), 137.0 (C2’, 1J=214.0), 132.1 (C7’a), 125.6 (C4), 124.0 (C6’, 1J=161.0, 3J=7.8), 123.0 (C5’, 1J=160.4, 3J=7.8), 120.1 (C4’, 1J=161.8, 3J=8.0), 119.4 (C5, 1J=161.9), 110.7 (C7’, 1J=165.0, 3J=8.3); 15N-NMR (DMSO-d6) δ: -181.2 (N1’), -134.4 (N3’), -70.3 (CH=N), -53.2 (N1); 1H-NMR (CDCl3) δ: 10.20 (s br, 1H, OH), 8.90 (s, 1H, CH=N), 8.58 (s br, 1H, C2-H), 8.41 (s, 1H, C2’-H), 8.34 (d , 1H, 3J5-6= 5.0, C6-H), 7.86 (ddd, 1H, 3J5’-4’= 8.0, 4J6’-4’= 1.3, 5J7’-4’= 0.7, C4’-H), 7.69 (ddd, 1H, 3J6’-7’= 7.9, 3J5’-7’= 1.3, C7’-H), 7.46 (ddd, 1H, C6’-H), 7.41 (ddd, 1H, C5’-H), 7.31 (d, 1H, C5-H); 13C-NMR (CDCl3) δ: 152.9 (C3), 152.1 (CH=N), 142.6 (C3’a), 141.4 (C6), 141.3 (C2), 135.9 (C2’), 131.0 (C7’a), 125.1 (C6’), 124.2 (C5’), 123.5 (C5), 121.9 (C4), 121.4 (C4’,), 110.2 (C7’); 15N-NMR (CDCl3) δ:-186.5 (N1’); 13C-CP/MAS NMR δ: 154.0 (C3), 140.7 (CH=N), 140.7 (C3’a), 140.1 (C6), 138.2 (C2), 137.3 (C2’), 133.7 (C7’a), 128.1 (C4), 122.8 (C6’), 122.0 (C5’), 119.1 (C4’), 118.6 (C5), 113.0 (C7’); 15N- CP/MAS NMR δ: -177.7 (N1’), -135.1 (N3’), -87.1 (CH=N), -67.6 (N1).
