Synthesis and Structural Characterization of 1- and 2-Substituted Indazoles: Ester and Carboxylic Acid Derivatives

Abstract
:Introduction
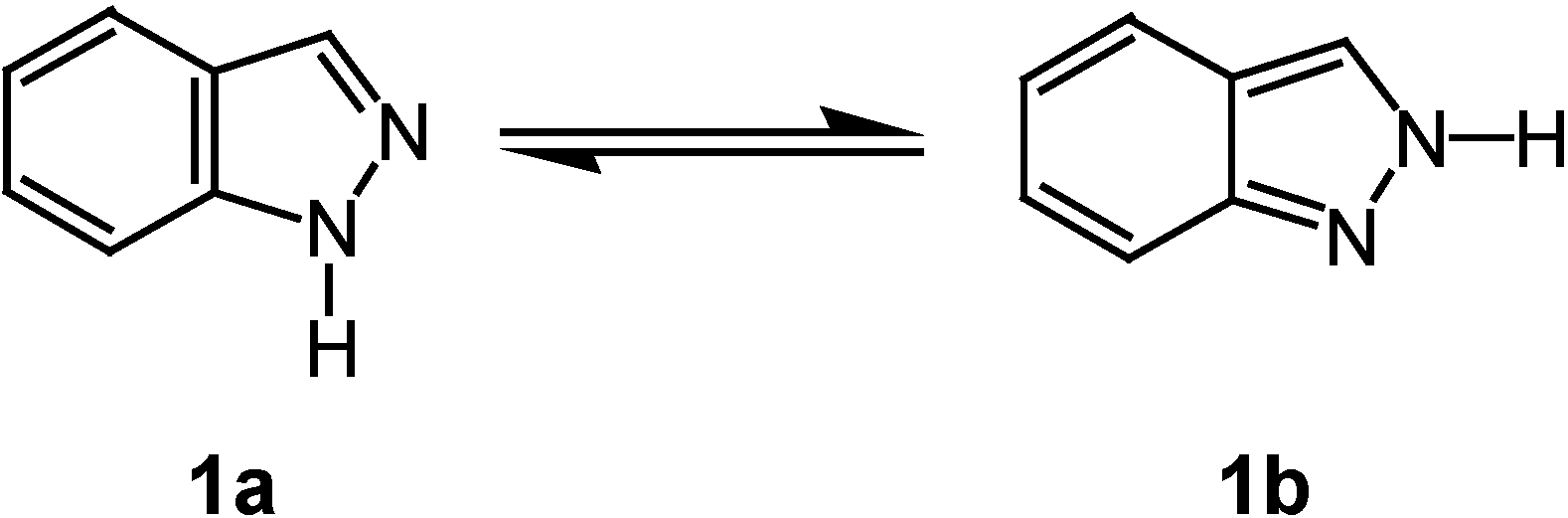
Results and Discussion
Synthesis


{kind=link}
{kind=link}
{kind=link}
| Series | n | Base (solvent) | X(CH2)nCO2R | 2 (%) | 3 (%) | 2 + 3 (%) | |
|---|---|---|---|---|---|---|---|
| X | R | ||||||
| a | 0 | Kt-BuO (THF) | Cl | Me | 99 | - | 99 |
| Kt-BuO (THF) | Cl | Et | (45)[a] | (10)[a] | |||
| b | 1 | Kt-BuO(THF) | Br | Et | 55 | 13 | 68 |
| K2CO3 (DMF) | Br | Et | 67 | 22 | 89 | ||
| Kt-BuO (THF) | Br | Et | (15)[a] | (15)[a] | |||
| c | 2 | NaH (THF) | Br | Et | 47 | 39 | 86 |
| K2CO3 (DMF) | Br | Et | 49 | 46 | 95 | ||
| Kt-BuO (THF) | Br | Et | (26)[a] | (18)[a] | |||
| d[b] | 3 | Kt-BuO (DMSO) | Br | Et | 49 | 12 | 61[c] |
| K2CO3 (DMF) | Br | Et | 59 | 37 | 96 | ||
| e[b] | 4 | K2CO3 (DMF) | Br | Et | 59 | 31 | 90 |
| f[b] | 5 | Kt-BuO (THF) | Br | Et | (39)[a] | (39)[a] | |
| K2CO3 (DMF) | Br | Et | 62 | 34 | 96 | ||
| g | 6 | K2CO3 (DMF) | Br | Et | 61 | 36 | 97 |
| h | 9 | K2CO3 (DMF) | Br | Me | 63 | 34 | 97 |
| i | 10 | K2CO3 (DMF) | Br | Me | 60 | 38 | 98 |

| N° | n | R | Yield (%) | mp (°C) | Formula | Analysis (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Calcd. | Found | ||||||||||
| C | H | N | C | H | N | ||||||
| 4b | 1 | Et | 97 | 186-188 1 | C9H8N2O2 | 61.36 | 4.58 | 15.90 | 61.32 | 4.52 | 15.85 |
| 4c | 2 | Et | 98 | 106-107 2 | C10H10N2O2 | 63.15 | 5.30 | 14.73 | 62.96 | 5.34 | 14.49 |
| 4d | 3 | Et | 93 | 60-62 | C11H12N2O2 | 64.69 | 5.92 | 13.72 | 64.71 | 5.97 | 13.74 |
| 4e | 4 | Et | 99 | 82-83 | C12H14N2O2 | 66.04 | 6.47 | 12.84 | 66.11 | 6.58 | 12.86 |
| 4f | 5 | Et | 100 | 69-70 | C13H16N2O2 | 67.22 | 6.94 | 12.06 | 67.42 | 6.99 | 11.97 |
| 4g | 6 | Et | 74 | 54-58 | C14H18N2O2 | 68.27 | 7.37 | 11.37 | 68.46 | 7.25 | 11.36 |
| 4h | 9 | Me | 93 | 78-81 | C17H24N2O2 | 70.80 | 8.39 | 9.71 | 70.67 | 8.49 | 9.65 |
| 4i | 10 | Me | 95 | 73-74 | C18H26N2O2 | 71.49 | 8.67 | 9.26 | 71.39 | 8.91 | 9.29 |

| N° | n | R | Yield (%) | mp (°C) | Formula | Analysis (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Calcd. | Found | ||||||||||
| C | H | N | C | H | N | ||||||
