An Improved Synthesis of Some 5-Substituted Indolizines Using Regiospecific Lithiation

Abstract
:Introduction
- –
- optimization of the protocol for direct lithiation of 2- and 1,2- substituted indolizines by varying temperature and time of metallation;
- –
- discovery of a novel and easy route to 5-formyl and 5-haloindolizines by means of direct lithiation followed by electrophilic quench;
- –
- the first Suzuki type cross-coupling reaction between 5-iodoindolizines and a boronic acid;
- –
- follow-up of the chemistry of 5-formylindolizines and their derivatives;
- –
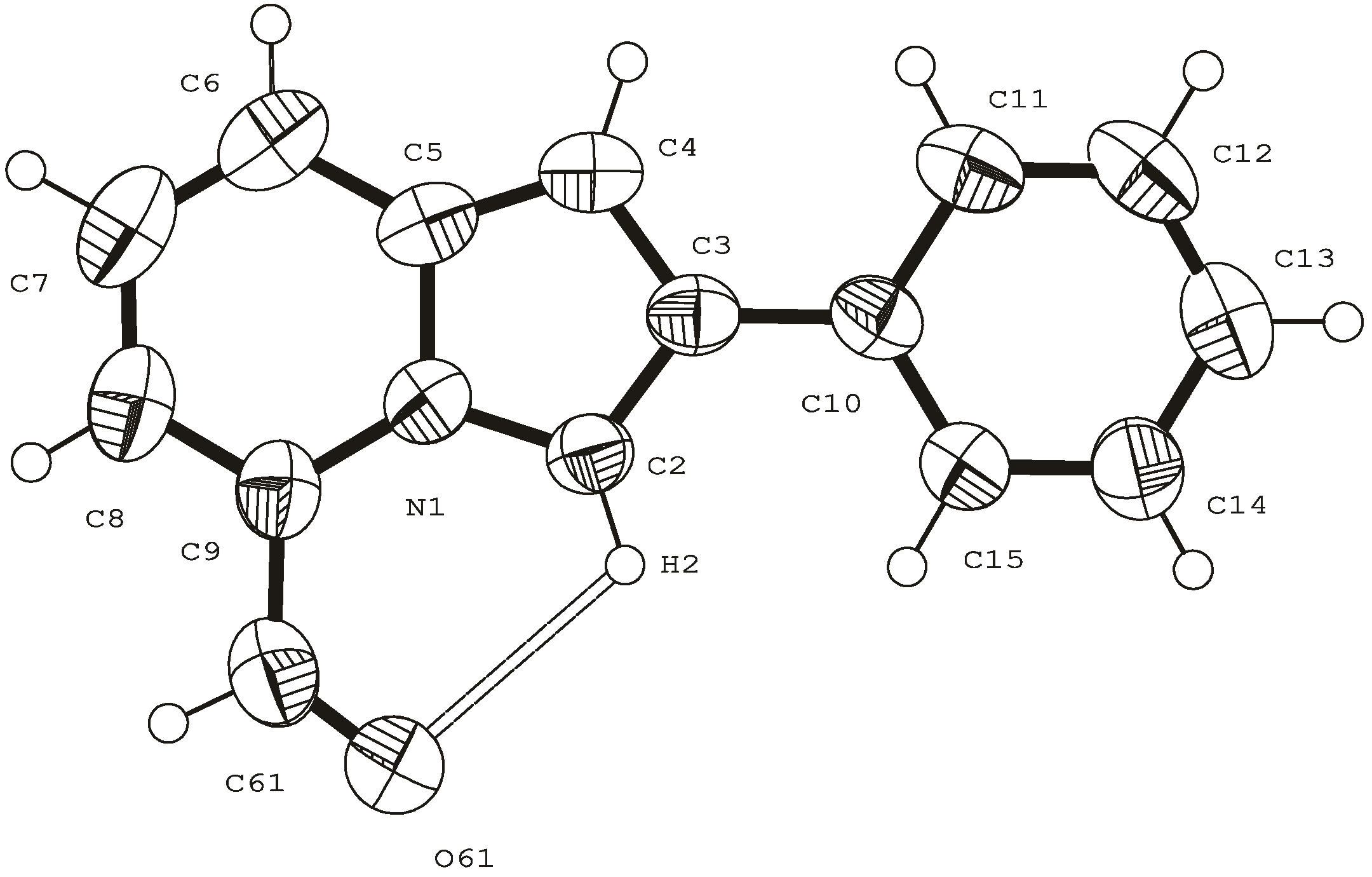
- direct proof of the structure of 5-formyl-2-phenylindolizine by X-ray analysis.
Results and Discussion
Preparation of indolizines.


{kind=link}
| Substances 1 | R1 | R2 |
|---|---|---|
| 1a | H | Ph |
| 1b | H | tBu |
| 1c | Me | tBu |
Direct lithiation of indolizines.

