An Efficient Protocol for the Solid-phase Synthesis of Malondiamides

Abstract
:Introduction
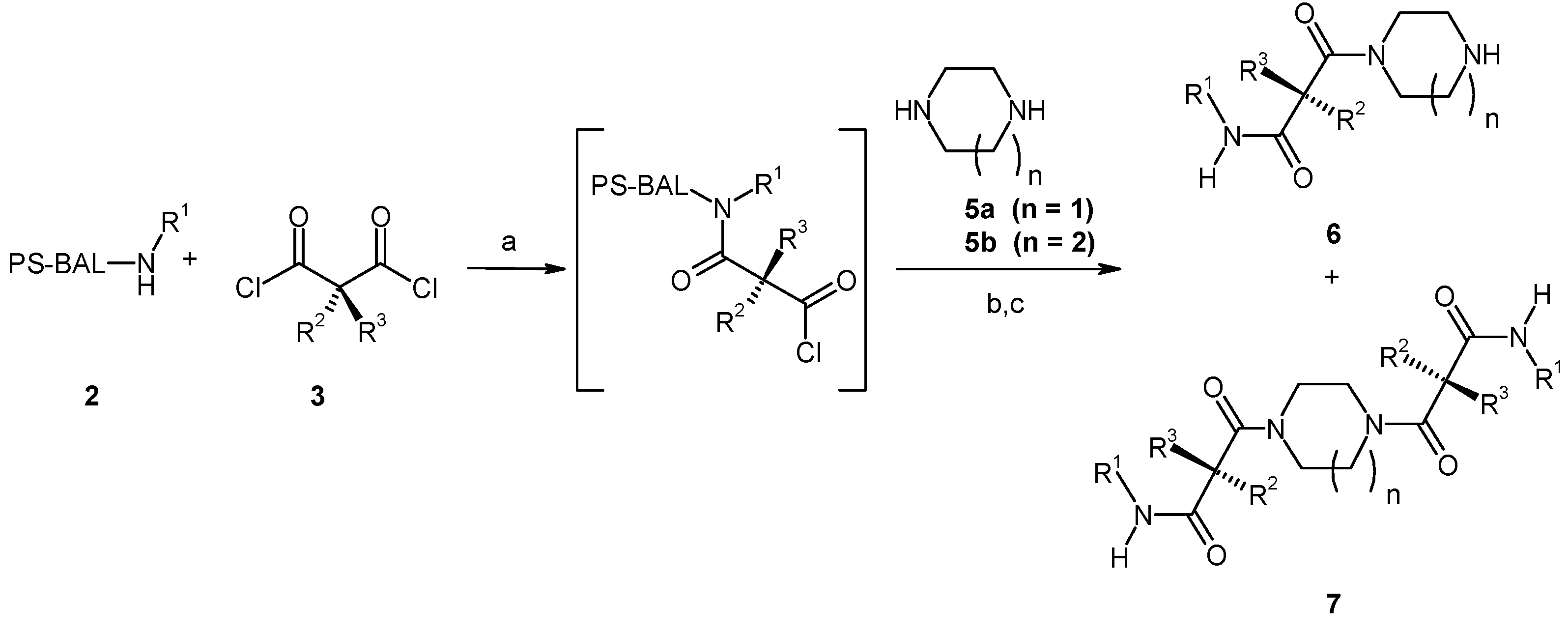
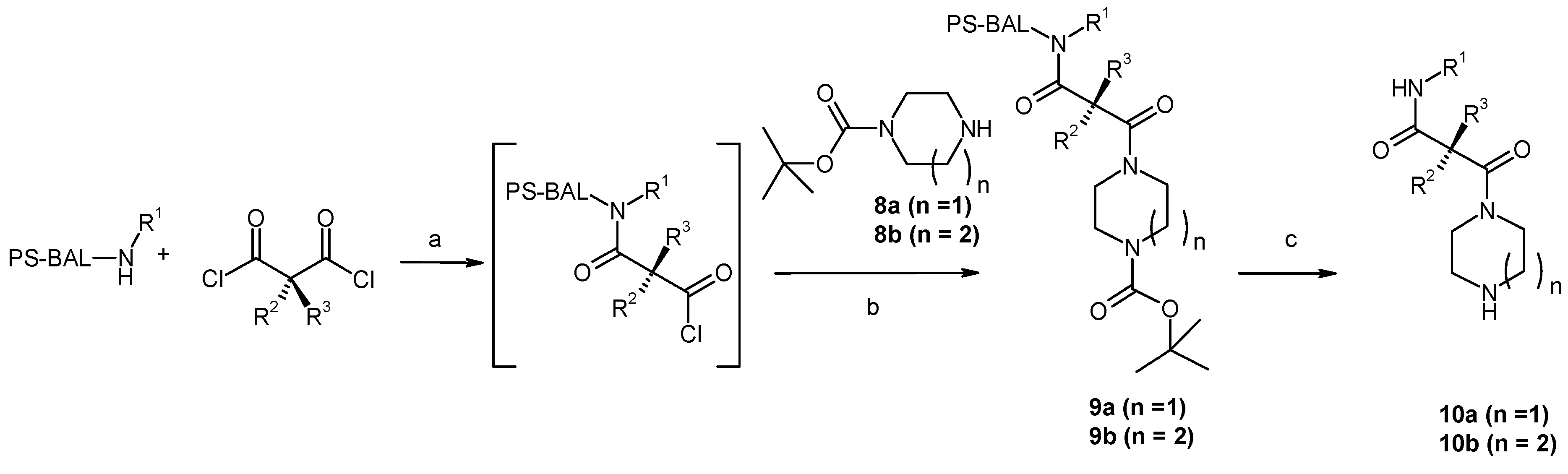
Results and Discussion
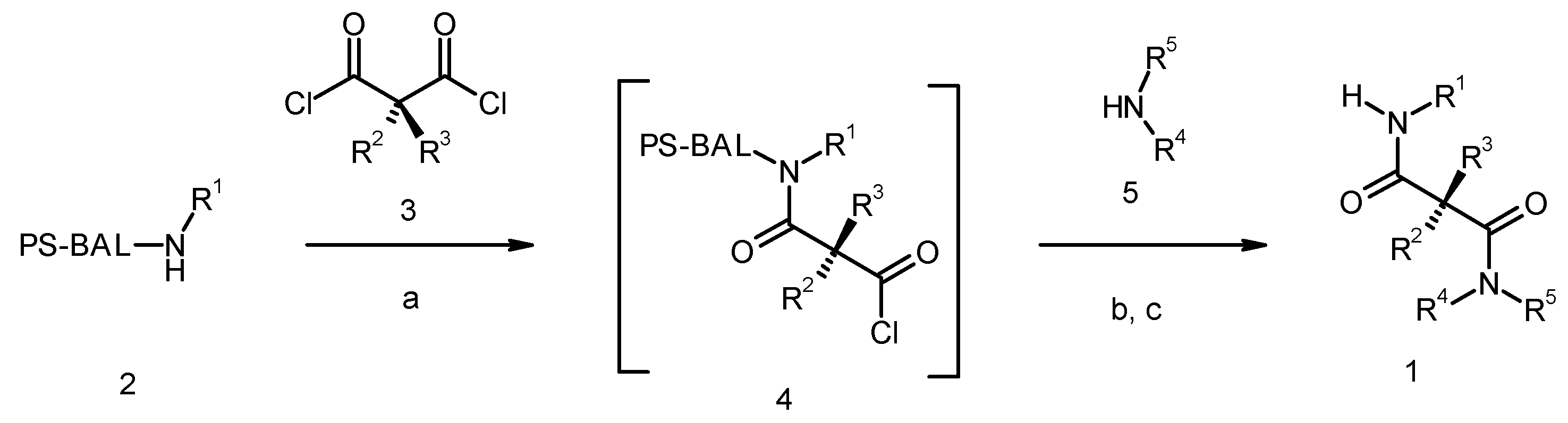

{kind=link}
{kind=link}
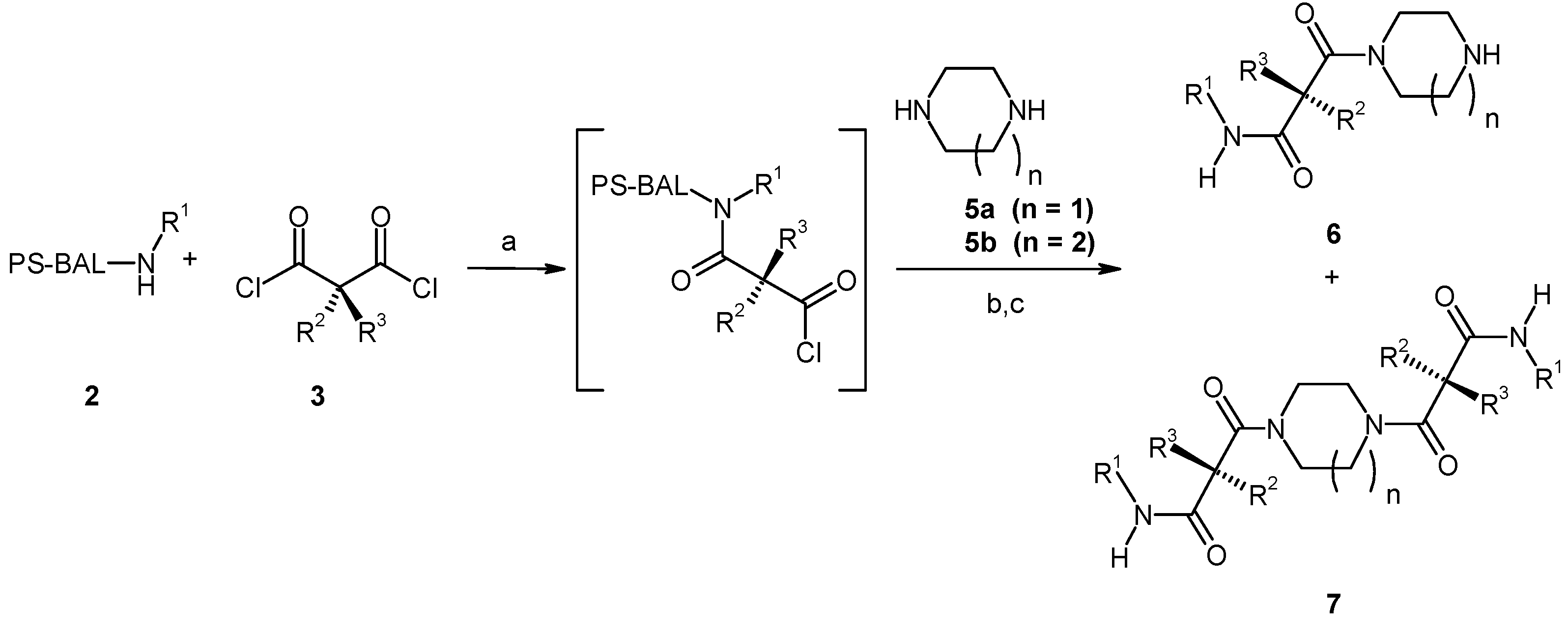{kind=link}
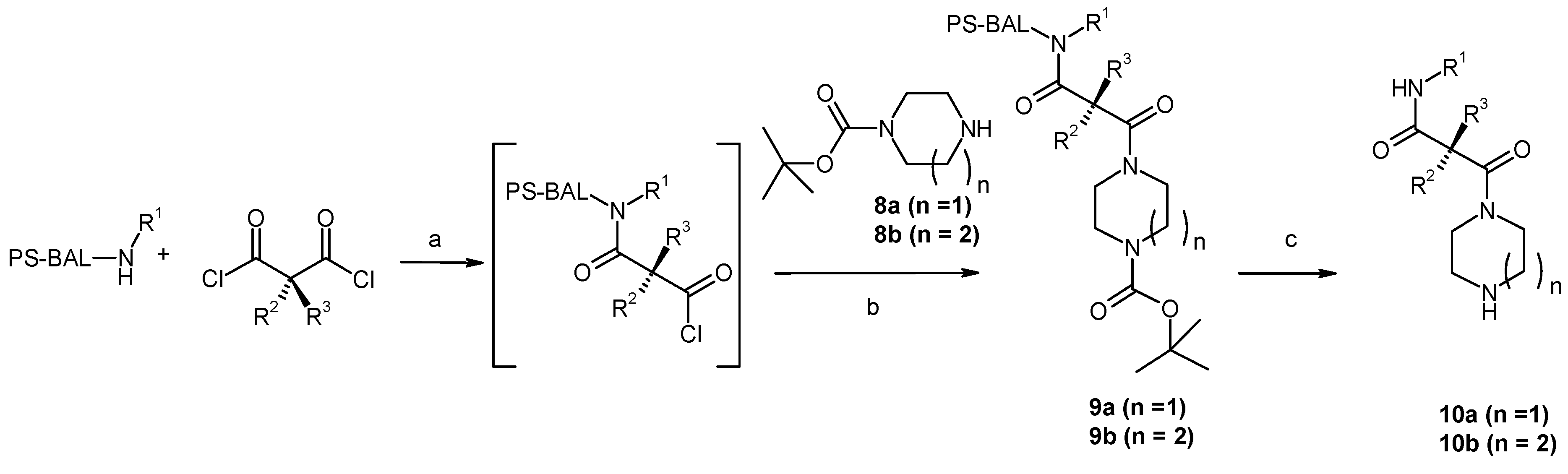{kind=link}
