Experimental
General
All commercial chemicals and solvents were used as received. Melting points were determined in open tubes on a Büchi apparatus and are uncorrected. IR spectra were recorded on a Perkin-Elmer spectrophotometer. Microanalyses were performed in the Microanalysis Laboratory of ENSCM (Montpellier). 1H and 13C- Nuclear Magnetic Resonance spectra were determined on a Brüker AC 250 spectrometer. Chemical shifts are recorded in ppm (δ) and coupling constants in Hertz, relative to tetrametylsilane used as internal standard. Multiplicity is indicated as s (singlet), d (doublet), q (quadruplet), m (multiplet) and combinations of these signals. Fast-atom bombardment mass spectra (FAB) were recorded in positive or negative mode with glycerol (G), thioglycerol (GT), or 3-nitrobenzyl alcohol (NOBA) as matrix. Optical rotations for solutions in CHCl3 were measured with a POLAX model 2L digital polarimeter. All reactions were monitored by Thin Layer Chromatography (TLC) on silica gel Merck 60 F254 precoated aluminium plates, developed by spraying with ninhydrin solution. Column chromatography was performed using silica gel 60 (230-400 mesh).
General synthetic procedure for carbamoylation–sulfamoylation:
A solution of N-chlorosulfonyl
tert-butylcarbamate (0.05 mol) was prepared by addition of
tert- butanol (4.8 mL in 50 mL of dried dichloromethane) into a solution of CSI (7.1 g in the same solvent). The resulting solution of Boc-sulfamoyl chloride (25 mL) and triethylamine (17.40 g, 17.1 mL, 85 mmol) in dichloromethane (100 mL) was added into a suspension of amino ester (0.05 mol) in the same solvent (120 mL) at 0°C. The reaction was complete in 45 minutes. The reaction mixture was then diluted with dichloromethane (100 mL) and washed with two portions of 0.1 N HCl solution. The organic layer was dried (Na
2SO
4) and concentrated
in vacuo to give the crude product, which was purified by column chromatography eluting with dichloromethane to give compounds
1-5 in 75-80% yield. The synthesis by this general method of compounds
1-
4 from CSI,
tert-butyl alcohol and the methyl esters of the amino acids L-alanine, L-valine, L-leucine and L-aspartic acid was previously described [
18,
19].
(S)-Dimethyl [N-(N- tertiobutyloxycarbonyl)-sulfamoyl] glutamate (5). Yield=76%; TLC: Rf=0.34 (CHCl2-MeOH 9:1); m.p=105-106°C; [α]D =+17 (c=1; MeOH). IR (KBr) ν cm-1: 1744, 1706 (C=O), 1380 and 1160 (SO2), 3305 and 3353 (NH). 1H-NMR (CDCl3) δ ppm: 7.72 (s, 1H), 5.55 (d, J=8.4 Hz, 1H), 4.42 (m, 1H), 3.72-3.80 (2s, 6H), 3.00 (ddd, J=16.5, 6.4, 6.4 Hz, 2H), 3.20 (ddd, J=16.5, 6.4, 6.4Hz, 2H), 2,20 (m, 2H), 1.50 (s, 9H); 13C-NMR (CDCl3 δ ppm: 173, 171, 151, 85, 56, 53, 52, 48, 41, 29, 28, 27; M.S: (NOBA, FAB< 0): 353 [M-H] -, 253, 707. M=354; Anal. Calcd. for C12H22 N2O8S: C, 40.67; H, 6.21; N, 7.90; S, 9.03; found C,40.73; H, 6.24; N, 7.94; S, 8.92.
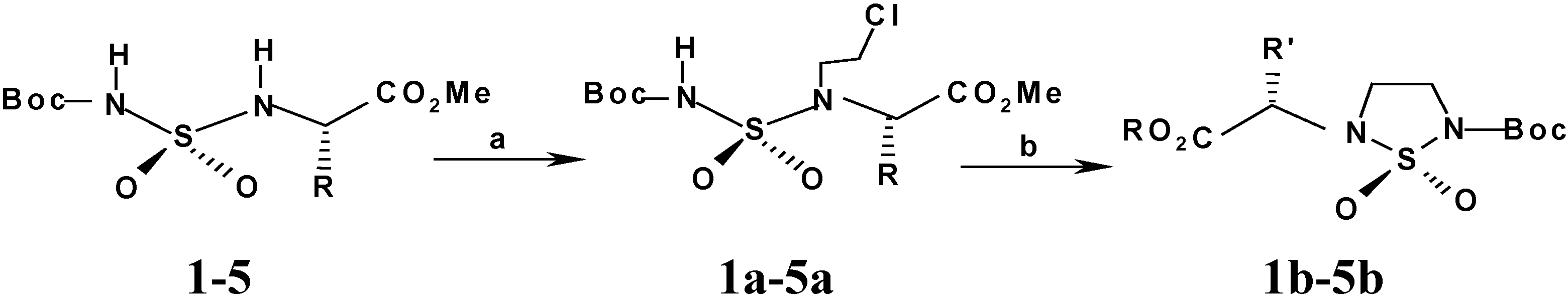
General procedure for the synthesis of N-Boc, N’-(2-chlroalkyl) sulfamides 1a-5a
A solution of N-tert-butyloxycarbonylsulfamoylamino esters 1-5 (30 mmol), triphenyl-phosphine (7.6 g) and chloroethanol (2.4g; 2 mL) in THF (25 mL) was added dropwise (20 min, 5°C) to a solution of equimolar quantities of diethyl (diisopropyl) azodicarboxylate (30 mmol; 5.22 g or 6.06 g) in the same solvent (25 mL). The reaction medium was stirred under an atmosphere of dry nitrogen for about 45 min. TLC revealed that the substituted compound formed is less polar than its precursor (UV, ninhydrin). Oxydoreduction compounds were removed by filtration after precipitation into diethyl ether. The filtrate was concentrated and the crude residue was purified by column chromatography eluting with dichloromethane. Substituted compounds 1a-5a were recovered in 65-75% yield.