Preparation of 5-substituted indolizines

| Indolizine | E | Product | Yield,% | Mp, °C |
|---|---|---|---|---|
| 1a | CHO | 2a | 74 | 128-130 |
| 1b | CHO | 2b | 94 | 81-83 |
| 1c | CHO | 2c | 95 | 37-39 |
| 1a | C(O)Ph | 2d | 55 | 284-286 (lit. 286) [1] |
| 1b | I | 2e | 96 | 57-59 |
| Chemical shifts, ppm (J, Hz) | |||||||
|---|---|---|---|---|---|---|---|
| No. | H1, s | H3, s | H6, d (J67) | H7, m | H8, d (J78) | CHO, s | HR1,R2 |
| 2a | 7.04 | 9.18 | 7.83 (8.7) | 6.95 | 7.52 (6.9) | 9.88 | m: 7.71; 7.39; 7.24 (2-Ph) |
| 2b | 6.61 | 8.71 | 7.71 (8.6) | 6.86 | 7.42 (7.2) | 9.81 | s, 1.37 (2-tBu) |
| 2c | - | 8.65 | 7.71 (8.7) | 6.78 | 7.35 (7.4) | 9.75 | s, 2.47 (1-CH3); s, 1.42 (2- tBu) |
Reactivity of the 5-COR group


| 5-Formylindolizine | Oxime | Yield,% | Mp, °C |
|---|---|---|---|
| 2a | 3a | 89 | 82-84 |
| 2b | 3b | 96 | 49-51 |
| 2c | 3c | quant. | 69-71 |
| No. | Chemical shifts, ppm (J, Hz) | |||||||
|---|---|---|---|---|---|---|---|---|
| H1, s | H3, s | H6, d (J67) | H7, m | H8, d (J78) | CH=N, s | OMe, s | HR1,R2 | |
| 3a | 6.92 | 8.84 | 7.56 (9.1) | 6.81 | 6.93 (6.9) | 8.40 | 4.13 | m: 7.68; 7.38; 7.23 (2-Ph) |
| 3b | 6.50 | 8.34 | 7.43 (8.9) | 6.72 | 6.82 (6.7) | 8.31 | 4.07 | s, 1.37 (2-tBu) |
| 3c | - | 8.30 | 7.43 (8.7) | 6.65 | 6.76 (6.7) | 8.26 | 4.05 | s, 2.46 (1-CH3); s, 1.41 (2- tBu) |
Reduction of the 5-formyl group.

| 5-Formylindolizine | Product | Yield,% | Mp, °C |
|---|---|---|---|
| 2a | 4a | quant. | 110-112 |
| 2b | 4b | quant. | 82 |
| 2c | 4c | quant. | 76-78 |
| No. | Chemical shifts, ppm (J, Hz) | |||||||
|---|---|---|---|---|---|---|---|---|
| H1, s | H3, s | H6, d (J67) | H7, m | H8, d (J78) | OCH2, d (JCH2OH) | -OH, t (JCH2OH) | HR1,R2 | |
| 4a | 6.73 | 7.77 | 7.34* | 6.70 | 6.58 (6.6) | 4.72** | 5.33** | m: 7.68; 7.39*; 7.19 (2-Ph) |
| 4b | 6.31 | 7.19 | 7.21 (9.3) | 6.22 | 6.49 (6.3) | 4.64 (4.5) | 5.24 (4.5) | s, 1.35 (2-tBu) |
| 4c | - | 7.10 | 7.19 (9.4) | 6.56 | 6.42 (6.9) | 4.60 (5.4) | 5.19 (5.4) | s, 2.40 (1-CH3); s, 1.40 (2- tBu) |
Suzuki-type cross-coupling reaction between 5-iodoindolizine and arylboronic acid.