| 5b | 1 | Et | 96 | 254-256 1 | C9 H8N2O2 | 61.36 | 4.58 | 15.90 | 61.24 | 4.51 | 15.90 |
| 5c | 2 | Et | 98 | 147-149 2 | C10H10N2O2 | 63.15 | 5.30 | 14.73 | 63.05 | 5.37 | 14.70 |
| 5d | 3 | Et | 97 | 132-134 | C11H12N2O2 | 64.69 | 5.92 | 13.72 | 64.71 | 5.99 | 13.64 |
| 5e | 4 | Et | 98 | 112-114 | C12H14N2O2 | 66.04 | 6.47 | 12.84 | 65.92 | 6.18 | 12.74 |
| 5f | 5 | Et | 59 | 86-87 | C13H16N2O2 | 67.22 | 6.94 | 12.06 | 66.97 | 7.31 | 11.66 |
| 5g | 6 | Et | 62 | 77-78 | C14H18N2O2 | 68.27 | 7.37 | 11.37 | 68.15 | 7.38 | 11.27 |
| 5h | 9 | Me | 90 | 68 | C17H24N2O2 | 70.80 | 8.39 | 9.71 | 70.69 | 8.68 | 9.76 |
| 5i | 10 | Me | 93 | 82 | C18H26N2O2 | 71.49 | 8.67 | 9.26 | 71.53 | 8.68 | 9.15 |
Spectroscopic Characterization
| N° | n | R | C7 | C4 | C5 | C6 | C3 | C3a | C7a | CO | NCH2 | CH2 | OCH2CH3 | OCH3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 110.0 | 120.4 | 120.1 | 125.8 | 133.4 | 122.8 | 139.9 | |||||||
| 2a | 0 | Me | 114.3 | 121.1 | 124.0 | 129.2 | 140.2 | 125.7 | 139.7 | 151.0 | 54.3 | |||
| 2b | 1 | Et | 108.6 | 121.1 | 120.8 | 126.6 | 134.1 | 124.1 | 140.0 | 167.8 | 50.1 | 61.6, 13.9 | ||
| 2c | 2 | Et | 109.0 | 120.9* | 120.5* | 126.3 | 133.5 | 123.9 | 139.5 | 171.1 | 44.1 | 34.5 | 60.8, 14.0 | |
| 2d | 3 | Et | 108.8 | 121.0* | 120.4* | 126.1 | 133.0 | 123.9 | 139.4 | 172.7 | 47.6 | 24.9, 30.9 | 60.4, 14.1 | |
| 2e | 4 | Et | 108.9 | 121.1* | 120.4* | 126.1 | 132.8 | 124.0 | 139.3 | 173.2 | 48.4 | 22.2, 29.2, 33.7 | 60.3, 14.2 | |
| 2f | 5 | Et | 108.8 | 121.0* | 120.3* | 126.0 | 132.7 | 123.9 | 139.3 | 173.4 | 48.5 | 24.5, 26.3, 29.4, 34.0 | 60.1, 14.1 | |
| 2g | 6 | Et | 108.9 | 121.1* | 120.3* | 126.0 | 132.7 | 123.9 | 139.3 | 173.7 | 48.7 | 24.7, 26.5, 28.7, 29.6, 34.2 | 60.2, 14.2 | |
| 2h | 9 | Me | 108.9 | 121.0 | 120.3 | 126.0 | 132.6 | 123.9 | 139.3 | 174.2 | 48.8 | 24.8, 26.8, 29.0, 29.1, 29.2, 29.8, 34.0 | 51.4 | |
| 2i | 10 | Me | 109.0 | 121.1* | 120.3* | 126.0 | 132.6 | 124.0 | 139.3 | 174.3 | 48.9 | 24.9, 26.8, 29.05, 29.13, 29.26, 29.31, 29.8, 34.1 | 51.4 | |
| 3b | 1 | Et | 117.6 | 120.3 | 122.1 | 126.4 | 124.4 | 122.2 | 149.2 | 167.2 | 54.5 | 62.2, 14.1 | ||
| 3c | 2 | Et | 117.1 | 120.1 | 121.51 | 125.9 | 123.5 | 121.5 | 148.9 | 170.7 | 48.7 | 34.9 | 60.8, 13.9 | |