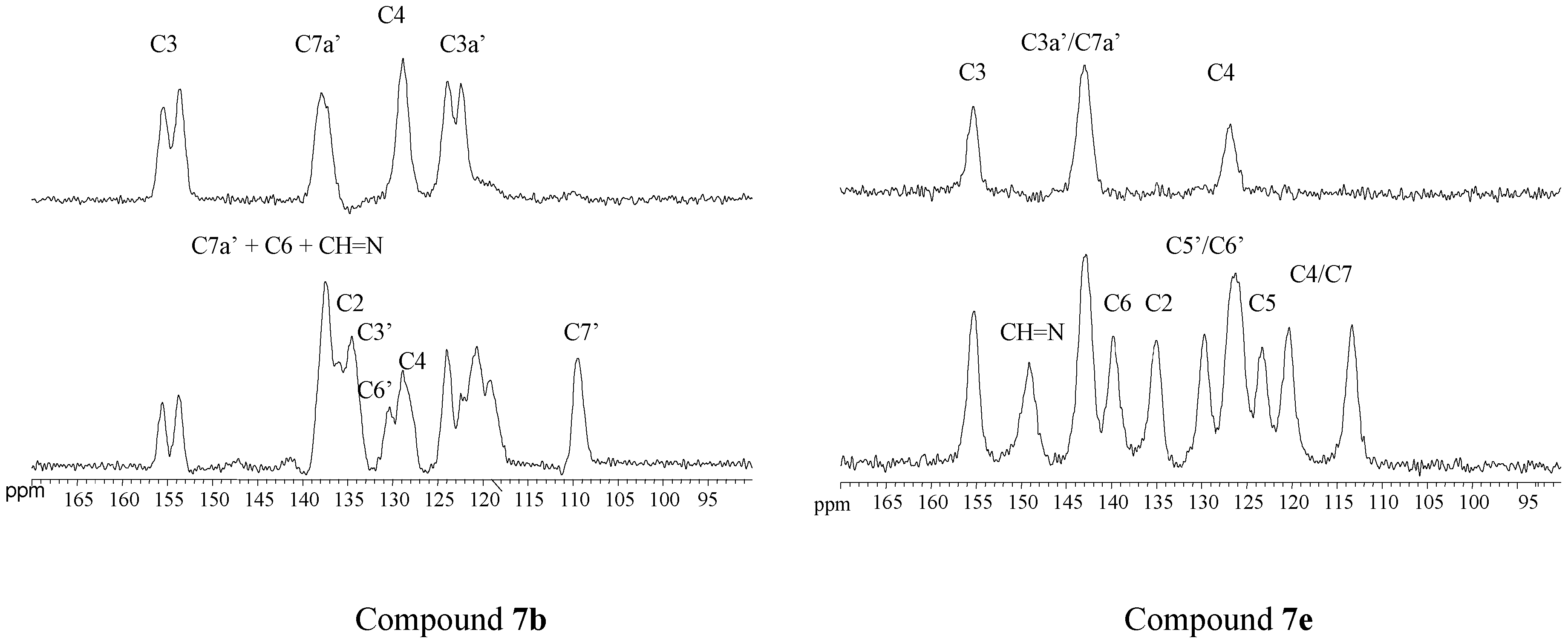
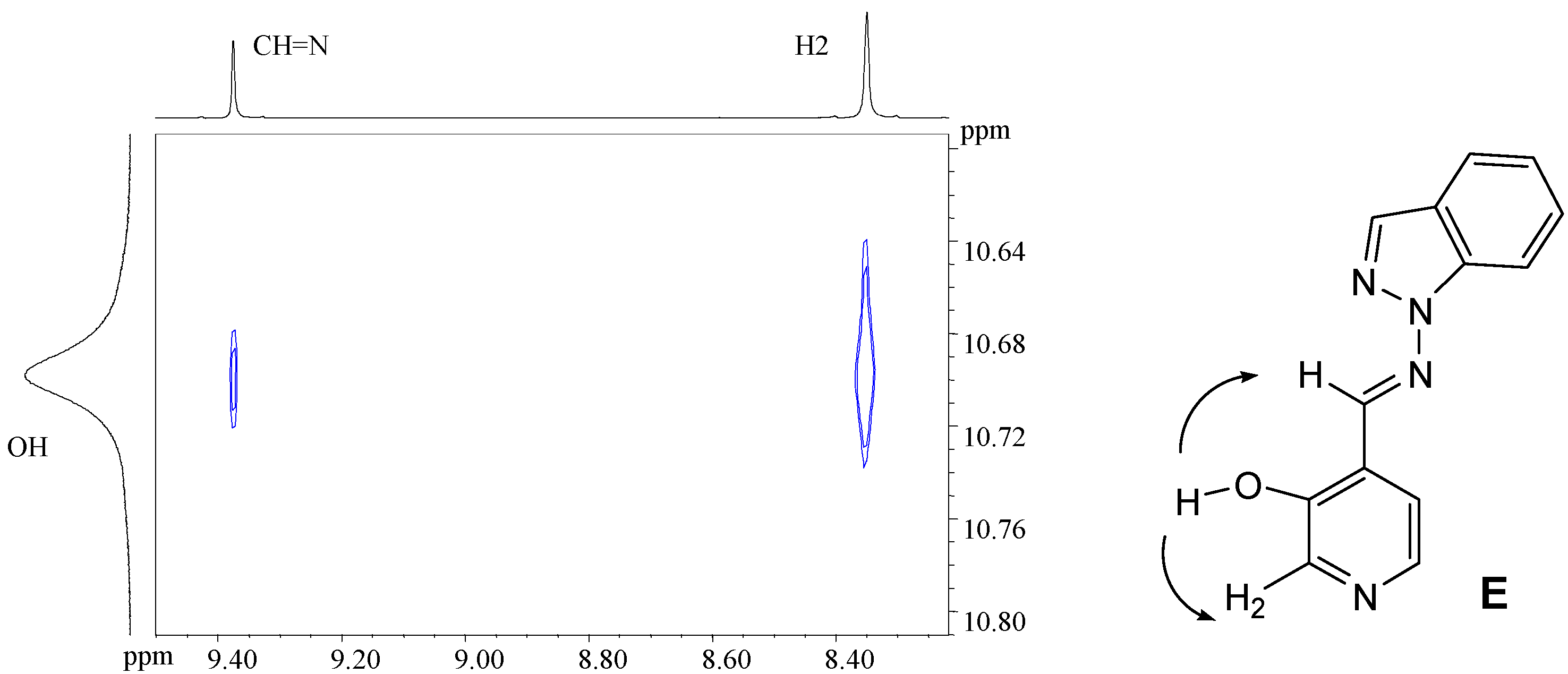
4-[(E)-(1H-indazol-1-ylimino)methyl]pyridin-3-ol (7b). TLC [Rf 0.82 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (from CHCl3-C2H5OH), mp 190 ºC (microscope) and 186.0 ºC (DSC); Anal. Calcd for C13H10N4O: C, 65.54; H, 4.23; N, 23.52. Found: C, 63.25; H, 4.24; N, 22.76; 1H-NMR (DMSO-d6) δ: 10.69 (s br, 1H, OH), 9.37 (s, 1H, CH=N), 8.35 (s, 1H, C2-H), 8.34 (t, 1H, 4J4’-3’=5J7’-3’=0.8, C3’-H), 8.16 (d, 1H, 3J5-6= 5.0, C6-H), 7.89 (d, 1H, C5-H), 7.87 (ddd, 1H, 3J6’-7’= 8.1, 4J5’-7’= 0.9, C7’-H), 7.84 (ddd, 1H, 3J5’-4’=8.0, 4J6’-4’= 1.0, C4’-H), 7.56 (ddd, 1H, 3J5’-6’= 6.9, C6´-H), 7.29 (ddd, H5’); 13C-NMR (DMSO-d6) δ: 152.4 (C3), 140.5 (C6, 1J=181.6, 3J=11.1), 139.8 (CH=N, 1J=172.0), 139.6 (C2, 1J=178.5, 3J=11.3), 137.6 (C7’a, 3J=3J=9.1), 134.2 (C3’, 1J=192.8), 128.3 (C6’, 1J=159.6, 3J=7.7, 2J=2.2), 125.8 (C4), 123.5 (C3’a), 122.6 (C5’, 1J=161.3, 3J=7.0), 121.5 (C4’, 1J=163.8, 3J=8.2), 118.9 (C5, 1J=163.9, 3J=9.8, 2J=4.1), 110.0 (C7’, 1J=167.5, 