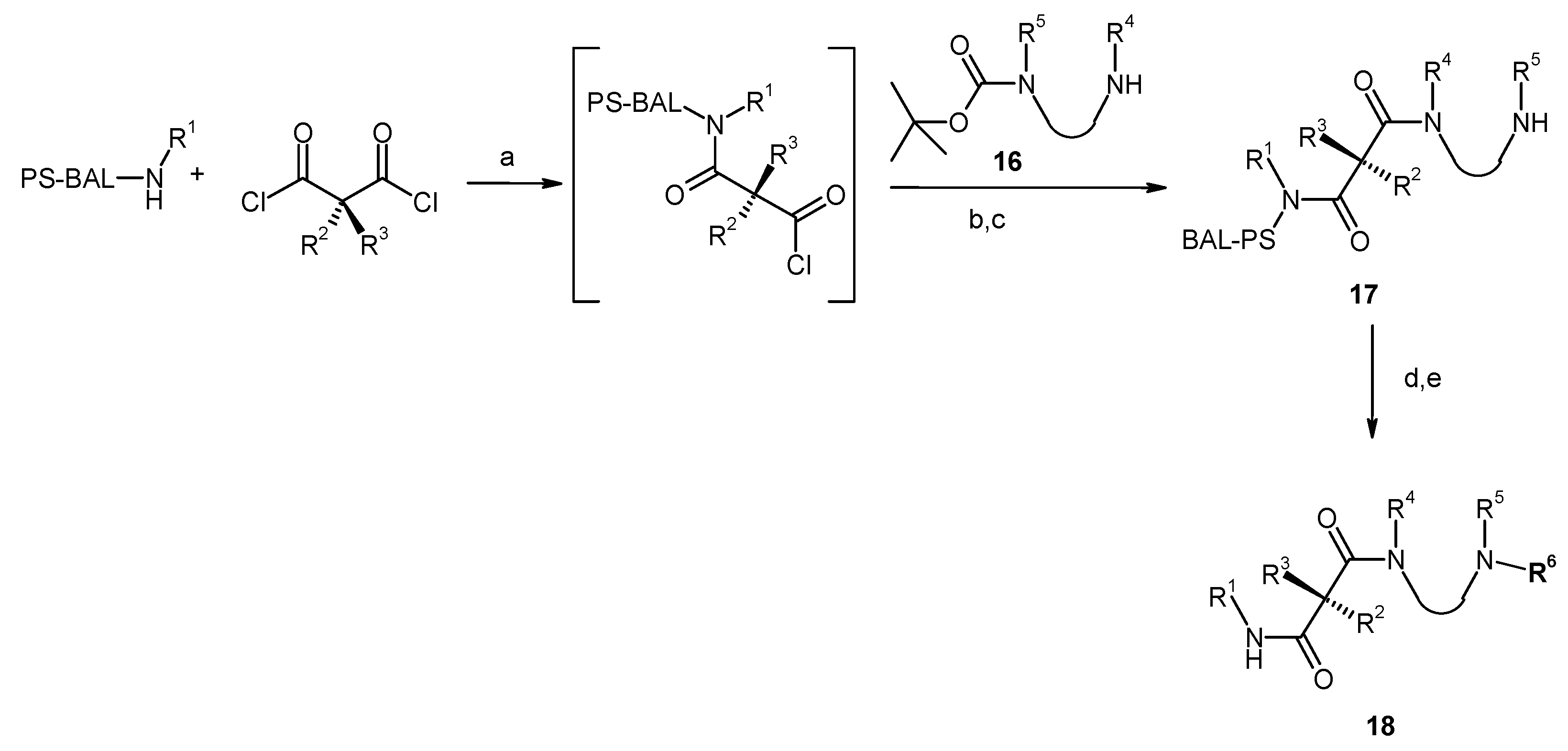{kind=link}
| Entry | Compound | Rt a | Exact Mass Formula | Mass b | Relative Peak Area(%) c | |
|---|---|---|---|---|---|---|
| 1 | 11 | 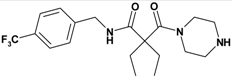 | 3.05 | 385.43 C19H26F3NO2 | 386.3 | 94 |
| 2 | 12 | 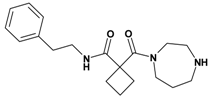 | 3.27 | 329.45 C19H27N3O2 | 330.4 | 93 |
| 3 | 13 |  | 2.59 | 390.49 C20H30N4O4 | 391.6 | 93 |