(S) Methyl [N,N’-tert-butyloxycarbonyl, N’-chloroethyl)sulfamoyl] alaninate (1a). Yield=65%; TLC: Rf=0.52 (CHCl3); m.p.=88-90 °C; [α]D = -17 (c=1, MeOH); IR (KBr) ν cm-1: 1744, 1706 (C=O), 1380 and 1160 (SO2), 3305 (NH); 1H-NMR (CDCl3) δ ppm: 5.55 (d, J=8.42 Hz, 1H), 4.42 (m, 1H), 3,70 (s, 3H), 3,65 (t, J=6.8 Hz, 2H), 3.95 (t, J=6.8 Hz, 2H), 1.50 (s, 9H ), 1.30 (d, J=7.2 Hz, 3H). 13C-NMR (CDCl3 δ ppm: 170, 150, 84, 54, 52 49, 29, 27, 25; M.S: (NOBA, FAB<0): 343 [M-H]-, 243, 687. M=344-346; Anal. Calcd. for C11H21N2O6SCl: C, 38.31; H, 6.09; N, 8.12; S, 9. 28; found: C, 38.33; H, 6.09; N, 8.09; S, 9.22.
(S) Methyl [N-( N’-tert-butyloxycarbonyl, N’-chloroethyl)sulfamoyl] valinate (2a). Yield=68%; TLC: Rf=0.65 (CHCl3); m.p.=60-62 °C; [α]D=-9 (c=1; MeOH) ; IR (KBr) ν cm-1: 1730, 1700 (C=O), 1390 and 1160 (SO2), 3310 (NH); 1H-NMR (CDCl3) δ ppm: 5.80 (d, J=8.42 Hz, 1H), 3.95 (t, J=6,8 Hz, 2H), 3.87 (m, 1H), 3.68 (s, 3H), 3.65 (t, J=6.8 Hz, 2H), 2.10 (m, 1H ), 1.52 (s, 9H), 0.98 (2d, J=6,9 Hz, 6H, 2CH3); 13C-NMR (CDCl3) δ ppm: 172, 152, 85,55, 52, 48, 41, 28, 27, 22; M.S: (NOBA, FAB>0): 373 [M+H] +, 273, 747. M=372-274; Anal. Calcd. for C13H25 N2O6SCl: C, 41.88; H, 6.71; N, 7.51; S, 8.59; found: C, 41.93; H, 6.74; N, 7.46; S, 8.51.
(S) Methyl [N-(N’-tert-butyloxycarbonyl, N’-chloroethyl)-sulfamoyl] leucinate (3a). Yield=75%; TLC: Rf =0.60 (CHCl3); m.p.=67-69°C; [α]D=-23 (c=1; MeOH); IR (KBr) ν cm-1: 1740, 1712 (C=O), 1390 and 1160 (SO2), 3267 (NH); 1H-NMR (CDCl3) δ ppm: 5.80 (d, J=8.8 Hz, 1H), 3,95 (t, J=6,8 Hz, 2H), 3.87 (q, J=6.8 Hz, 1H), 3.68 (s, 3H), 3,65 (t, J=6.8 Hz, 2H), 1,87 (m, 2H), 1.55 (s, 9H), 1.48 (m, 1H), 0.98-0.88 (2d, J=3.9 Hz, 6H); 13C-NMR (CDCl3) δ ppm: 172, 152, 85, 56, 53, 48, 43, 41, 28, 25, 24, 22; M.S: (NOBA, FAB>0): 387 [M+H] +, 287, 773. M=386-388; Anal. Calcd. for C14H27N2O6SCl: C, 43.46; H, 6.98; N, 7.24; S, 8.28; found: C, 43.49; H, 6.94; N, 7.20; S, 8.21.