| 3d | 3 | Et | 117.2 | 119.9 | 121.47 | 125.7 | 122.8 | 121.5 | 148.8 | 172.4 | 52.3 | 25.5, 30.7 | 60.4, 14.0 | |
| 3e | 4 | Et | 117.2 | 120.0 | 121.5 | 125.7 | 122.6 | 121.6 | 148.7 | 173.0 | 53.2 | 21.9, 29.8, 33.5 | 60.3, 14.1 | |
| 3f | 5 | Et | 117.3 | 119.9 | 121.5 | 125.7 | 122.5 | 121.6 | 148.8 | 173.4 | 53.4 | 24.3, 26.0, 30.2, 33.9 | 60.2, 14.1 | |
| 3g | 6 | Et | 117.3 | 120.0 | 121.5 | 125.7 | 122.5 | 121.7 | 148.8 | 173.6 | 53.6 | 24.7, 26.3, 28.5, 30.4, 34.1 | 60.2, 14.2 | |
| 3h | 9 | Me | 117.3 | 120.0* | 121.4* | 125.6 | 122.4 | 121.7 | 148.7 | 174.2 | 53.7 | 24.8, 26.6, 28.96, 28.98, 29.0, 29.1, 30.6, 34.0 | 51.4 | |
| 3i | 10 | Me | 117.4 | 120.0* | 121.5* | 125.7 | 122.5 | 121.7 | 148.8 | 174.3 | 53.8 | 24.9, 26.6, 29.04, 29.12, 29.24, 29.28, 30.6, 34.1 | 51.4 |
| N° | n | Solvent | C7 | C4 | C5 | C6 | C3 | C3a | C7a | CO | NCH2 | CH2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4b | 1 | MeOD | 110.4 | 122.1 | 122.1 | 128.1 | 135.0 | 125.4 | 141.8 | 171.5 | 50.6 | |
| 4c | 2 | MeOD | 110.6 | 122.0* | 121.9* | 127.8 | 134.5 | 125.2 | 141.0 | 174.6 | 45.2 | 35.1 |
| 4d | 3 | MeOD | 110.3 | 122.2* | 121.9* | 127.8 | 134.1 | 125.2 | 141.0 | 174.6 | 48.6 | 26.2, 31.6 |
| 4d | 3 | CDCl3 | 108.9 | 121.3* | 120.7* | 126.6 | 132.9 | 123.7 | 139.4 | 177.6 | 47.4 | 24.7, 30.8 |
| 4e | 4 | MeOD | 110.3 | 122.1* | 121.8* | 127.7 | 133.8 | 125.2 | 140.9 | 177.0 | 49.2 | 23.3, 30.3, 34.3 |
| 4e | 4 | CDCl3 | 108.8 | 121.2* | 120.5* | 126.4 | 132.7 | 123.7 | 139.3 | 178.3 | 48.2 | 21.9, 29.0, 33.4 |
| 4f | 5 | MeOD | 110.4 | 122.2 | 121.8 | 127.7 | 133.8 | 125.1 | 140.9 | 177.4 | 49.3 | 25.6, 27.3, 30.6, 34.7 |
| 4f | 5 | CDCl3 | 108.9 | 121.2 | 120.5 | 126.3 | 132.7 | 123.8 | 139.3 | 178.9 | 48.5 | 24.2, 26.2, 29.4, 33.8 |
| 4f | 5 | DMSO | 109.6 | 120.8 | 120.3 | 125.9 | 132.4 | 123.4 | 139.2 | 174.4 | 47.9 | 24.1, 25.8, 29.2, 33.6 |
| 4g | 6 | MeOD | 110.4 | 122.1* | 121.8* | 127.7 | 133.7 | 125.1 | 140.9 | 177.5 | 49.4 | 25.8, 27.4, 29.7, 30.7, 34.7 |
| 4h | 9 | MeOD | 110.4 | 122.2* | 121.8* | 127.7 | 133.7 | 125.1 | 140.9 | 177.7 | 49.5 | 26.0, 27.7, 30.11, 30.18, 30.2, 30.3, 30.9, 34.9 |
| 4i | 10 | MeOD | 110.4 | 122.1 | 121.8 | 127.7 | 133.7 | 125.1 | 140.9 | 177.7 | 49.5 | 26.0, 27.7, 30.1, 30.27, 30.34, 30.4, 30.9, 34.9 |