3J=8.4); 15N-NMR (DMSO-d6) δ: -157.5 (N1’), -81.8 (N2’), -70.1 (CH=N), -54.4 (N1); 1H-NMR (CDCl3) δ: 10.37 (s, 1H, OH), 9.18 (s, 1H, CH=N), 8.51 (s, 1H, C2-H), 8.28 (d, 1H, 3J5-6= 4.9, C6-H), 8.11 (t, 1H, 4J4’-3’=5J7’-3’=0.9, C3’-H), 7.75 (td, 1H, 3J5’-4’=8.1, 4J6’-4’= 0.9, C4’-H), 7.70 (qd, 1H, 3J6’-7’= 8.4, 4J5’-7’= 0.9, C7’-H), 7.55 (ddd, 1H, 3J5’-6’= 7.0, C6´-H), 7.29 (d, 1H, C5-H), 7.29 (ddd, H5’); 13C-NMR (CDCl3) δ: 152.6 (C3), 141.2 (C6, 1J=181.2, 3J=11.5), 146.2 (CH=N, 1J=172.4, 3J=6.3), 140.6 (C2, 1J=181.1), 137.5 (C7’a), 134.6 (C3’, 1J=191.4, 3J=2.5), 128.7 (C6’, 1J=161.0, 3J=7.7), 123.6 (C5, 1J=158.0), 123.1 (C5’, 1J=162.6), 122.9 (C4), 124.0 (C3’a), 121.5 (C4’, 1J=163.5, 3J=8.1), 109.5 (C7’, 1J=167.3, 3J=7.9); 15N-NMR (CDCl3) δ: -162.8 (N1’), -86.2 (N2’), -83.9 (CH=N), -59.7 (N1); 13C-CP/MAS NMR δ: 155.6/153.8 (C3), 137.9 (C7’a), 137.5 (C6 and CH=N), 135.9 (C2), 134.5 (C3’), 130.3 (C6’), 128.9 (C4), 123.9/122.4 (C3’a), 121 (C5’ and C4’), 119.1 (C5), 109.4 (C7’); 15N- CP/MAS NMR δ: -154.5 (N1’), -88.7 (N2’), -80.8 (CH=N), -68.9 (N1).
