Chromate Oxidation of α-Nitro Alcohols to α-Nitro Ketones: Significant Improvements to a Classic Method

Abstract
:Introduction
Results and Discussion
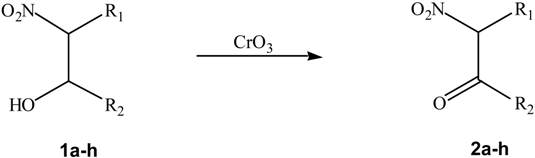

| 2b | C2H5 | CH3 | 95 |
| 2c | CH3 | (CH2)2CH3 | 93 |
| 2d | CH3 | (CH2)3CH3 | 93 |
| 2e | CH3 | (CH2)5CH3 | 87 |
| 2f | CH3 | (CH2)6CH3 | 85 |
| 2g | CH3 | CH2CH(CH3)2 | 90 |
Conclusions
Experimental
General
General Procedure for the Oxidation Reaction.
Acknowledgements
References and Notes
- Sato, N; Matsura, T. J. Chem. Soc. Perkin Trans. 1. 1996, 2345.
- Ballini, R; Bosica, G; Parrini, M. Tetrahedron Lett. 1998, 39, 7963.
- Mélot, J-M; Françoise, T-B; Foucaud, A. Tetrahedron Lett. 1986, 27, 493.
- Hurd, C. D; Nilson, M. E. J. Org. Chem. 1955, 20, 927.
- Seebach, D; Beck, A. K; Mukhopadhyay, T; Thomas, E. Helv. Chim. Acta 1982, 65, 1101.
- Corey, E. J; Suggs, J. W. Tetrahedron Lett. 1975, 16, 2647.
- Omura, K; Swern, D. Tetrahedron 1978, 34, 1651.Mancuso, A. J; Swern, D. Synthesis 1981, 3, 165.
- Crumbie, R. L; Nimitz, J. S; Mosher, H. S. J. Org. Chem. 1982, 47, 4040.
- Henry; Battice, De. Bull. Acad. Roy. Belg. 1898, 36, 149, [Chem. Zentr. 1898, II, 887].Henry. Ber. 1899, 32, 865.
- The 1H-NMR spectrum of 2b is interesting in that the resonance at 5.1 ppm appears as a doublet of doublets. The connectivity of the –CH(NO2)CH2CH3 was verified by a COSY experiment: the dd at δ 5.1 correlates with the m at δ 2.1, which correlates with the t at δ 1.05. A simple rigid search of the rotation barrier about the C2-C3 bond using CAChe molecular modeling software gave a rotational barrier of 82 kcal/mole. This indicates that the barrier to rotation is large and explains the observed dd at 5.1 ppm for 2b.
- Sample Availability: Available from the authors.
© 2005 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes
Share and Cite
Elmaaty, T.A.; Castle, L.W. Chromate Oxidation of α-Nitro Alcohols to α-Nitro Ketones: Significant Improvements to a Classic Method. Molecules 2005, 10, 1458-1461. https://doi.org/10.3390/10121458
Elmaaty TA, Castle LW. Chromate Oxidation of α-Nitro Alcohols to α-Nitro Ketones: Significant Improvements to a Classic Method. Molecules. 2005; 10(12):1458-1461. https://doi.org/10.3390/10121458
Chicago/Turabian StyleElmaaty, Tarek Abou, and Lyle W. Castle. 2005. "Chromate Oxidation of α-Nitro Alcohols to α-Nitro Ketones: Significant Improvements to a Classic Method" Molecules 10, no. 12: 1458-1461. https://doi.org/10.3390/10121458
APA StyleElmaaty, T. A., & Castle, L. W. (2005). Chromate Oxidation of α-Nitro Alcohols to α-Nitro Ketones: Significant Improvements to a Classic Method. Molecules, 10(12), 1458-1461. https://doi.org/10.3390/10121458