(S) Methyl [N-(N’-tert-butyloxycarbonyl, N’-chloroethyl)-sulfamoyl] aspartate (4a). Yield=70%; TLC: Rf =0.67 (CHCl3); oil; [α]D=-33 (c=1; MeOH) ; IR (KBr) ν cm-1: 1755, 1715 (C=O) 1370 and 1130 (SO2), 3269 (NH); 1H-NMR (CDCl3) δ ppm: 5.85 (d, J=8.4 Hz, 1H), 4.30 (q, J=8.4 Hz, 1H), 3.90 (t, J=6.7 Hz, 2H), 3.70-3.80 (2s, 6H), 3.68 (t, J=6.7Hz, 2H), 3.50 (2dd, J= 4.1, 8.4 Hz, 2H), 1.50 (s, 9H); 13C-NMR (CDCl3 δ ppm: 171, 170, 150, 85, 55, 51, 52, 49, 42, 28, 27; M.S: (NOBA, FAB>0): 407 [M+H] +, 307, 805. M=406-404; Anal. Calcd. for C13H23 N2O8SCl: C,38.75; H, 5.71; N, 6.95; S, 7.95; found: C, 38. 73; H, 5.74; N, 6.94; S, 7.92.
(S) Methyl [N-(N’-tert-butyloxycarbonyl, N’-chloroethyl)-sulfamoyl] glutamate (5a). Yield=72%; TLC: Rf =0.65 (CHCl3); oil; [α]D =-21 (c=1; MeOH) ; IR (KBr) ν cm-1: 1746, 1715 (C=O) 1395 and 1156 (SO2), 3310 (NH); 1H-NMR (CDCl3) δ ppm: 6.10 (d, J=8.5 Hz, 1H), 4.30 (m, 1H), 3.90 (t, J=6.7Hz, 2H), 3.68 (t, J=6.9 Hz 2H), 3.60-3.65 (2s, 6H), 2.48 (m, 2H), 2.10 (m, 2H),1.50 (s, 9H); 13C-NMR (CDCl3 δ ppm: 173, 171, 151, 85, 56, 53, 52, 48, 41, 29, 28, 27; M.S: (NOBA, FAB>0): 417 [M+H] +, 317, 833. M=416-418; Anal. Calcd. for C14H25 N2O8SCl: C, 40.33; H, 6.02; N, 6.72; S, 7.68; found: C, 40.42; H, 6.04; N,6.75; S, 7.62.
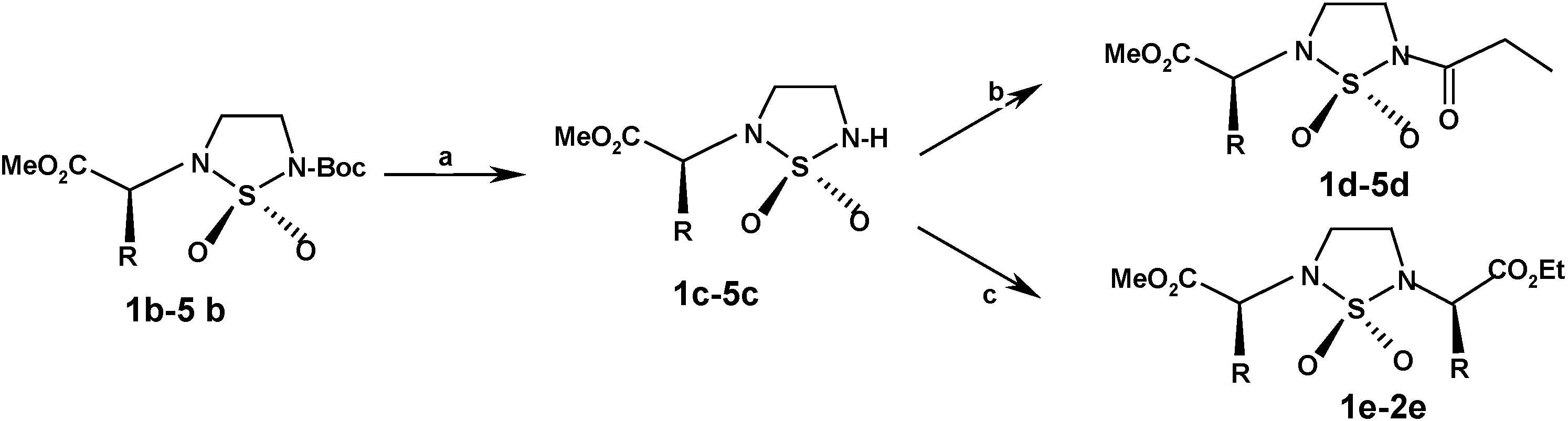
General procedure for preparation of N-Boc-N-substituted)-1,2,5-thiadiazolidine 1,1-dioxides 1b-5b.
Cyclization with K2CO3 in DMSO
The 2-chloroalkyl compounds (10 mmol) were dissolved in dimethysulfoxide (DMSO) and anhydrous K2CO3 (1.5 equiv.) was added in one portion. The resulting mixture was stirred at room temperature for 8 h, diluted with dichloromethane (200 mL) and acidified with 5% HCl. The organic layer was washed with water, dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography on silica gel. Recristallization of the crude product from CH2Cl2- petroleum ether (1:5) afforded the pure expected cyclosulfamides 1b-5b in 82-92% yields.