4-[(E)-(2H-indazol-2-ylimino)methyl]pyridin-3-ol (7c). TLC [Rf 0.79 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (from C2H5OH), mp 270 ºC (microscope) and 267.7 ºC with decomposition at 284.7 (DSC); Anal. Calcd for C13H10N4O: C, 65.54; H, 4.23; N, 23.52. Found: C, 64.75; H, 4.39; N, 23.30; 1H-NMR (DMSO-d6) δ: 10.94 (s, 1H, OH), 9.77 (s, 1H, CH=N), 8.70 (d, 1H, 5J7’-3’=0.9, C3’-H), 8.40 (s, 1H, C2-H), 8.20 (d, 1H, 3J5-6=5.0, C6-H), 7.81 (d, 1H, C5-H), 7.75 (td, 1H, 3J5’-4’=8.5, 4J6’-4’=5J7’-4’=1.1, C4’-H), 7.66 (qd, 1H, 3J6’-7’=8.8, 4J5’-7’=1.0, C7’-H), 7.36 (ddd, 1H, 3J5’-6’=6.6, C6’-H), 7.11 (ddd, 1H, C5’-H); 13C-NMR (DMSO-d6) δ: 153.8 (C3), 148.9 (CH=N, 1J=176.1), 146.0 (C7’a, 3J=3J=3J=7.3), 140.5 (C6, 1J=180.7, 3J=11.7), 140.2 (C2, 1J=179.8, 3J=11.1), 127.9 (C6’, 1J=158.5, 3J=7.3), 124.4 (C4), 123.7 (C3’, 1J=197.1), 122.2 (C5’, 1J=159.9, 3J=8.2), 121.3 (C3’a), 121.2 (C4’, 1J=163.9, 3J=7.4), 119.5 (C5, 1J=164.5, 3J=8.5), 117.2 (C7’, 1J=163.1, 3J=7.0); 15N-NMR (DMSO-d6) δ: -125.5 (N2’), -63.8 (CH=N), -50.5 (N1); 1H-NMR (CDCl3) δ: 10.14 (s, 1H, OH), 9.61 (s, 1H, CH=N), 8.56 (s, 1H, C2-H), 8.23 (d, 1H, 5J7’-3’=0.9, C3’-H), 8.33 (d, 1H, 3J5-6=4.9, C6-H), 7.69 (m, 2H, C4’-H and C7’-H), 7.39 (ddd, 1H, 3J5’-6’=6.6 3J7’-6’=8.9, 5J4’-6’=1.0, C6’-H), 7.37 (d, 1H, C5-H), 7.15 (ddd, 1H, 3J4’-5’=8.4, 4J7’-5’=0.8, C5’-H); 13C-NMR (CDCl3) δ: 154.5 (CH=N), 153.0 (C3), 147.0 (C7’a), 141.6 (C6), 141.2 (C2), 128.6 (C6’), 124.5 (C5), 123.1 (C5’), 122.5 (C3’), 121.8 (C3’a), 121.7 (C4), 120.7 (C4’), 117.5 (C7’); 15N-NMR (CDCl3) δ: -133 (N2’); 13C-CP/MAS NMR δ: 156.1 (C3), 146.9 (CH=N), 145.0 (C7’a), 141.3 (C6), 139.4 (C2), 129.3 (C4), 127.4 (C6’), 123.5 (C3’), 121.7 (C5’ and C3’a), 121.1 (C4’), 119.6 (C5), 118.4 (C7’); 15N-CP/MAS NMR δ: -122.9 (N2’), -110.6 (N1’), -84.0 (CH=N), ‑69.9/-66.7 (N1).