| 4 | 14 | 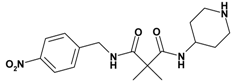 | 2.22 | 348.41 C17H24N4O4 | 348.3 | 92 |
| 5 | 15 | 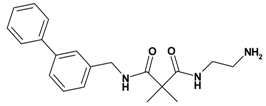 | 6.75 | 339.44 C20H25N3O2 | 340.5 | 92 |
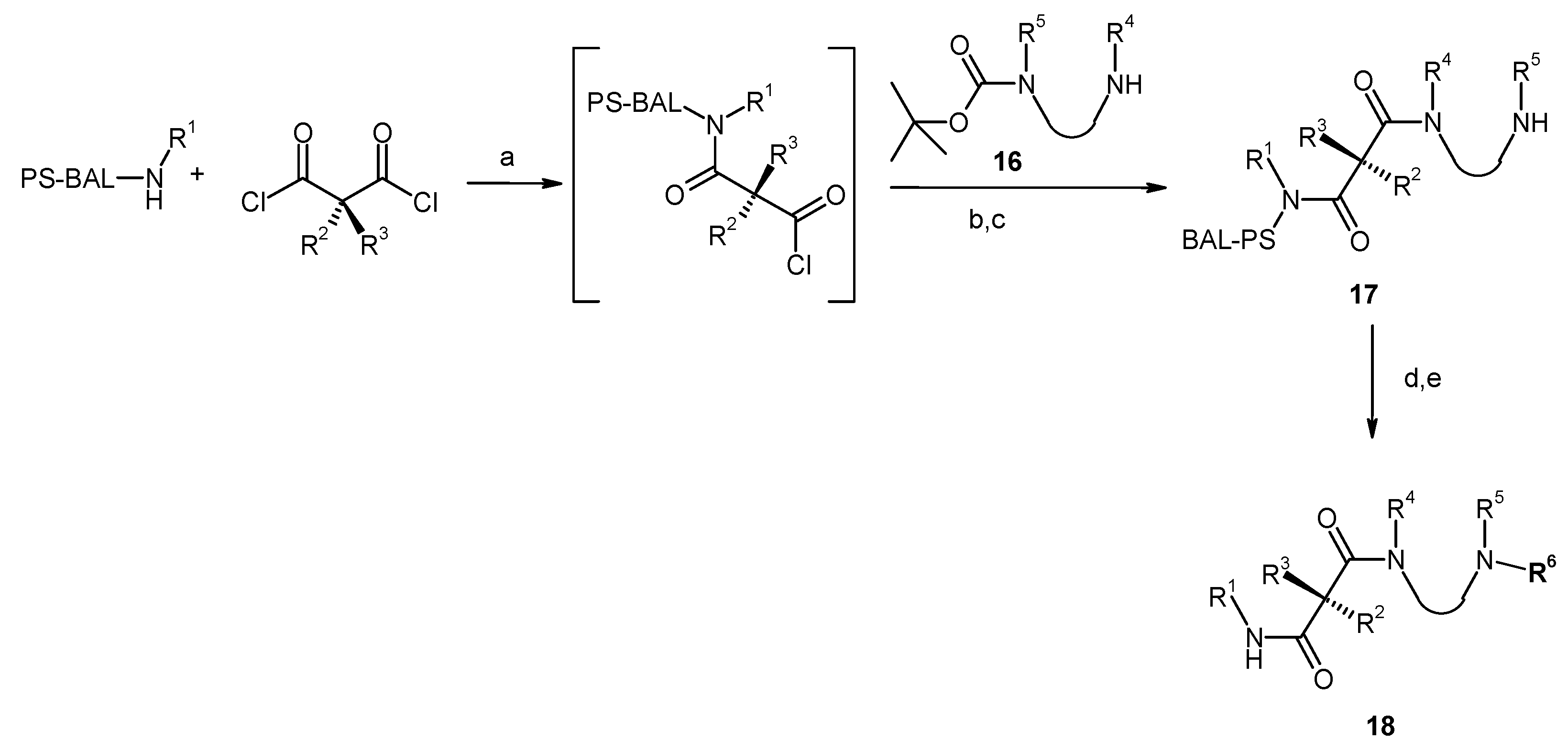
| Entry | Compound | Rt a | Exact Mass Formula | Mass b | Relative Peak Area(%) c | |
|---|---|---|---|---|---|---|