| 5b | 1 | DMSO | 117.0 | 120.8 | 121.2 | 125.8 | 125.5 | 121.6 | 148.2 | 169.3 | 54.2 | |
| 5c | 2 | MeOD | 117.4 | 121.6 | 122.7 | 127.6 | 125.9 | 122.9 | 150.1 | 174.0 | 49.9 | 35.6 |
| 5d | 3 | MeOD | 117.4 | 121.6* | 122.7* | 127.5 | 125.5 | 123.1 | 150.1 | 176.2 | 53.4 | 27.0, 31.5 |
| 5e | 4 | MeOD | 117.3 | 121.5* | 122.6* | 127.5 | 125.4 | 123.1 | 149.9 | 176.9 | 53.9 | 23.0, 31.0, 34.2 |
| 5f | 5 | MeOD | 117.3 | 121.5 | 122.6 | 127.4 | 125.4 | 123.1 | 149.9 | 177.3 | 54.1 | 25.5, 27.1, 31.3, 34.6 |
| 5g | 6 | MeOD | 117.3 | 121.5* | 122.6* | 127.4 | 125.3 | 123.0 | 149.9 | 177.5 | 54.2 | 25.8, 27.2, 29.6, 31.4, 34.7 |
| 5h | 9 | MeOD | 117.3 | 121.5 | 122.6 | 127.4 | 125.3 | 123.1 | 149.9 | 177.7 | 54.3 | 26.0, 27.5, 30.0, 30.1, 30.2, 30.3, 31.6, 34.9 |
| 5i | 10 | MeOD | 117.3 | 121.5* | 122.6* | 127.4 | 125.3 | 123.1 | 149.9 | 177.7 | 54.3 | 26.1, 27.5, 30.1, 30.2, 30.3, 30.38, 30.41, 31.6, 35.0 |
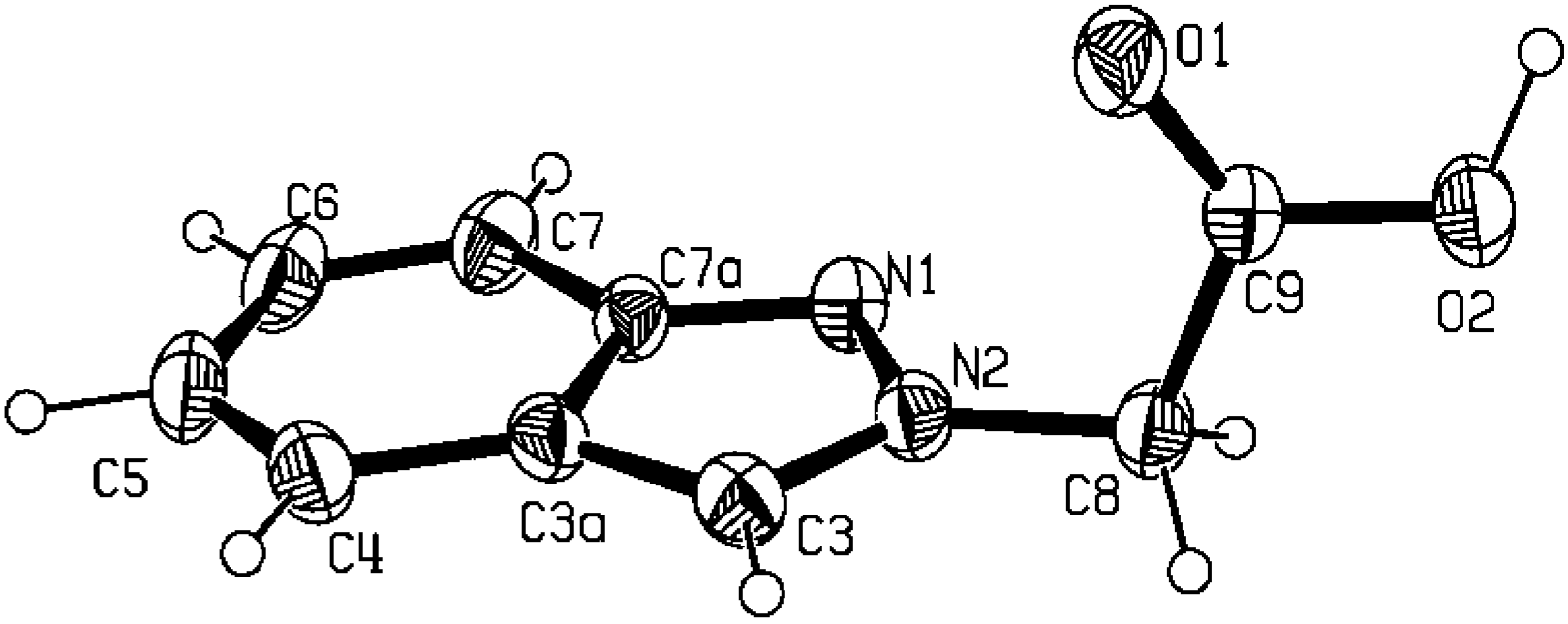
Molecular and crystal structure of 2-indazol-2-yl-acetic acid (5b)
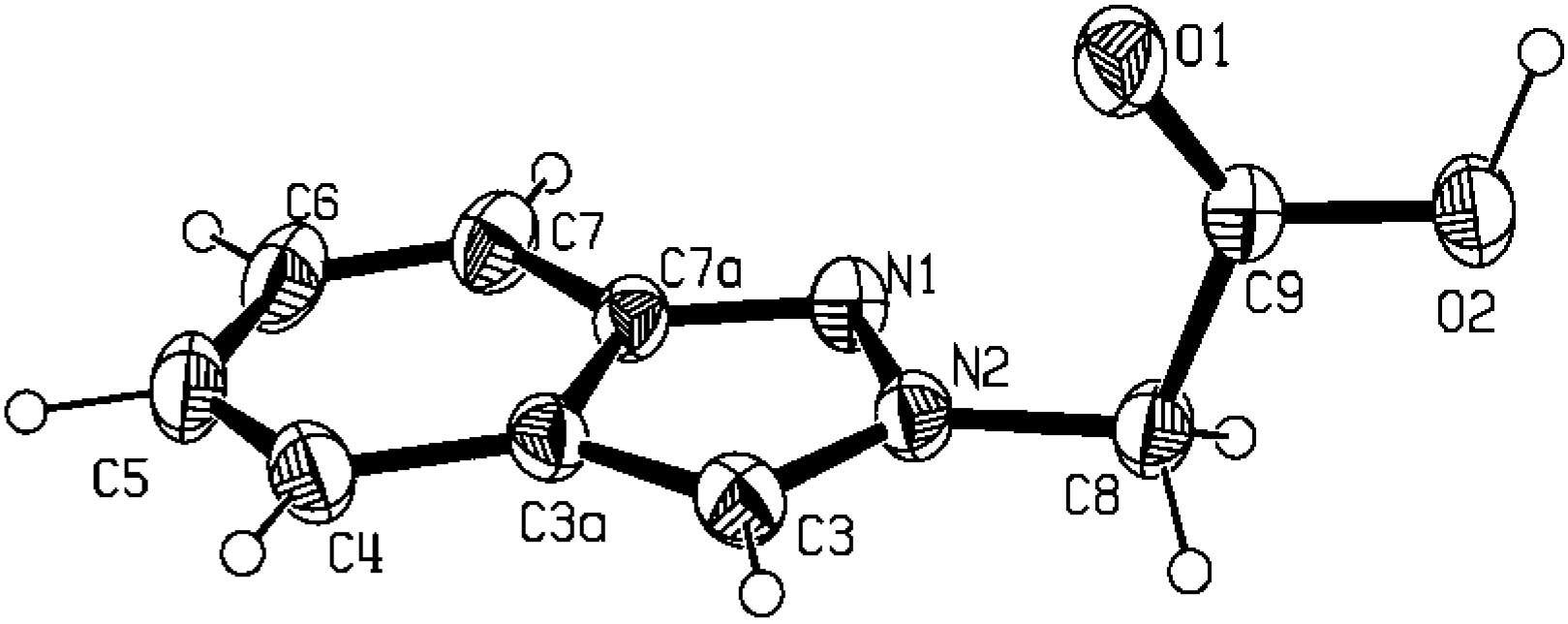
| N1-C7a | 1.349(2) | C3a-C4 | 1.421(2) |
| N1-N2 | 1.3526(19) | C8-C9 | 1.516(2) |
| N2-C3 | 1.334(2) | C5-C4 | 1.355(3) |
| N2-C8 | 1.446(2) | C5-C6 | 1.408(3) |
| C7a-C3a | 1.413(2) | C7-C6 | 1.360(3) |
| C7a-C7 | 1.413(3) | C9-O1 | 1.200(2) |
| C3a-C3 | 1.384(3) | C9-O2 | 1.307(2) |
| C7a-N1-N2 | 104.08(12) | N2-C8-C9 | 112.22(14) |
| C3-N2-N1 | 113.32(14) | C4-C5-C6 | 121.66(18) |
| C3-N2-C8 | 127.88(16) | C5-C4-C3a | 118.2(2) |
| N1-N2-C8 | 118.73(14) | C6-C7-C7a | 117.5(2) |
| N1-C7a-C3a | 111.07(15) | N2-C3-C3a | 107.08(16) |
| N1-C7a-C7 | 128.05(17) | C7-C6-C5 | 122.1(2) |
| C3a-C7a-C7 | 120.86(16) | O1-C9-O2 | 125.35(16) |
| C3-C3a-C7a | 104.44(14) | O1-C9-C8 | 124.33(15) |
| C3-C3a-C4 | 135.92(18) | O2-C9-C8 | 110.32(14) |
| C7a-C3a-C4 | 119.64(17) |
 main pattern found in carboxylic acid derivatives (C-O-H…O-C-O-H…O contacts) [25]. The one-dimensional zigzag chain along the b axis obtained with a C(6) synthon as well as the weak π…π contacts [~ 2.75(4) Å] within neighbouring chains are shown in the packing diagrams of Figure 3 (a, b and c, respectively). The supramolecular motif found in the crystal structure of compound 5b agrees with the data in the literature, either for 1H-unsubstituted indazoles [26] where N-H…N hydrogen bonds are responsible for the supramolecular pattern, or with pyrazole [27] or even with pyrazole carboxylic acid derivatives [28] where the usual hydrogen bond ring pattern [25] is not found, due to the strength of this heteromeric intermolecular interaction [29].