(N2-(2’S)-Propionic acid methyl ester, N5-tert-butyloxycarbonyl)-1,2,5-thiadiazolidine 1,1-dioxide (1b). Yield=86%; TLC: Rf =0.58 (CHCl3); Mp=132-134°C; [α]D=-34. (c=1; MeOH); IR (KBr) ν cm-1: 1750 1712, 1378 and 1167 (SO2); 1H-NMR (CDCl3) δ ppm: 4.15 (q, J=7.8 Hz, 1H), 3.80 (t, J=6.4 Hz 2H), 3.70 (s, 3H), 3.65 (t, J=6.4 Hz, 2H), 1.50 (d, J=7.8 Hz, 1H), 1.52 (s, 9H); 13C-NMR (CDCl3 δ ppm: 172, 150, 84, 56, 52, 43, 39, 28, 27; M.S: (NOBA, FAB< 0): 307 [M-H]-, 207; M=308; Anal. Calcd. For C11H20N2O6S: C, 42.85; H, 6.49; N, 9.09; S, 10.39; found: C, 42.83; H, 6.44; N, 9.04; S, 10,32.
(N2-(2’S)-3’-methylbutyric acid methyl ester, N5-tert-butyloxycarbonyl)-1,2,5-thiadiazolidine-1,1- dioxide (2b). Yield=90%; TLC: Rf =0.60 (CHCl3); m.p.=154-155°C; [α]D=-38 (c=1; MeOH); IR (KBr) ν cm-1: 1745, 1718 (C=O), 1390 and 1160 (SO2); 1H-NMR (CDCl3) δ ppm: 4.15 (d, J=7.2 Hz, 1H), 3,95 (t, J=6.4 Hz, 2H), 3.80 (t, J=6.4 Hz, 2H), 3.70 (s, 3H), 2.20 (m, 1H), 1.57 (s, 9H),1.30 (d, J= 7.2Hz, 3H), 0,98 (2d, J=6.9 Hz, 6H); 13C-NMR (CDCl3 δ ppm: 172, 149, 84, 56, 53, 43, 39, 28, 26, 23, 22; M.S: (NOBA, FAB>0): 337 [M+H] +, 237; M=336; Anal. Calcd. for C13H24N2O6S: C, 46.43; H, 7.14; N, 8.33; S, 9.52; found: C, 46.48; H, 7.17; N, 8.34; S, 9.44.
(N2-(2’S)-4’-Methylpentanoic acid methyl ester, N5-tert-butyloxycarbonyl)-1,2,5-thiadiazolidine-1,1- dioxide (3b). Yield= 87%; TLC: Rf =0.58 (CHCl3); m.p.=138-139 °C; [α]D =-53 (c=1; MeOH); IR (KBr) ν cm-1: 1747, 1728 (C=O), 1360 and 1120 (SO2); 1H-NMR (CDCl3) δ ppm: 4.30 (t, J=8.4 Hz), 3.95 (t, J=6.4 Hz, 2H), 3.72 (s, 3H), 3.55 (t, J=6.4 Hz, 2H), 1.55-1.65 (m, 3H), 1.51 (s, 9H), 0.95-1.00 (2d, J=6.9 Hz, 6H) ;13C-NMR (CDCl3 δ ppm: 171, 149, 86, 54, 53, 43, 39, 37, 28, 25, 23, 21 ; M.S: (NOBA, FAB>0): 351 [M+H] +, 251 ; M=350; Anal. Calcd. for C14H26N2O6S: C, 48.00; H, 7.43; N, 8.00; S, 9. 14; found: C, 48.07; H, 7.48; N, 7.94; S, 9.07.
(N2-(2’S)-Bis(1’,3’-methoxycarbonyl)ethyl), N5-tert-butyloxycarbonyl)-1,2,5-thiadiazolidine-1,1-dioxide (4b). Yield=82%; Rf =0.61(CHCl3); oil; [α]D =+38 (c=1; MeOH); IR (KBr) ν cm-1: 1754, 1751, 1710 (C=O) 1390 and 1150 (SO2); 1H-NMR (CDCl3) δ ppm: 4.30 (m, 1H), 3.70-3.80 (2s, 6H), 3.95 (t, J=6.4Hz, 2H, 3.55 (t, J=6.4 Hz, 2H), 3.50 (ddd, J=17.2, 7.1, 4.5 Hz, 2H), 1,50 (s, 9H); 13C-NMR (CDCl3 δ ppm: 177, 171, 150, 85, 57 53, 52, 43, 39, 28, 25; M.S: (NOBA, FAB>0): 367 [M+H] +, 267; M=366; Anal. Calcd. for C13H22N2O8S: C, 42.62; H, 6.01; N, 7.65; S, 8.74; found : C, 42.63; H, 6.04; N, 7.69; S, 8.69.