| 1 | 19 | 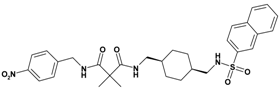 | 5.90 | 580.71 C30H36N4O6S | 581.4 | 96 |
| 2 | 20 | 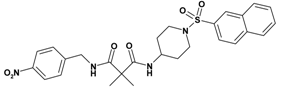 | 5.67 | 538.63 C27H30N4O6S | 539.4 | 95 |
| 3 | 21 d | 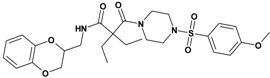 | 6.13 | 545.66 C27H35N3O7S | 546.5 | 92 |
| 4 | 22 | 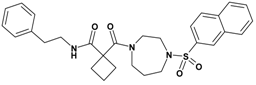 | 6.65 | 519.67 C29H33N3O4S | 520.5 | 94 |
| 5 | 23 | 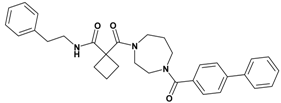 | 6.39 | 509.65 C32H35N3O3 | 510.4 | 93 |
| 6 | 24 | 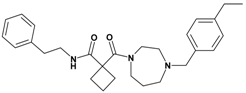 | 4.89 | 447.63 C28H37N3O2 | 448.4 | 93 |
| 7 | 25 |  | 8.27 | 644.10 C32H33ClF3N5O4 | 644.4 646.5 | 100 |
| 8 | 26 | 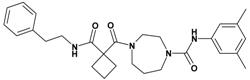 | 5.92 | 476.62 C28H36N4O3 | 477.7 | 94 |
| 9 | 27 | 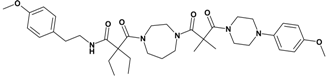 | 7.67 | 663.86 C37H53N5O6 | 664.5 | 69 |

Conclusions
Experimental
General Procedure
Acknowledgments
References and Notes
- Vögtle, M. M.; Marzinzik, A. L. A Short and Efficient Protocol for the Solid-Phase Synthesis of Retro-Inverso Surrogates. Synlett 2005, 496–500. [Google Scholar]
- The ratio was determined by HPLC at 254 nm and 214 nm and 1H-NMR.