4-[(E)-(1H-1,2,3-benzotriazol-1-ylimino)methyl]pyridin-3-ol (7d). TLC [Rf 0.70 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (CHCl3), mp 209 ºC (microscope) and 199.9 ºC and 204.7 ºC with decomposition at 237.1 ºC (DSC); Anal. Calcd for C12H9N5O: C, 60.25; H, 3.79; N, 29.27. Found: C, 60.01; H, 4.28; N, 28.12; 1H-NMR (DMSO-d6) δ: 11.00 (s br, 1H, OH), 9.75 (s, 1H, CH=N), 8.41 (s, 1H, C2-H), 8.22 (d, 1H, 3J5-6=5.0, H6), 8.13 (td, 1H, 3J5’-4’=8.4, 4J6’-4’=5J7’-4’=1.0, C4’-H), 7.96 (td,1H, 3J6’-7’=8.3, 4J5’-7’=1.0, C7’-H) 7.93 (d, 1H, C5-H), 7.70 (ddd, 1H, 3J5’-6’=6.9, C6’-H), 7.51 (ddd, 1H, C5’-H); 13C-NMR (DMSO-d6) δ: 153.1 (C3), 147.2 (CH=N, 1J=173.4), 144.8 (C3’a, 3J=9.8, 3J=4.5), 140.5 (C6, 1J=181.3, 3J=11.5), 140.2 (C2, 1J=179.4, 3J=11.4), 130.4 (C7’a, 3J=10.6, 3J=6.5), 129.2 (C6’, 1J=163.5, 3J=7.8), 125.3 (C5’, 1J=164.0, 3J=7.7), 124.6 (C4), 119.8 (C4’, 1J=166.5, 3J=7.8), 119.2 (C5, 1J=163.2), 110.5 (C7’, 1J=168.9, 3J=8.2); 15N-NMR (DMSO-d6) δ: -121.6 (N1’), -77.1 (CH=N), -49.7 (N1); 1H-NMR (CDCl3) δ: 10.20 (s br, 1H, OH), 9.64 (s, 1H, CH=N), 8.59 (s, 1H, C2-H), 8.35 (d, 1H, 3J5-6=4.6, C6-H), 8.11 (d, 1H, 3J5’-4’=8.3, C4’-H), 7.76 (d, 1H, 3J6’-7’=8.2, C7’-H), 7.65 (t, 1H, 3J5’-6’=7.5, C6’-H), 7.49 (t, 1H, C5’-H), 7.43 (d, 1H, C5-H); 13C-NMR (CDCl3) δ: 153.2 (C3), 152.3 (CH=N, 1J=172.8, 3J=6.1), 145.5 (C3’a), 141.1 (C6, 1J=183.0, 3J=11.8), 141.0 (C2, 1J=181.5, 3J=11.3), 130.3 (C7’a), 129.4 (C6’, 1J=163.4, 3J=8.1), 125.4 (C5’, 1J=163.7, 3J=7.9), 123.8 (C5, 1J=164.1, 3J=8.6), 122.4 (C4), 120.5 (C4’, 1J=166.5, 3J=7.8), 109.6 (C7’, 1J=169.5, 3J=8.3); 15N-NMR (CDCl3) δ: -126.9 (N1’), -89.4 (CH=N), -55.8 (N1); 13C-CP/MAS NMR δ: 154.8 (C3), 145.5 (CH=N), 143.9 (C3’a), 141.1 (C6), 138.2 (C2), 130.8 (C7’a), 128.7 (C6’), 123.6 (C5’ and C4), 118.7 (C5), 118.0 (C4’), 109.7 (C7’); 15N-CP/MAS NMR δ: -159.0 (N1’), -80.5 (CH=N), -67.6 (N3’), -55.3 (N1), -17.6 (N2’).