main pattern found in carboxylic acid derivatives (C-O-H…O-C-O-H…O contacts) [25]. The one-dimensional zigzag chain along the b axis obtained with a C(6) synthon as well as the weak π…π contacts [~ 2.75(4) Å] within neighbouring chains are shown in the packing diagrams of Figure 3 (a, b and c, respectively). The supramolecular motif found in the crystal structure of compound 5b agrees with the data in the literature, either for 1H-unsubstituted indazoles [26] where N-H…N hydrogen bonds are responsible for the supramolecular pattern, or with pyrazole [27] or even with pyrazole carboxylic acid derivatives [28] where the usual hydrogen bond ring pattern [25] is not found, due to the strength of this heteromeric intermolecular interaction [29].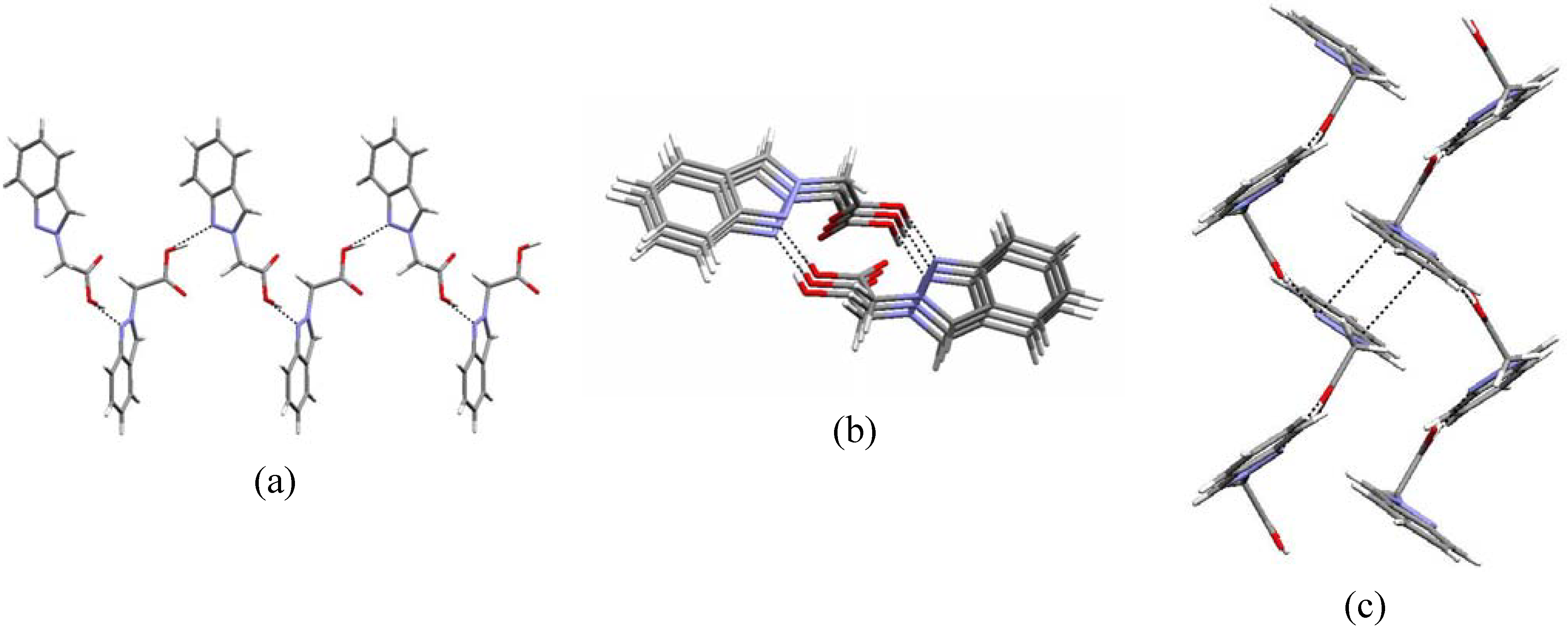
Conclusions
Experimental
General
Indazole-1-carboxylic acid methyl ester (2a)
Indazol-1-yl-acetic acid ethyl ester (2b) and indazol-2-yl-acetic acid ethyl ester (3b)
3-Indazol-1-yl-propionic acid ethyl ester (2c) and 3-indazol-2-yl-propionic acid ethyl ester (3c)
4-Indazol-1-yl-butyric acid ethyl ester (2d) and 4-indazol-2-yl-butyric acid ethyl ester (3d)
5-Indazol-1-yl-pentanoic acid ethyl ester (2e) and 5-indazol-2-yl-pentanoic acid ethyl ester (3e)
6-Indazol-1-yl-hexanoic acid ethyl ester (2f) and 6-indazol-2-yl-hexanoic acid ethyl ester (3f)
7-Indazol-1-yl-heptanoic acid ethyl ester (2g) and 7-indazol-2-yl-heptanoic acid ethyl ester (3g)
10-Indazol-1-yl-decanoic acid methyl ester (2h) and 10-indazol-2-yl-decanoic acid methyl ester (3h)
11-Indazol-1-yl-undecanoic acid methyl ester (2i) and 11-indazol-2-yl-undecanoic acid methyl ester (3i)
Indazol-1-yl-acetic acid (4b)
3-Indazol-1-yl-propionic acid (4c)
4-Indazol-1-yl-butyric acid (4d)
5-Indazol-1-yl-pentanoic acid (4e)
6-Indazol-1-yl-hexanoic acid (4f)
7-Indazol-1-yl-heptanoic acid (4g)
10-Indazol-1-yl-decanoic acid (4h)
11-Indazol-1-yl-undecanoic acid (4i)
