The 5-Endo-trig Cyclization of D-Glucose Derived γ-Alkenylamines with Mercury (II) Salts: Synthesis of 1-Deoxycastanospermine and its 8a-epi-Analogue†

{kind=link}
{kind=link}
{kind=link}
Abstract
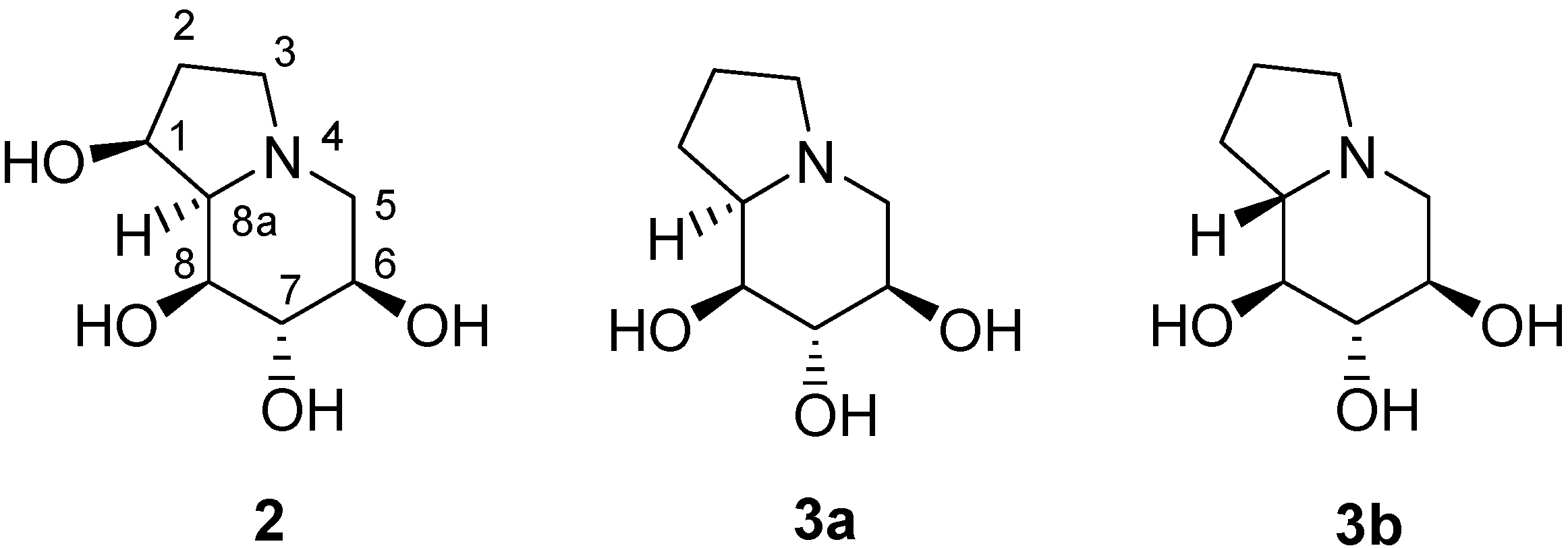
:Introduction
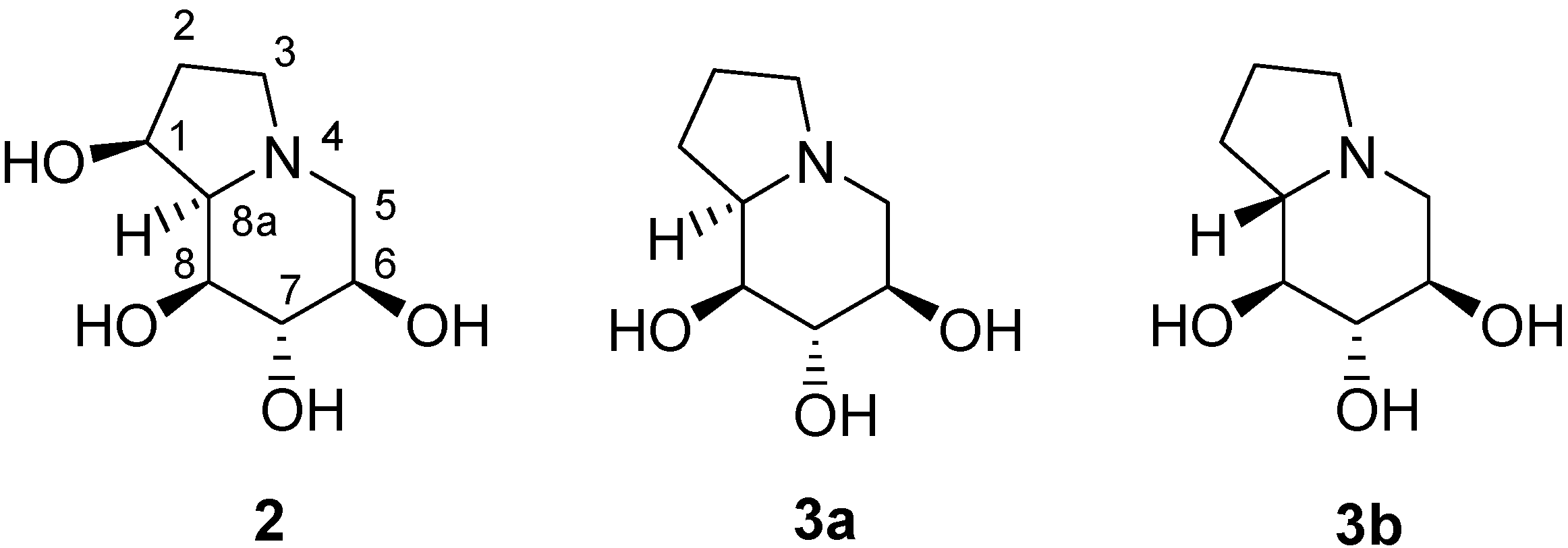
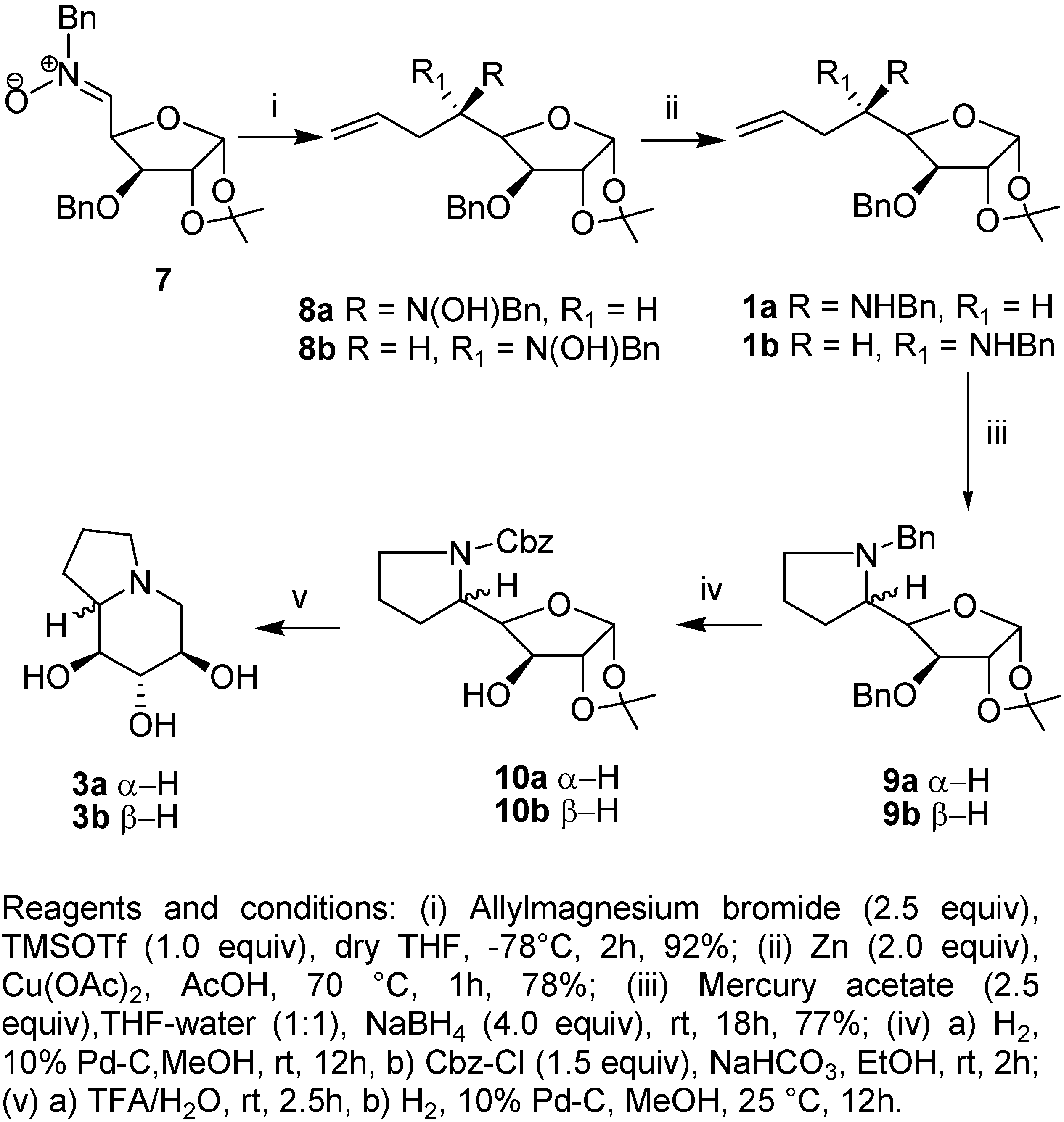
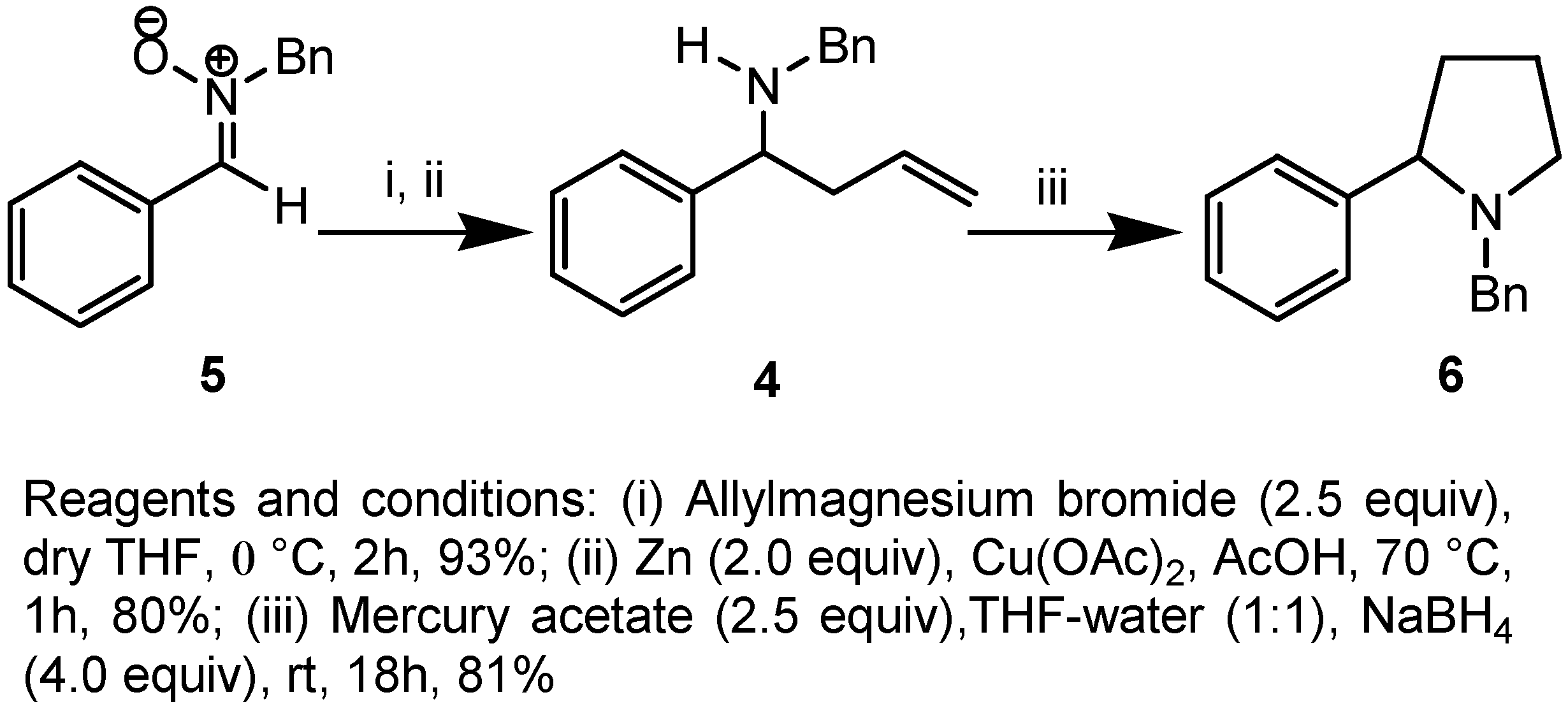
Results and Discussion