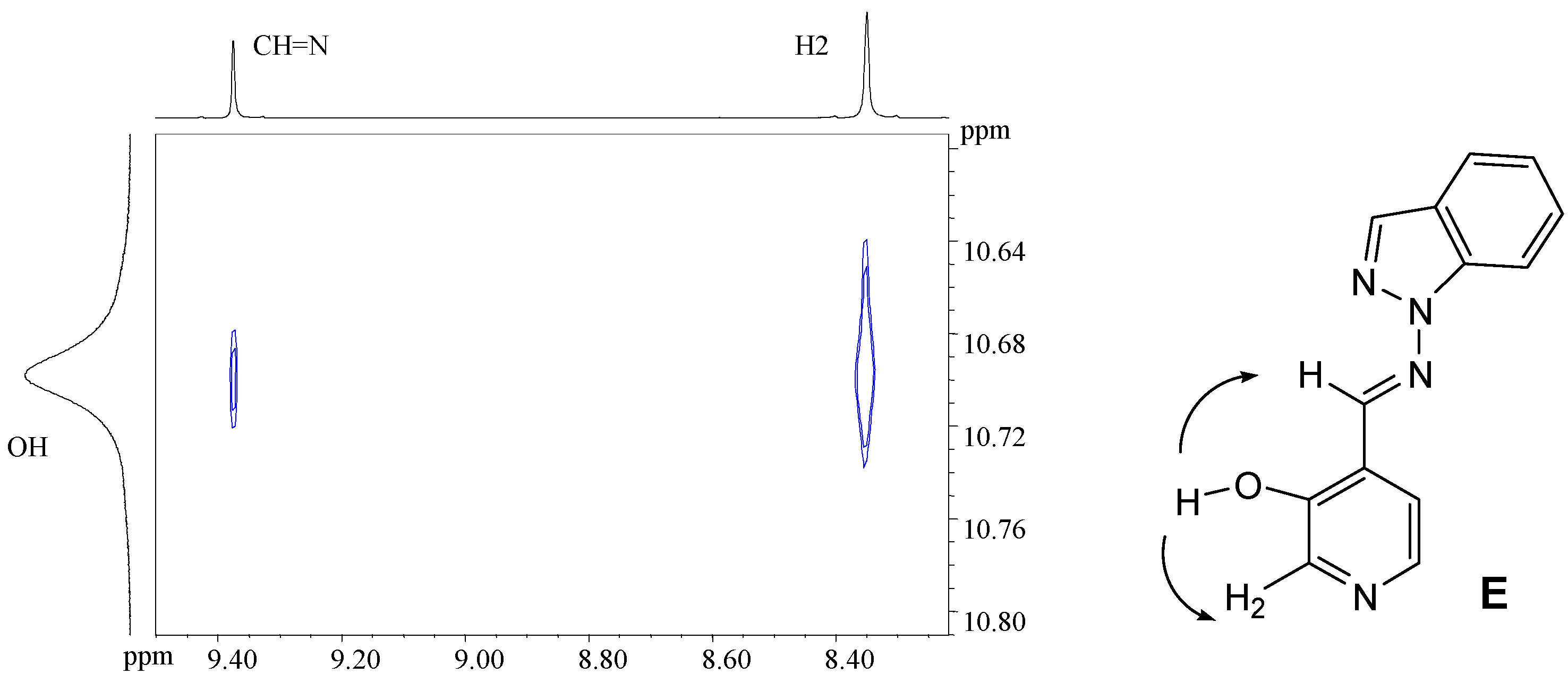
4-[(E)-(2H-1,2,3-benzotriazol-2-ylimino)methyl]pyridin-3-ol (7e). TLC [Rf 0.80 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (CHCl3), this compound changes its appearance at 250°C and then at 280°C it decomposes, and 259.6 ºC (DSC); Anal. calcd for C12H9N5O: C, 60.25; H, 3.79; N, 29.27. Found: C, 59.90; H, 3.95; N, 28.80; 1H-NMR (DMSO-d6) δ: 11.20 (s br, 1H, OH), 9.86 (s, 1H, CH=N), 8.43 (s, 1H, C2-H), 8.20 (d, 1H, 3J5-6=5.0, C6-H), 7.97 (m, 2H, C4’-H and C7’-H), 7.84 (d, 1H, C5-H), 7.52 (m, 2H, C5’-H and C6’-H); 13C-NMR (DMSO- d6) δ: 153.9 (C3, 3J=2J=5.2), 153.2 (CH=N, 1J=173.0, 3J=3.1), 142.7 (C3’a and C7’a), 140.5 (C6 and C2, 1J=180.2, 3J=11.3), 128.0 (C5’ and C6’, 1J=162.0, 3J=8.3), 123.5 (C4, 3J=3J=2J=6.0), 119.3 (C5, 1J=164.3, 3J=9.7, 2J=3.8), 118.1 (C4’ and C7’, 1J=169.2, 3J=5.2); 15N-NMR (DMSO- d6) δ: -90.2 (CH=N), -47.2 (N1); 1H-NMR (CDCl3) δ: 10.02 (s vbr, 1H, OH), 9.68 (s, 1H, CH=N), 8.62 (s, 1H, C2-H), 8.36 (d, 1H, 3J5-6=4.8, C6-H), 7.90 (m, 2H, C4’-H and C7’-H), 7.49 (m, 2H, C5’-H and C6’-H), 7.42 (d, C5-H); 13C-NMR (CDCl3) δ: 157.5 (CH=N, 1J=169.7), 153.5 (C3), 143.6 (C3’a and C7’a), 141.8 (C2, 1J=182.1, 3J=11.0), 141.2 (C6, 1J=182.7, 3J=11.2), 128.4 (C5’ and C6’, 1J=161.4, 3J=8.2), 124.7 (C5, 1J=162.9), 121.0 (C4), 118.4 (C4’ and C7’, 1J=170.1, 3J=4.9); 13C-CP/MAS NMR δ: 155.2 (C3), 149.1 (CH=N), 142.8 (C3’a and C7’a), 139.8 (C6), 135.0 (C2), 129.7(C5’), 126.2, (C6’ and C4), 123.2 (C5), 120.3 (C4’), 113.3 (C7’); 15N-CP/MAS NMR δ: -90.6 (N1’), -88.0 (N3’), -84.5 (CH=N), -83.6 (N2’), -60.7 (N1).