(N2-(2’S’)-Bis(1’,4’-methoxycarbonyl)propyl, N5-tert-butyloxycarbonyl)-1,2,5-thiadiazolidine-1,1- dioxide (5b). Yield=92%; TLC: Rf =0.58 (CHCl3); oil; [α]D =+38 (c=1; MeOH); IR (KBr) ν cm-1: 1748, 1745, 1720 (C=O), 1360 and 1120 (SO2) ; 1H-NMR (CDCl3) δ ppm: 4.30 (m, 1H), 3.95 (t, 2H, J=6,4Hz), 3.68 (t, J=6,4 Hz, 2H), 3.72-3.65 (2s, 6H), 2.48 (m, 2H), 2.10 (m, 2H),1.50 (s, 9H) ; 13C-NMR (CDCl3 δ ppm: 173, 170, 150, 85, 56, 53, 52, 43, 41, 30, 28, 27 ; M.S: (NOBA, FAB>0): 381 [M+H] +, 281; M=380; Anal. Calcd. for C14H24N2O8S : C, 44.21; H, 6.31; N, 7.37; S, 8.42; found C, 44.30; H, 6.34; N, 7.34; S, 8.36.
Deprotection
A solution of trifluoroacetic acid (50% in dried dichloromethane; 3 equiv) was added dropwise into a strirred solution of N,N’-substituted cyclosufamides 1b-5b (20 mmol) in dried dichloromethane (15 mL) at 0°C. The reaction medium was stirred during two hours, concentrated under reduced pressure and coevaporated with diethyl ether. The residue was purified by flash chromatography. Elution with CH2Cl2-MeOH (95:5) gave deprotected cyclic sulfamides 1c-5c in 85%-90% yield.
N2-(2’S)-(Propionic acid methyl ester) 1,2,5-thiadiazolidine 1,1-dioxide (1c). Yield=90%; TLC: Rf =0.45 (CHCl3); m.p.=124-125 °C; [α]D=-34. (c=1; MeOH) ; IR (KBr) ν cm-1: 1751 (C=O) 1375 and 1160 (SO2), 3345 (NH); 1H-NMR (CDCl3) δ ppm: 6.24 (t, J=6.7 Hz, 1H), 4.15 (q, 1H, J=7,8 Hz, 1H), 3,80 (t, J=6.7Hz, 2H), 3.70 (s, 3H), 3.65 (m, 2H), 1.50 (d, J=7.8 Hz, 3H); 13C-NMR (CDCl3 δ ppm: 172, 56, 52, 41, 39, 28; M.S: (NOBA, FAB< 0): 207 [M-H] -, 415; M=208; Anal. Calcd. for C6H12N2O4S: C, 34.61; H, 5.77; N, 13.46; S, 15.38; found C, 34.63; H, 5.84; N, 13.44; S, 15.32.
N2-(2’S)-(3-methylbutyric acid methyl ester) 1,2,5-thiadiazolidine 1,1-dioxide (2c). Yield=85%; TLC: Rf =0.55 (CHCl3); m.p.=134-135°C; [α]D =-44 (c=1; MeOH); IR (KBr) ν cm-1: 1745 (C=O), 1390 and 1160 (SO2), 3340 (NH); 1H-NMR (CDCl3) δ ppm: 6.30 (tJ=6.8Hz, 1H), 4.15 (d, J=7.2 Hz, 1H), 3.90 (t, J= 6,7 Hz, 2H), 3.70 (s, 3H), 3.60 (m, 2H), 1.30 (m, 1H), 0,98 (2d, J=6.9 Hz, 6H); 13C-NMR (CDCl3 δ ppm: 171, 56, 53, 41, 39, 26, 23, 22; M.S: (NOBA, FAB>0): 237 [M+H] +, 473; M=236; Anal. Calcd. for C8H16N2O4S: C, 40.67; H, 6.78; N, 11.86; S, 13.60; found: C, 40.73; H, 6.84; N, 11.94; S,13.52
N2-(2’S)-4’-methylpentanoic acid methyl ester) 1,2,5-thiadiazolidine 1,1-dioxide (3c). Yield= 90%; TLC: Rf =0.53 (CHCl3); m.p.=128-130 °C; [α]D=+76 (c=1; MeOH); IR (KBr) ν cm-1: 1747, 1360 and 1120 (SO2), 3320 (NH); 1H-NMR (CDCl3) δ ppm: 6.30 (tJ=6.8Hz,1H), 4.30 (m, 1H), 3.85 (t, J=6.4 Hz, 2H), 3.72 (s, 3H), 3.55 (m, 2H), 1.55-1.65 (m, 3H), 0,98 (2d, J=6.9 Hz, 6H);13C-NMR (CDCl3 δ ppm: 177, 57, 53, 41, 39, 28, 25, 22, 21; M.S: (NOBA, FAB>0): 251 [M+H] +, 501. M=250; Anal. Calcd. For C9H18N2O4S: C, 43.20; H,7.20; N, 11.20; S, 12.80; found: C, 43.23; H, 7.24; N, 11.17; S, 12.72.