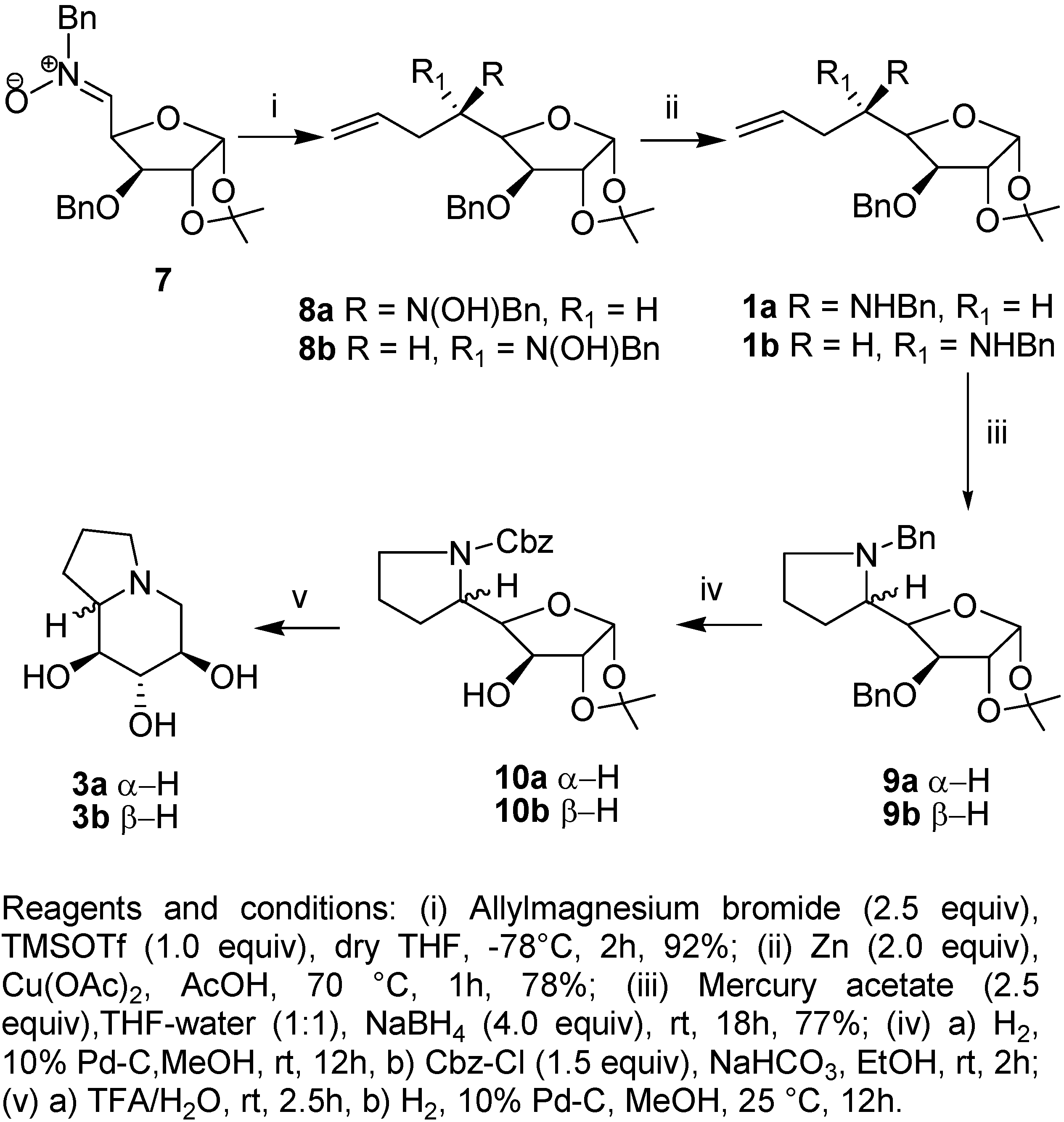
Conclusions
Experimental
General
Preparation of N-benzyl-N-(1-phenylbut-3-enyl)amine (4).
Preparation of N-benzyl-2-phenyl pyrrolidine (6).
Preparation of 5,6,7,8-tetradeoxy-5,8-(N-benzylimino)-1,2-O-isopropylidene-α-D–gluco-oct-1,4- furanose (9a).
Preparation of 5,6,7,8-tetradeoxy-5,8-(N-benzylimino)-1,2-O-isopropylidene-β-L-ido-oct-1,4 furanose (9b).