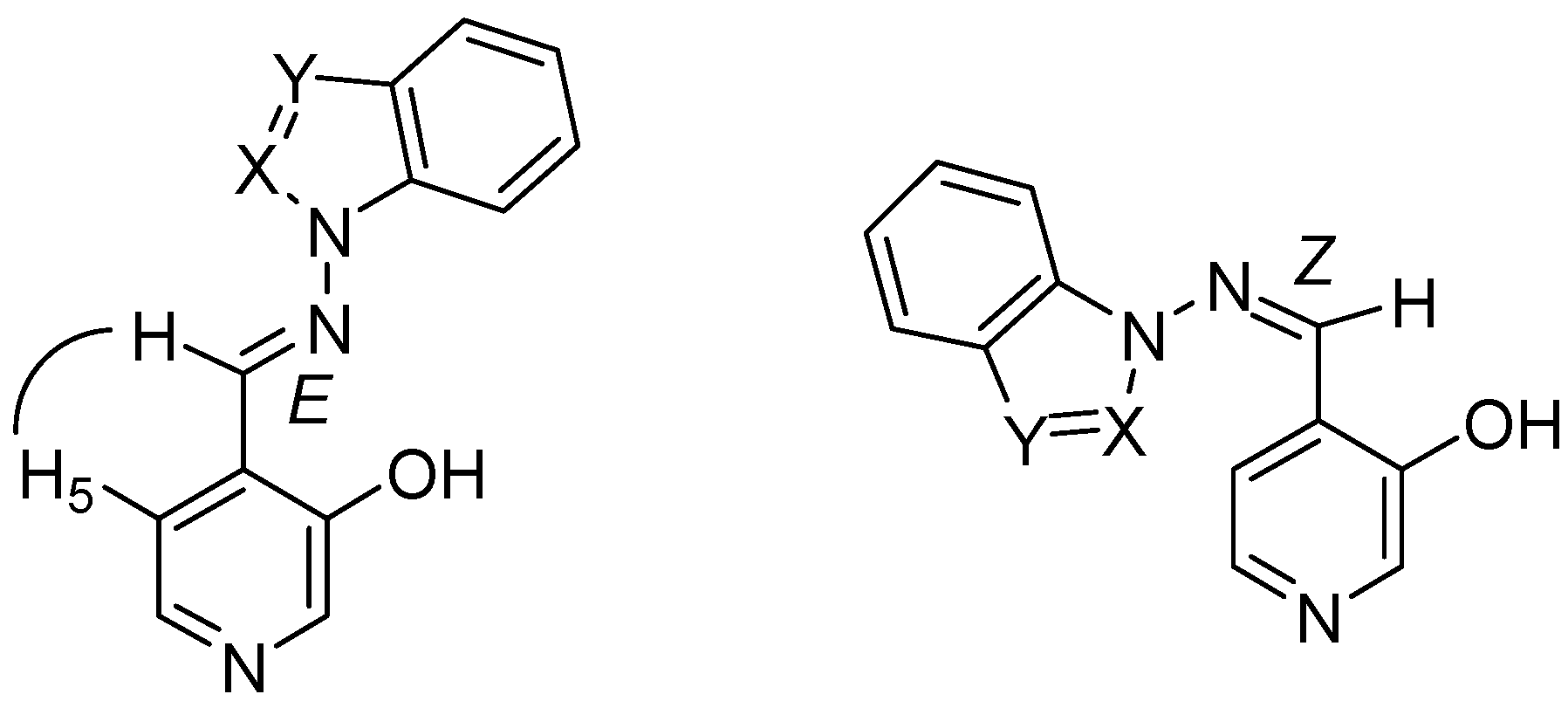

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