Conclusions
Experimental
General
Procedure for direct lithiation of indolizine 1 and preparing 5-formylindolizines 2.
Typical procedure for preparation of O-Me oximes 3.
Typical procedure for reduction of 5-formylindolizines 2.
Preparation of 5-iodo-2-tert-butylindolizine 2e via direct lithiation.
Cross-coupling reaction between 5-iodo-2-tert-butylindolizine 2e and p-methoxybenzeneboronic acid.
Acknowledgments
References
- Renard, M.; Gubin, J. Metallation of 2-Phenylindolizine. Tetrahedron Lett. 1992, 33, 4433–4434. [Google Scholar] [CrossRef]
- Bobrovskii, S. I.; Babaev, E. V.; Bundel, Yu. G. Base-promoted deutherium exchange in the indolizine nucleus. Chem. Heterocycl. Comp. (Engl. Transl.) 1987, 9, 1032. [Google Scholar]
- Babaev, E. V.; Efimov, A.V. Novel approach to synthesis of indolizine nucleus and the class of 5-aminoindolizines via recyclization of 5-methyloxazolo[3,2-a]pyridinium cation. Chem. Heteroc. Compounds (Engl. Transl.) 1997, 33, 7, 875-876. [Google Scholar]
- Yasuyoshi, M.; Toshihiko, Y.; Masazumi, I. A Novel Synthesis of Pyrrolo[2,1,5-de]quinolizinones (Cycl[3.3.2]azinones). Heterocycles 1987, 26(1), 199. [Google Scholar]
- Xin, J.; Ting, H.; Yuefei, H.; Hongwen, H. Novel Synthetic Routes to Nitrogen-Bridged Tricyclic Derivatives of Pyrrolo [2,1,5-cd]indolizine and Pyrrolo[2,1,5-de]quinolizine Derived from 2-Acyl-N-(acylmethyl)pyridinium Halides. J. Chem. Soc. Perkin Trans. I. 2001, 15, 1820. [Google Scholar]
- Chichibabin, A. E. Tautomerism in the pyridine series. Chem. Ber. 1927, 60B, 1607–17. [Google Scholar]
- Armarego, W. L. F. Ionization and ultraviolet spectra of indolizines. J. Chem. Soc. 1964, 4226–33. [Google Scholar]
- Reid, D. H.; Webster, R. G.; McKenzie, S. Studies of heterocyclic compounds. Part 25. Stable indolizine and pyrrolo[2,1-b]thiazole carboselenaldehydes. J. Chem. Soc. Perkin Trans. 1 1979, 2334–9. [Google Scholar]
- Babaev, E. V.; Torocheshnikov, V. N.; Bobrovsky, S. I. NMR spectra of indolizines and their sigma-complexes. Chem. Heterocycl. Comp. (Engl. Transl.) 1995, 31, 1079–1087. [Google Scholar]
- Babaev, E.V.; Efimov, A.V.; Maiboroda, D.A. Hetarenes with a nitrogen bridging atom. 1. Phenacylation of 2-substituted 6-methylpyridines. Chem. Heterocycl. Comp. (Engl. Transl.) 1995, 31, 962–968. [Google Scholar]
- Babaev, E. V.; Pasichnichenko, K. Yu.; Rybakov, V. B.; Zhukov, S. G. Heterocyclic compounds with a bridge nitrogen atom. 14. Cycloaddition of acetylenedicarboxylic acid ester to 2-chloro-N-phenacylpyridinium ylide. Crystal structure of dimethyl ester of 5-chloro-3-(p-nitrobenzoyl)-indolizine-1,2-dicarboxylic acid. Chem. Heterocycl. Comp. 2000, 36, 1192–1197. [Google Scholar]
- Terentyev, P. B.; Vinogradova, S. M.; Kost, A. N. Reaction of 2- and 4-vinylpyridines with phenacylpyridinium ylides. Chem. Heterocycl. Comp. (in Russian) 1980, 5, 651–656, [Chemical Abstr. 1980, 93: 220542]. [Google Scholar]
- Borrows, E. T.; Holland, D. O.; Pyrrocolines, V. The action of Grignard reagents and of iodine on 3-acetyl-2-phenylpyrrocoline. J. Chem. Soc. 1947, 670. [Google Scholar]
- Ogata, K. Heterocyclic compounds. II. Synthesis of indolizine derivatives. Yakugaku Zasshi 1969, 89, 1020. [Google Scholar]
- Gundersen, L.; Negussie, A. H.; Rise, F.; Oestby, O. B. Antimycobacterial Activity of 1-Substituted Indolizines. Arch. Pharm. 2003, 336, 191. [Google Scholar] [CrossRef]
- Jones, G.; Sliskovic, D. R. Triazolopyridines. Part 2. Preparation of 7-substituted triazolo[1,5-a]pyridines by directed lithiation. J. Chem. Soc. Perkin Trans. I. 1982, 967. [Google Scholar]
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Kuznetsov, A.G.; Bush, A.A.; Rybakov, V.B.; Babaev, E.V. An Improved Synthesis of Some 5-Substituted Indolizines Using Regiospecific Lithiation. Molecules 2005, 10, 1074-1083. https://doi.org/10.3390/10091074
Kuznetsov AG, Bush AA, Rybakov VB, Babaev EV. An Improved Synthesis of Some 5-Substituted Indolizines Using Regiospecific Lithiation. Molecules. 2005; 10(9):1074-1083. https://doi.org/10.3390/10091074
Chicago/Turabian StyleKuznetsov, Alexey G., Alexander A. Bush, Viktor B. Rybakov, and Eugene V. Babaev. 2005. "An Improved Synthesis of Some 5-Substituted Indolizines Using Regiospecific Lithiation" Molecules 10, no. 9: 1074-1083. https://doi.org/10.3390/10091074
APA StyleKuznetsov, A. G., Bush, A. A., Rybakov, V. B., & Babaev, E. V. (2005). An Improved Synthesis of Some 5-Substituted Indolizines Using Regiospecific Lithiation. Molecules, 10(9), 1074-1083. https://doi.org/10.3390/10091074