Preparation of 5,6,7,8-tetradeoxy-5,8-(N-benzoxycarbonylimino)-1,2-O-isopropylidene-α-D-gluco-oct-1,4-furanose (10a).
Preparation of 5,6,7,8-Tetradeoxy-5,8-(N-benzoxycarbonylimino)-1,2-O-isopropylidene-β-L-ido-oct-1,4-furanose (10b).
Preparation of (6S,7R,8R,8aR)-6,7,8-Trihydroxy-indolizidine (3a).
Preparation of (6S,7R,8R,8aS)-6,7,8-Trihydroxy-indolizidine (3b).
Acknowledgements
References and Notes
- Gasc, M. B. Tetrahedron 2002, 58, 705–711.Cardillo, G.; Orena, M. Tetrahedron 2002, 58, 3386–3395.Gasc, M. B.; Lattes, A.; Perie, J. J. Tetrahedron 1983, 39, 703.Perrie, J. J.; Laval, J. P.; Roussel, J.; Lattes, A. Tetrahedron 1971, 28, 675.
- Tokuda, M.; Yasufumi, Y.; Suginome, H. Chem. Lett. 1988, 1280–1290.Fraga, A. M.; Pencnha, E. P.; Verli, H. Tetrahedron Lett. 2002, 43, 1607–1611.Schumacher-W, M. H. G.; Peterson, S.; Peter, M. G. Liebigs Ann. Chem. 1994, 555–561.Le Moigne, F.; Tordo, P. J. Org. Chem. 1994, 59, 3365.Arnone, A.; Bravo, P.; Donadelli, A.; Resnati, G. J. Chem. Soc., Chem. Commun. 1993, 984.Stipa, P.; Finet, J. P.; Le Moigne, F.; Tordo, P. J. Org. Chem. 1993, 58, 4465.Chikkanna, D.; Han, H. Synlett 2004, 2311.Weis, C. D.; Newkome, G. R. J. Org. Chem. 1990, 55, 5801–5802.