N2-(2’S’)-bis(1’,3’-methoxycarbonyl)ethyl] 1,2,5-thiadiazolidine 1,1-dioxide (4c). Yield=88%; Rf =0.56 (CHCl3); oil; [α]D =-56 (c=1; MeOH); IR (KBr) ν cm-1: 1750-1755 (2C=O) 1390 and 1150 (SO2), 3325 (NH); 1H-NMR (CDCl3) δ ppm: 6.80 (tJ=6.7Hz,1H), 4.25 (2d, J=7.1, 4.6 Hz, 1H), 3.70-3.80 (2s, 6H), 3.70 (t, J=6.7 Hz, 2H), 3.55 (m, 2H), 3.50 (ddd, J= J=17.2, 7.1, 4.5 Hz, 2H) ; 13C-NMR (CDCl3 δ ppm: 177, 175, 57, 53, 52, 41, 39, 28; M.S: (NOBA, FAB>0): M=267 [M+H]+, 533; M=266; Anal. Calcd. for C8H14O6N2S: C, 36.09; H, 5.26; N, 10.52; S, 12.03; found: C, 36.13; H 5.24; N, 10.54; S, 12.00.
N2-(2’S’)-bis(1’,4’-methoxycarbonyl)propyl] 1,2,5-thiadiazolidine 1,1-dioxide (5c). Yield=89%; TLC: Rf =0.54 (CHCl3); oil; [α]D= +67 ( c=1; MeOH); IR (KBr) ν cm-1: 1745-1738 (2C=O) 1360 and 1120 (SO2), 3145 (NH) ; 1H-NMR (CDCl3) δ ppm: 6.60 (tJ=6.8Hz,1H), 4,30 (m, 1H), 3,70 (t, J=6.4 Hz, 2H), 3.65 (m, 2H), 3.72-3.65 (2s, 6H), 2.50 (m, 2H), 2.10 (m, 2H); 13C-NMR (CDCl3 δ ppm: 172, 170, 56, 53, 52, 42, 40, 30, 28 ; M.S: (NOBA, FAB>0): 281 [M+H] +, 561. M=280; Anal. Calcd. For C9H16N2O6S: C, 38.57; H, 5.71; N, 10.00; S, 11.43; found: C,38.63; H, 5.74; N, 10.04; S, 11.36.
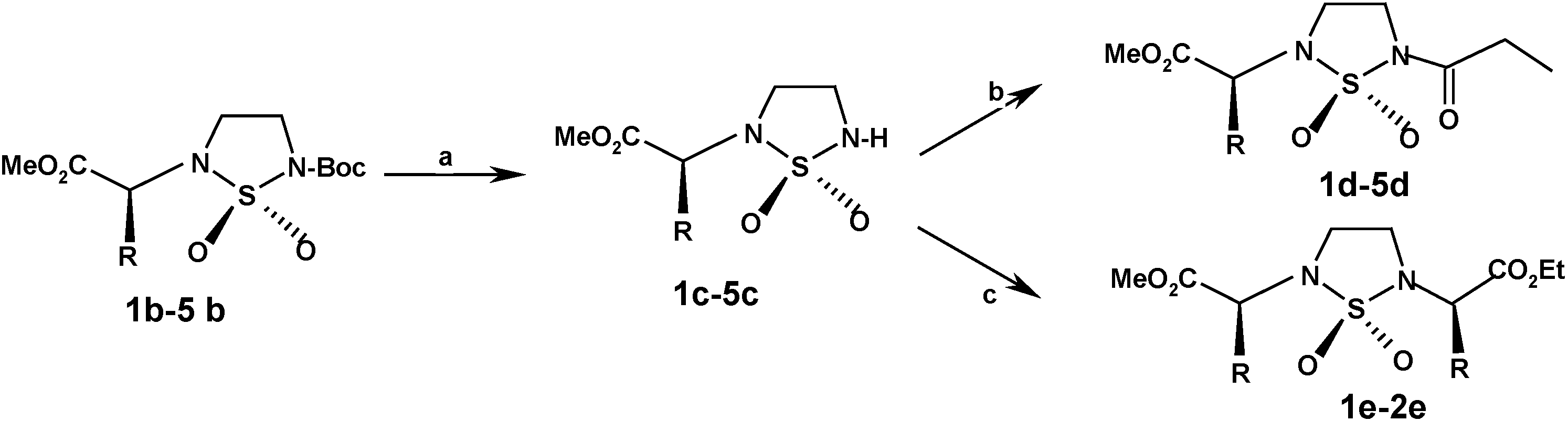
Propionylation
To a stirring solution of N-substituted cyclosulfamide 1c-5c (20 mmol), in dichloromethane (50 mL) was added triethylamine (1.1 equiv., 22 mmol, 2.22 g, 1.60 mL), and catalytic quantities of dimetylaminopyridine (DMAP). Propionyl chloride (1.5 equiv., 30 mmol, 2.41 g, 2.57 mL) diluted in the same solvent (15 mL) was added slowly to the resulting solution. When the addition was completed, the reaction mixture was stirred under an atmosphere of dry nitrogen. TLC reveals the formation of a substituted compound less polar than its precursor. The reaction mixture was concentrated in vacuo. The residue diluted with dichoromethane (50 mL), acidified with 0.1 N HCl solution and washed with water. The organic layer was dried with (Na2SO4) and concentrated under reduced pressure to give the crude product. The residue was purified on silica gel by column chromatography eluting with dichloromethane to give the N2, N5 substituted cyclosulfamides 1d-5d in 75-90 % yields.