Indazol-2-yl-acetic acid (5b)
3-Indazol-2-yl-propionic acid (5c)
4-Indazol-2-yl-butyric acid (5d)
5-Indazol-2-yl-pentanoic acid (5e)
6-Indazol-2-yl-hexanoic acid (5f)
7-Indazol-2-yl-heptanoic acid (5g)
10-Indazol-2-yl-decanoic acid (5h)
11-Indazol-1-yl-undecanoic acid (5i)
X-ray data analysis of compound 5b
| Empirical formula | C9 H8 N2O2 |
| Formula weight | 176.17 |
| Temperature | 293(2) K |
| Wavelength | 1.54184 A |
| Crystal system, space group | Monoclinic, P21/n |
| Unit cell dimensions | a = 9.615(2) Å |
| b = 8.524(2) Å β = 92.420(10)° | |
| c = 10.109(6) Å | |
| Volume | 827.8(6) A3 |
| Z, Calculated density | 4, 1.414 Mg/m3 |
| Absorption coefficient | 0.855 mm-1 |
| F(000) | 368 |
| Crystal size | 0.5 × 0.4 × 0.2 mm |
| Theta range for data collection | 6.80 to 59.39 deg. |
| Limiting indices | -10<=h<=0, -9<=k<=0, -11<=l<=11 |
| Reflections collected / unique | 1281 / 1201 [R(int) = 0.0134] |
| Completeness to theta = 59.39 | 99.2% |
| Absorption correction | None |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 1201 / 0 / 151 |
| Goodness-of-fit on F2 | 1.134 |
| Final R indices [I>2sigma(I)] | R1 = 0.0411, wR2 = 0.0980 |
| R indices (all data) | R1 = 0.0571, wR2 = 0.1054 |
| Extinction coefficient | 0.017(2) |
| Largest diff. peak and hole | 0.210 and -0.223 e. Å-3 |
Acknowledgments
References
- (a)Elguero, J. Comprehensive Heterocyclic Chemistry: Pyrazoles and their benzo derivatives; Katritzky, A. R., Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 5, pp. 167–303. [Google Scholar](b)Elguero, J. Comprehensive Heterocyclic Chemistry II: Pyrazoles; Katritzky, A. R., Rees, C. W., Scriven, E. F. V., Eds.; Pergamon Press: Oxford, 1996; Vol. 3, pp. 1–75. [Google Scholar]
- (a)Stadlbauer, W. Houben-Weyl, Methoden der Organischen Chemie: Indazole (Benzopyrazole); Schaumann, E., Ed.; Georg-Thieme-Verlag Stuttgart: New York, 1994; Vol. E8b, Hetarenes III/2; pp. 764–864. [Google Scholar](b)Stadlbauer, W. Science of Synthesis: Indazoles; Neier, R., Ed.; Georg-Thieme-Verlag Stuttgart: New York, 2002; Vol. 2.12.4, Hetarenes; pp. 227–324. [Google Scholar]
- Ikeda, Y.; Takano, N.; Matsushita, H.; Shiraki, Y.; Koide, T.; Nagashima, R.; Fujimura, Y.; Shindo, M.; Suzuki, S.; Iwasaki, T. Arzneim.-Forsch. 1979, 29, 511–520.