- The 5-endo-trig radical- and/or iodo-cyclization of γ-alkenylamines are known to give pyrrolidine ring skeletons. See: Chatgilialoglu, C.; Ferreri, C.; Guerra, M.; Timokhin, V.; Froudakis, G.; Gimisis, T. J. Am. Chem. Soc. 2002, 124, 10765. [CrossRef]Knight, D. W.; Redfern, A. L.; Gilmore, J. J. Chem. Soc., Perkin Trans. 1. 2001, 2874.Lee, W. S.; Jang, K. C.; Kim, J. H.; Park, K. H. J. Chem. Soc., Chem. Commun. 1999, 251.
- Jachak, S. M.; Dhavale, D. D.; Karche, N. P.; Trombini, C. Tetrahedron 2004, 60, 3009.Jachak, S. M.; Dhavale, D. D.; Karche, N. P.; Trombini, C. Synlett 2004, 1549.
- Burgess, K.; Henderson, I. Tetrahedron 1992, 48, 4045.Izquierdo, I.; Plaza, M. T.; Robles, R.; Mota, A. J. Tetrahedron: Asymmetry 1998, 9, 1015.Jirousek, M. R.; Cheung, A. W-H.; Babine, R. E.; Sass, P. M.; Schow, S. R.; Wick, M. M. Tetrahedron Lett. 1993, 34, 3671.Marek, D.; Wadouachi, A.; Uzan, R.; Beaupere, D.; Nowogrocki, G.; Laplace, G. Tetrahedron Lett. 1996, 37, 49.Zhao, H.; Mootoo, D. R. J. Org. Chem. 1996, 61, 6762.Maggini, M.; Prato, M.; Ranelli, M.; Scorrano, G. Tetrahedron Lett. 1992, 33, 6537.Kang, S. H.; Kim, J. S. J. Chem. Soc., Chem. Commun. 1998, 1353.Kefalas, P.; Grierson, D. S. Tetrahedron Lett. 1993, 34, 3555.Ina, H.; Kibayashi, C. J. J. Org. Chem. 1993, 58, 52. [CrossRef]Furneaux, R. H.; Mason, J. M.; Tyler, P. C. Tetrahedron Lett. 1994, 35, 3143.Overkleeft, H. S.; Pandit, U. K. Tetrahedron Lett. 1996, 37, 547.Mulzer, J.; Dehmlow, H. J. J. Org. Chem. 1992, 57, 3194. [CrossRef]Burgess, K.; Chaplin, D. A.; Henderson, I.; Pan, Y. T.; Elbein, A. D. J. Org. Chem. 1992, 57, 1103. [CrossRef]Kim, N.-S.; Choi, J.-R.; Cha, J. K. J. Org. Chem. 1993, 58, 7096. [CrossRef]Furneaux, R. H.; Masson, J. M.; Tyler, P. C. Tetrahedron Lett. 1995, 36, 3055. [CrossRef]
- Truscheit, E.; Frommer, W.; Junge, B.; Muller, L.; Schmidt, D. D.; Wingender, W. Angew. Chem., Int. Ed. Engl. 1981, 20, 744. [CrossRef]Furneaux, R. H.; Gainsford, G. J.; Mason, J. M.; Tyler, P. C.; Hartley, O.; Winchester, B. G. Tetrahedron 1997, 57, 245.
- Humphries, M. J.; Matsumoto, K.; White, S. L. Cancer Res. 1986, 46, 5215.Humphries, M. J.; Matsumoto, K.; White, S. L.; Olden, K. Cancer Res. 1986, 46, 5215.