[N2-(2’S’)-(propionic acid methyl ester), N5-propionyl] 1,2,5-thiadiazolidine 1,1-dioxide (1d). Yield=90%; TLC: Rf=0.59 (CHCl3); m.p.=88-89°C; [α]D=-17 (c=1; MeOH); IR (KBr) ν cm-1: 1750-1715 (C=O), 1375 and 1160 (SO2) ; 1H-NMR (CDCl3) δ ppm: 4.15 (q, J=7.8 Hz, 1H), 3.80 (t, J=6.2 Hz, 2H), 3.70 (s, 3H), 3.65 (t, J=6.7 Hz, 2H), 2.85 (q, J=7.4 Hz, 2H), 1.50 (d, 3H, J=7.8 Hz), 1.15 (t, 3H, J=7.4 Hz, 3H); 13C-NMR (CDCl3) δ ppm: 172, 170, 56, 53, 41, 39, 29, 28, 12; M.S: (NOBA, FAB>0): 265 [M+H] +, 208; M=264; Anal. Calcd. for C9H16O5N2S: C, 40.91; H, 6.06;N, 10.60; S, 12.12; found: C, 40.98; H, 6.17; N, 10.65; S, 12.05.
[N2-(2’S)-(3’-methylbutyric acid methyl ester), N5-propionyl] 1,2,5-thiadiazolidine 1,1-dioxide (2d). Yield=88%; TLC: Rf =0,62 (CHCl3); m.p.=94-95°C; [α]D =-14 (c=1; MeOH). IR (KBr) ν cm-1: 1748-1712 (C=O), 1389-1163 (SO2); 1H-NMR (CDCl3) δ ppm: 4.15 (d, J=7.2 Hz, 1H), 3.95 (t, J=6.7 Hz, 2H), 3.8 (t, J=6,7 Hz, 2H), 3.70 (s, 3H), 2.85 (q, 2H, J=7.4 Hz, 2H), 1.30 (m, 1H), 1.16 (t, J=7,4 Hz, 3H), 0.98 (2d, J=6.9 Hz, 6H); 13C-NMR (CDCl3 δ ppm: 172, 170, 56, 53, 43, 39, 26, 28, 23, 22, 13; M.S: (NOBA, FAB>0): M=373 [M+H] +, 174 ,745; M=372; Anal. Calcd. for C11H20N2O5S; C, 45.20; H, 6.85; N, 9.59; S, 10.96; found: C, 45.23; H, 6.19; N, 9.54; S, 10.89.
[N2-(2’S)-4’-methylpentanoic acid methyl ester), N5-propionyl] 1,2,5-thiadiazolidine 1,1-dioxide (3d). Yield=85%; TLC: Rf =0.65 (CHCl3); m.p.=106-108°C; [α]D=+54 (c=1; MeOH); IR (KBr) ν cm-1: 1747-1718 (C=O), 1362 and 1125 (SO2); 1H-NMR (CDCl3) δ ppm: 4.10 (m, 1H), 3.92 (t, J= 6.7 Hz, 2H), 3.75 (t, J=6.7 Hz, 2H), 3.72 (s, 3H), 2.85 (q, J=7,4 Hz, 2H), 1,55-1,65 (m, 12H), 1.15 (t, J=7.4 Hz, 3H), 0.98-1.00 (2d, J=6.9 Hz, 6H); 13C-NMR (CDCl3 δ ppm: 175 ,170, 57, 53, 41,39, 28, 29, 25, 23, 21, 12; M.S: (NOBA, FAB>0): 307 [M+H] +, 250; M=306; Anal. Calcd. for C12H22N2O5S: C, 47.06; H, 7.19; N, 9.15; S, 10.46; found: C, 47.12; H, 7.25; N, 9.08; S, 10.42.
[N2-(2’S’)-bis(1’,3’-methoxycarbonylethyl), N5-propionyl] 1,2,5-thiadiazolidine 1,1-dioxide (4d). Yield=80%, TLC: Rf =0.67 (CHCl3); oil; [α]D =-87 (c=1; MeOH); IR (KBr) ν cm-1: 1749, 1753 and 1715 (C=O) 1380 and 1150 (SO2); 1H-NMR (CDCl3) δ ppm: 4.30 ( 2d, J=7.3, 4.6 Hz, 1H), 3.70-3.80 (2s, 6H), 3.85 (t, J=6.8 Hz, 2H), 3.60 (t, J=6.8 Hz, 2H), 3.50 (ddd, J=J=17.2, 7.3, 4.6 Hz, 2H), 2.85 (q, J=7,4 Hz, 2H), 1.18 (t, J=7,4 Hz, 3H); 13C-NMR (CDCl3 δ ppm: 173, 172, 165, 57, 53, 52, 43, 40, 29, 25, 12; M.S: (NOBA, FAB>0): M=323 [M+H] +, 266. M=322; Anal. Calcd. for C11H18N2O7S: C, 40.99; H, 5.54; N, 8.69; S, 9.34; found: C, 41.03; H, 5.64; N, 8.80; S, 9.30.