- Picciola, G.; Ravenna, F.; Carenini, G.; Gentili, P.; Riva, M. Farmaco Ed. Sci. 1981, 36, 1037–1056.
- The Merck Index, 12th ed.; Budavari, S. (Ed.) Merck & Co.: Rahway, New Jersey, 1996.
- Mosti, L.; Menozzi, G.; Fossa, P.; Schenone, P.; Lampa, E.; Parrillo, C.; D’Amisco, M.; Rossi, F. Farmaco 1992, 47, 567–584.
- Andronati, S.; Sava, V.; Makan, S.; Kolodeev, G. Pharmazie 1999, 54, 99–101.
- Kharitonov, V. G.; Sharma, V. S.; Magde, D.; Koesling, D. Biochemistry 1999, 38, 10699–10706.
- Rodgers, J. D.; Johnson, B. L.; Wang, H.; Greenberg, R. A.; Erickson-Viitanen, S.; Klabe, R. M.; Cordova, B. C.; Rayner, M. M.; Lam, G. N.; Chang, C.-H. Bioorg. Med. Chem. Lett. 1996, 6, 2919–2924. [CrossRef]Sun, J.-H.; Teleha, C. A.; Yan, J.-S.; Rodgers, J. D.; Nugiel, D. A. J. Org. Chem. 1997, 62, 5627–5629.
- Morie, T.; Harada, H.; Kato, S. Synth. Commun. 1997, 27, 559–566.Bermudez, J.; Fake, C. S.; Joiner, G. F.; Joiner, K. A.; King, F. D.; Miner, W. D.; Sanger, G. J. J. Med. Chem. 1990, 33, 1924–1929.
- Nofre, C.; Tinti, J. M.; Quar, F. FR Patent 26099603, 1988.
- Catalán, J.; Valle, J. C.; Claramunt, R. M.; Boyer, G.; Laynez, J.; Gómez, J.; Jiménez, P.; Tomás, F.; Elguero, J. J. Phys. Chem. 1994, 41, 10606–10612.
- Escande, A.; Lapasset, J.; Faure, R.; Vicenta, E.-J.; Elguero, J. Tetrahedron 1974, 30, 2903–2909.Faure, R.; Vicent, E. J.; Elguero, J. Heterocycles 1983, 20, 1713–1716.Catalán, J.; Paz, J. L. G.; Elguero, J. J. Chem. Soc., Perkin Trans. 2 1996, 57–60.Foces-Foces, C.; Hager, O.; Jagerovic, N.; Jimeno, M. L.; Elguero, J. Chem. Eur. J. 1997, 3, 121–126.
- Black, P. J.; Heffernan, M. L. Aust. J. Chem. 1963, 14, 1051–1055.Jaffari, G. A.; Nunn, A. J. J. Chem. Soc., Perkin Trans. 1 1973, 2371–2374.Palmer, M. H.; Findlay, R. H.; Kennedy, S. M. F.; McIntyre, P. S. J. Chem. Soc., Perkin Trans. 2 1975, 1695–1700.Brown, F. J.; Yee, Y. K.; Cronk, L. A.; Hebbel, K. C.; Krell, R. D.; Snyder, D. W. J. Med. Chem. 1990, 33, 1771–1781.
- Yamazaki, T.; Baum, G.; Shechter, H. Tetrahedon Lett. 1974, 49-50, 4421–4424.
- Begtrup, M.; Claramunt, R. M.; Elguero, J. J. Chem. Soc., Perkin Trans. 2 1978, 99–104.
- Cheung, M.; Boloor, A.; Stafford, J. A. J. Org. Chem. 2003, 68, 4093–4095.