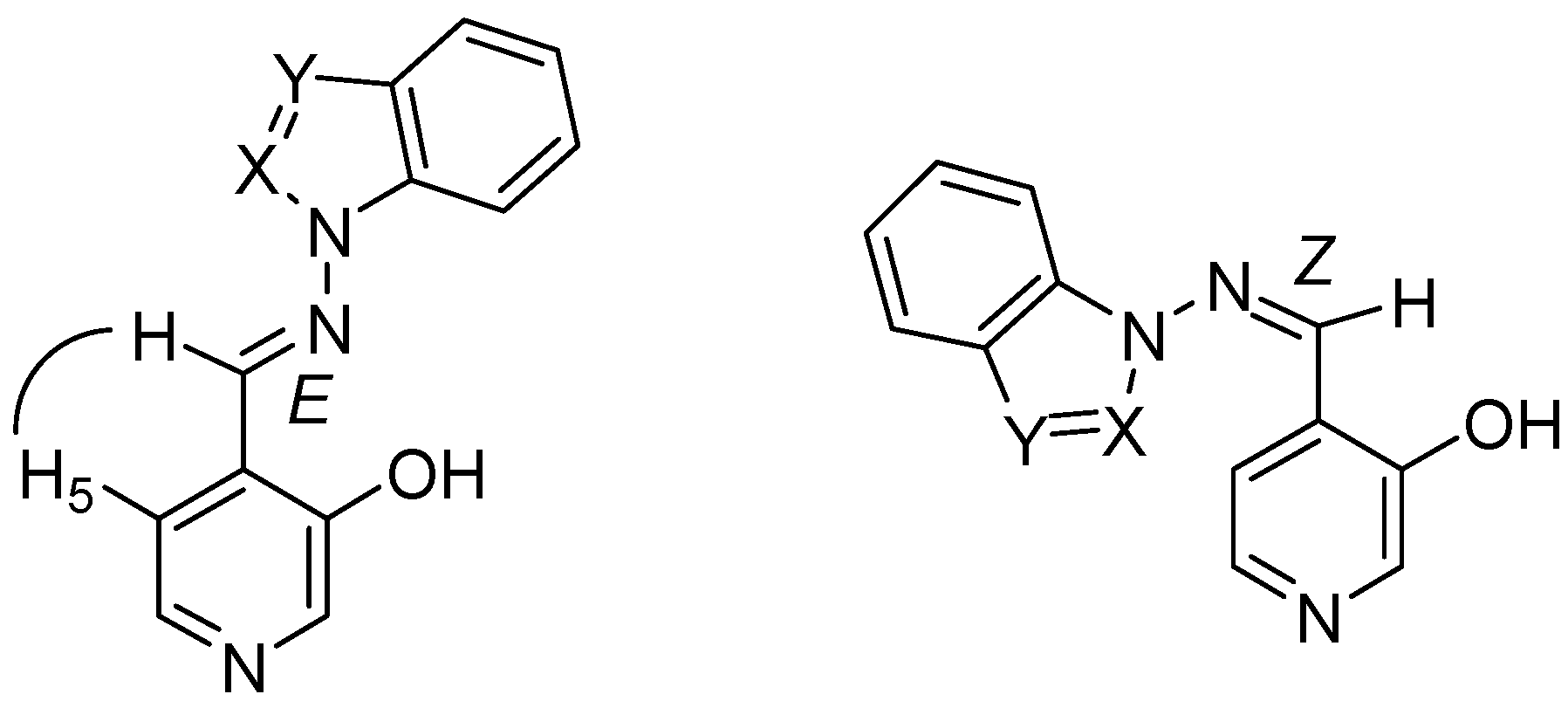{kind=link}
{kind=link}