- Karples, A.; Fleet, G. W. J.; Dwek, R. A.; Petursson, S.; Namgoong, S. K.; Ramsden, N. G.; Jacob, G. S.; Rademacher, T. W. Proc. Natl. Acad. Sci. U.S.A. 1998, 85, 9229.Walker, B. D.; Kowalski, M.; Goh, W. C.; Kozarsky, K.; Krieger, M.; Rosen, C.; Rohrschneider, L.; Haseltine, W. A.; Sodroski, J. Proc. Natl. Acad. Sci. 1987, 84, 8120.Sunkara, P. S.; Bowling, T. L.; Liu, P. S.; Sjoerdsma, A. Biochem. Biophys. Res. Commun. 1987, 148, 206.
- Dhavale, D.; Desai, V.; Sindkhedkar, M.; Mali, R.; Castellari, C.; Trombini, C. Tetrahedron: Asymm. 1997, 1475.Dhavale, D.; Saha, N.; Desai, V. J. Org. Chem. 1997, 62, 7482.Dhavale, D.; Saha, N.; Desai, V. J. Org. Chem. 1999, 64, 1715.Dhavale, D.; Saha, N.; Desai, V. J. Chem. Soc., Chem. Commun. 1999, 1719.Saha, N.; Desai, V.; Dhavale, D. Tetrahedron 2001, 57, 39.Dhavale, D. D.; Patil, N. T.; Tilekar, J. N. J. Org. Chem. 2001, 66, 1065.Dhavale, D. D.; Patil, N. T.; Tilekar, J. N. Tetrahedron Lett. 2001, 42, 747.Dhavale, D.; Patil, N.; John, S.; Sabharwal, S. Bioorg. Med. Chem. 2002, 10, 2155.Dhavale, D.; Saha, N.; Desai, V.; Tilekar, J. Arkivoc 2002, (VII), 91.Patil, N.; Tilekar, J.; Jadhav, H.; Dhavale, D. Tetrahedron 2003, 59, 1873.
- In general, the intramolecular exo-trig nucleophilic addition reaction to a double bond, for rings smaller than five membered, is favored over the endo-trig one. See: Johnson, C. D. Acc. Chem. Res. 1993, 476.Baldwin, J. E.; Thomas, R. C.; Lawrence, I. K.; Lee, S. J. Org. Chem. 1997, 42, 3846.
- Franco, S.; Junquera, F.; Merchan, F.; Mercino, P.; Tejero, T. Syn. Commun. 1994, 2537.
- The observed rotation value of 10a was consistent with the reported value. The 1H- and 13C-NMR spectra were also found to be in good agreement with the structure [9f].
- Sample availability: Contact the authors.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
Dhavale, D.; Jachak, S. The 5-Endo-trig Cyclization of D-Glucose Derived γ-Alkenylamines with Mercury (II) Salts: Synthesis of 1-Deoxycastanospermine and its 8a-epi-Analogue†. Molecules 2005, 10, 893-900. https://doi.org/10.3390/10080893
Dhavale D, Jachak S. The 5-Endo-trig Cyclization of D-Glucose Derived γ-Alkenylamines with Mercury (II) Salts: Synthesis of 1-Deoxycastanospermine and its 8a-epi-Analogue†. Molecules. 2005; 10(8):893-900. https://doi.org/10.3390/10080893
Chicago/Turabian StyleDhavale, D., and S. Jachak. 2005. "The 5-Endo-trig Cyclization of D-Glucose Derived γ-Alkenylamines with Mercury (II) Salts: Synthesis of 1-Deoxycastanospermine and its 8a-epi-Analogue†" Molecules 10, no. 8: 893-900. https://doi.org/10.3390/10080893
APA StyleDhavale, D., & Jachak, S. (2005). The 5-Endo-trig Cyclization of D-Glucose Derived γ-Alkenylamines with Mercury (II) Salts: Synthesis of 1-Deoxycastanospermine and its 8a-epi-Analogue†. Molecules, 10(8), 893-900. https://doi.org/10.3390/10080893