- Zhang, A. J.; Russel, D. H.; Zhu, J. P; Burgess, K. A method for removal of N-BOC protecting groups from substrates on TFA-sensitive resins. Tetrahedron Lett. 1998, 7439–7442. [Google Scholar]
- Bolton, G. L.; Hodges, J. C.; Rubin, J. R. Solid phase synthesis of fused bicyclic amino acid derivatives via intramolecular Pauson-Khand cyclization: Versatile scaffolds for combinatorial chemistry. Tetrahedron 1997, 6611–6634. [Google Scholar]
- Bicknell, A. J.; Hird, N. W. Synthesis of a highly functionalized rigid template by solid phase azomethine ylide cycloaddition. Biorg. Med. Chem. Lett. 1996, 2441–2444. [Google Scholar]
- Green, J. Solid Phase Synthesis of Lavendustin A and Analogs. J. Org. Chem. 1995, 4287–4290. [Google Scholar] Devraj, R.; Cushman, M. A Versatile Solid Phase Synthesis of Lavendustin A and Certain Biologically Active Analogs. J. Org. Chem. 1996, 9368–9373. [Google Scholar]
- Reaction conditions: 10 eq. 2-chloro-4-trifluoromethyl-pyrimidine-5-carboxylic acid p-chloro-benzyl ester, DMA/DIPEA 4:1, 24 h, RT; TFA95%/DCM 1:4.
- Lee, S. H.; Chung, S. H.; Lee, Y. S. Preparation of resin-bound ketimines via transimination and its application in the synthesis of hydantoin libraries. Tetrahedron Lett. 1998, 9469–9472. [Google Scholar]
- Zaragoza Dörwald, F. Organic Synthesis on Solid Phase; Wiley-VCH: Weinheim, 2000; and references therein. [Google Scholar]
- Sample availability: Contact the authors
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Vögtle, M.M.; Marzinzik, A.L. An Efficient Protocol for the Solid-phase Synthesis of Malondiamides. Molecules 2005, 10, 1438-1445. https://doi.org/10.3390/10121438
Vögtle MM, Marzinzik AL. An Efficient Protocol for the Solid-phase Synthesis of Malondiamides. Molecules. 2005; 10(12):1438-1445. https://doi.org/10.3390/10121438
Chicago/Turabian StyleVögtle, Markus M., and Andreas L. Marzinzik. 2005. "An Efficient Protocol for the Solid-phase Synthesis of Malondiamides" Molecules 10, no. 12: 1438-1445. https://doi.org/10.3390/10121438
APA StyleVögtle, M. M., & Marzinzik, A. L. (2005). An Efficient Protocol for the Solid-phase Synthesis of Malondiamides. Molecules, 10(12), 1438-1445. https://doi.org/10.3390/10121438