[N2-(2’S’)-bis(1’,4’-methoxycarbonylpropyl), N5-propionyl] 1,2,5-thiadiazolidine 1,1-dioxide (5d). Yield=75%, TLC: Rf =0.51 (CHCl3); oil; [α]D=+43 (c=1; MeOH); IR (KBr) ν cm-1: 1745, 1738 and 1713 (C=O), 1360 and 1120 (SO2); 1H-NMR (CDCl3) δ ppm: 4.30 (m, 1H), 3.85 (t, J=6.4Hz, 2H), 3.65 (t, J=6.4 Hz, 2H), 3.72-3.65 (2s, 6H), 2.48 (m, 2H), 2.10 (m, 2H), 2.85 (q, J=7,4 Hz, 2H), 1.14 (t, J=7.4 Hz, 3H); 13C-NMR (CDCl3) δ ppm: 173, 171, 165, 56, 53, 52, 42, 41, 30, 29, 25, 12; Anal. Calcd. For C12H20O7N2S: C, 42.86; H, 5.95; N, 8.33; S, 9.52; found: C, 42.92; H, 9.87; N, 8.28; S, 9.43.
Alkylation via the Mitsunobu Reaction
To a stirring solution of N-substituted cyclosulfamide 1d-2d (3.23 mmol) in THF (2 mL) was slowly added DEAD (3.23 mmol, 0.5 mL) via dropwise addition. A solution consisting of (L)-(-)- ethyl lactate (3.23 mmol, 0.37 mL) and PPh3 (3.23 mmol, 847 mg) in THF (3mL), was slowly transferred via cannula into the cyclosulfamide solution. The reaction medium was stirred under an atmosphere of dry nitrogen for about 45 min. TLC reveals (UV, ninhydrin) the formation of a substituted compound less polar than its precursor. Oxydoreduction compounds were removed by filtration after precipitation into diethylether. The filtrate was concentrated and the crude residue was purified by colunm chromatography eluting with dichloromethane. N,N’-Substituted cyclosufamides 1e-2e were recovered in 65-75% yield.
[N2-(2S)-(Methoxycarbonylethyl), N5-(2’R)-(propionic acid ethyl ester)]-1,2,5-thiadiazolidine 1,1- dioxide (1e). Yield=65%; TLC: Rf=0.64 (CHCl3); m.p.=88-89°C; [α]D=-65 (c=1; MeOH); IR (KBr) ν cm-1: 1750-1731 (C=O), 1360 and 1152 (SO2); 1H-NMR (CDCl3) δ ppm: 4.76 (q, J=7.3, 1H), 4.65 (q, J=7.2, 1H), 4.15 (q, J=7.2 Hz, 1H), 3.80 (t, J=6.7 Hz, 2H), 3.70 (s, 3H), 3.65 (t, J=6.7 Hz, 2H),1.44 (d, J=7.2, 3H), 1.39 (d, J=7.1 Hz, 3H), 1.28 (t, J=7.2, 3H); 13C-NMR (CDCl3 δ ppm: 172, 170, 56, 55, 53, 50, 41, 39, 29, 28, 12; M.S: (NOBA, FAB>0): M=309 [M+H] +. M=308; Anal. Calcd. for C11H20N2O6N2S C, 42.86, H, 6.49, N, 9.09. S,10.03. Found: C, 40.86, H,6.13, N,9.47. S, 10.03.
[N2-(2S)-(3-Methylmethoxycarbonylpropyl), N5-(2’R)-propionic acid ethyl ester)] 1,2,5-thiadiazol- idine-1,1-dioxide (2e). Yield =75%; TLC: Rf =0.61 (CHCl3); m.p.=80-81°C; [α]D =-12.0 (c=0,5, CHCl3); IR (KBr) ν cm-1: 1745, 1729, 1346, 1145; 1H-NMR (CDCl3) δ ppm: 4.70 (q, J=7.3 Hz, 1H), 4.20 (q, J=7.1Hz, 2H), 3.90 (d, J=3.3 Hz, 1H), 3.80-3.60 (m, 4H), 3.70 (s, 3H), 2.12 (m, 1H), 1.39 (d, J=7.1 Hz, 3H), 1.26 (t, J=7.3 Hz, 3H), 0,98 (d, J=6.8 Hz, 3H ), 0,88 (d, J=6.9 Hz,3H); 13C-NMR (CDCl3 δ ppm: 172, 169, 56, 55, 53, 52, 50, 41, 39, 28, 22, 21, 14; M.S: (NOBA, FAB>0): 337 [M+H]+. M=336; Anal. Calcd. for C13H24N2O6S: C, 46.43; H, 7.14; N, 8.33; C, 9.52. Found: C, 46.49; H, 7.18; N, 8.34; S, 9.43.


{kind=link}
{kind=link}
{kind=link}