- Elguero, J.; Fruchier, A.; Jacquier, R. Bull. Soc. Chim. Fr. 1966, 2075–2084.Buu-Hoï, N. P.; Hoeffinger, J.-P.; Jacquignon, P. Bull. Soc. Chim. Fr. 1967, 2019–2020.Elguero, J.; Fruchier, A.; Jacquier, R. Bull. Soc. Chim. Fr. 1969, 2064–2076.Elguero, J.; Fruchier, A.; Jacquier, R.; Scheidegger, U. J. Chim. Phys. Phys.-Chim. Biol 1971, 68, 1113–1121.Palmer, M. H.; Findlay, R. H.; Kennedy, S. M. F.; McIntyre, P. S. J. Chem. Soc., Perkin Trans. 2 1975, 1695–1700.Bouchet, P.; Fruchier, A.; Joncheray, G.; Elguero, J. Org. Magn. Reson. 1977, 9, 716–718.Stefaniak, L. Org. Magn. Reson. 1978, 11, 385–389.Elguero, J.; Fruchier, A.; Tjiou, E. M.; Trofimenko, S. Khim. Geterotsikl. Soedin. 1995, 9, 1159–1179.
- Kingsbury, W. D.; Gyurik, R. J.; Theodorides, V. J.; Parish, R. C.; Gallagher, G., Jr. J. Med. Chem. 1976, 19, 839–840. [CrossRef]
- Auwers, K. V.; Allardt, H. G. Ber. 1926, 59, 95–100.
- Levy, D. E.; Smyth, M. S.; Scarborough, R. M. WO Patent 03/022214, 2003.
- Costa, M. R. G.; Curto, M. J. M.; Davies, S. G.; Duarte, M. T.; Resende, C.; Teixeira, F. C. J. Organomet. Chem. 2000, 604, 157–169.
- Farrugia, L. J. J. Appl. Cryst. 1997, 30, 565, (based on ORTEP-III (v.1.0.3.) by Johnson C. K.; Burnett, M. N.).
- Allen, F. H. Acta Cryst. 2002, B58, 380–388.Bruno, I. J.; Cole, J. C.; Edginton, P. R.; Kessler, M.; Macrae, C. F.; MacCabe, P. M.; Pearson, J.; Taylor, R. Acta Cryst. 2002, B58, 389–397.
- Ferguson, G.; Gallaghe, J. F.; McAlees, A. J. Acta Cryst. 1995, C51, 454–458.Bernstein, J.; Davies, R. E.; Shimoni, L.; Chang, N.-L. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [CrossRef]Allen, F. H.; Motherwell, W. D. S.; Raithby, P. R.; Shields, G. P.; Taylor, R. New J. Chem. 1999, 25–34.
- Foces-Foces, C. Acta Cryst. 2005, E61, o337–o339.Hager, O.; Foces-Foces, C.; Jagerovic, N.; Elguero, J.; Trofimenko, S. Acta Cryst. 1996, C52, 2894–2896.
- Foces-Foces, C.; Echevarría, A.; Jagerovic, N.; Alkorta, I.; Elguero, J.; Langer, U.; Klein, O.; Minguet-Bomvehí, M.; Limbach, H.-H. J. Am. Chem. Soc. 2001, 123, 7898–7906.Foces-Foces, C.; Alkorta, I.; Elguero, J. Acta Cryst. 2000, B56, 1018–1028.Ching, N.; Pan, L.; Huang, X.; Li, J. Acta Cryst. 2000, C56, 1124–1125.
- Boa, A. N.; Crane, J. D. Acta Cryst. 2004, E60, o966–o967.
- Aakeröy, C. B.; Salmon, D. J. Cryst. Eng. Comm. 2005, 7, 439–448.
- Perrin, D. D.; Armarego, W. L. F.; Perrin, D. R. Purification of Laboratory Chemicals, 2nd ed.; Pergamon Press: Oxford, 1980. [Google Scholar]
- Konoike, T.; Araki, Y.; Hayashi, T.; Sakurai, K.; Tozyo, T. EP Patent 628569, 1994.
- Altomare, A.; Burla, M. C.; Camalli, M.; Cascarano, G. L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A. G. G.; Polidori, G.; Spagna, R. J. Appl. Cryst. 1999, 32, 115–119. [CrossRef]
- Sheldrick, G. M. SHELXL, A Program for Refining Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Farrugia, L. J. J. Appl. Cryst. 1999, 32, 837–837. [CrossRef]
- Sample Availability: Available from authors.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Teixeira, F.C.; Ramos, H.; Antunes, I.F.; Curto, M.J.M.; Duarte, M.T.; Bento, I. Synthesis and Structural Characterization of 1- and 2-Substituted Indazoles: Ester and Carboxylic Acid Derivatives. Molecules 2006, 11, 867-889. https://doi.org/10.3390/11110867
Teixeira FC, Ramos H, Antunes IF, Curto MJM, Duarte MT, Bento I. Synthesis and Structural Characterization of 1- and 2-Substituted Indazoles: Ester and Carboxylic Acid Derivatives. Molecules. 2006; 11(11):867-889. https://doi.org/10.3390/11110867
Chicago/Turabian StyleTeixeira, Fátima C., Hélène Ramos, Inês F. Antunes, M. João M. Curto, M. Teresa Duarte, and Isabel Bento. 2006. "Synthesis and Structural Characterization of 1- and 2-Substituted Indazoles: Ester and Carboxylic Acid Derivatives" Molecules 11, no. 11: 867-889. https://doi.org/10.3390/11110867
APA StyleTeixeira, F. C., Ramos, H., Antunes, I. F., Curto, M. J. M., Duarte, M. T., & Bento, I. (2006). Synthesis and Structural Characterization of 1- and 2-Substituted Indazoles: Ester and Carboxylic Acid Derivatives. Molecules, 11(11), 867-889. https://doi.org/10.3390/11110867