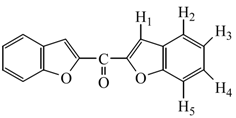
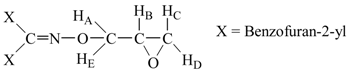
Results and Discussion
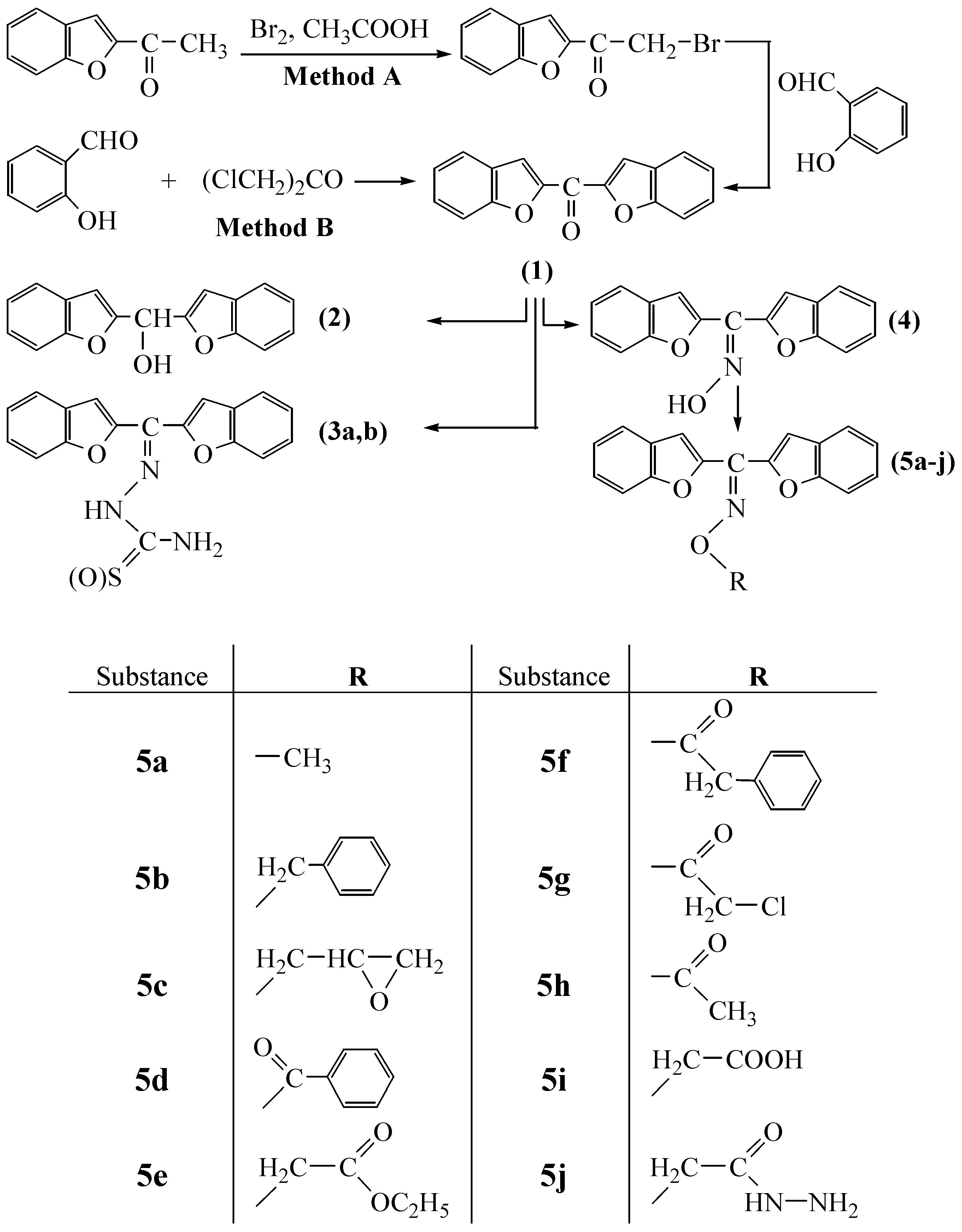
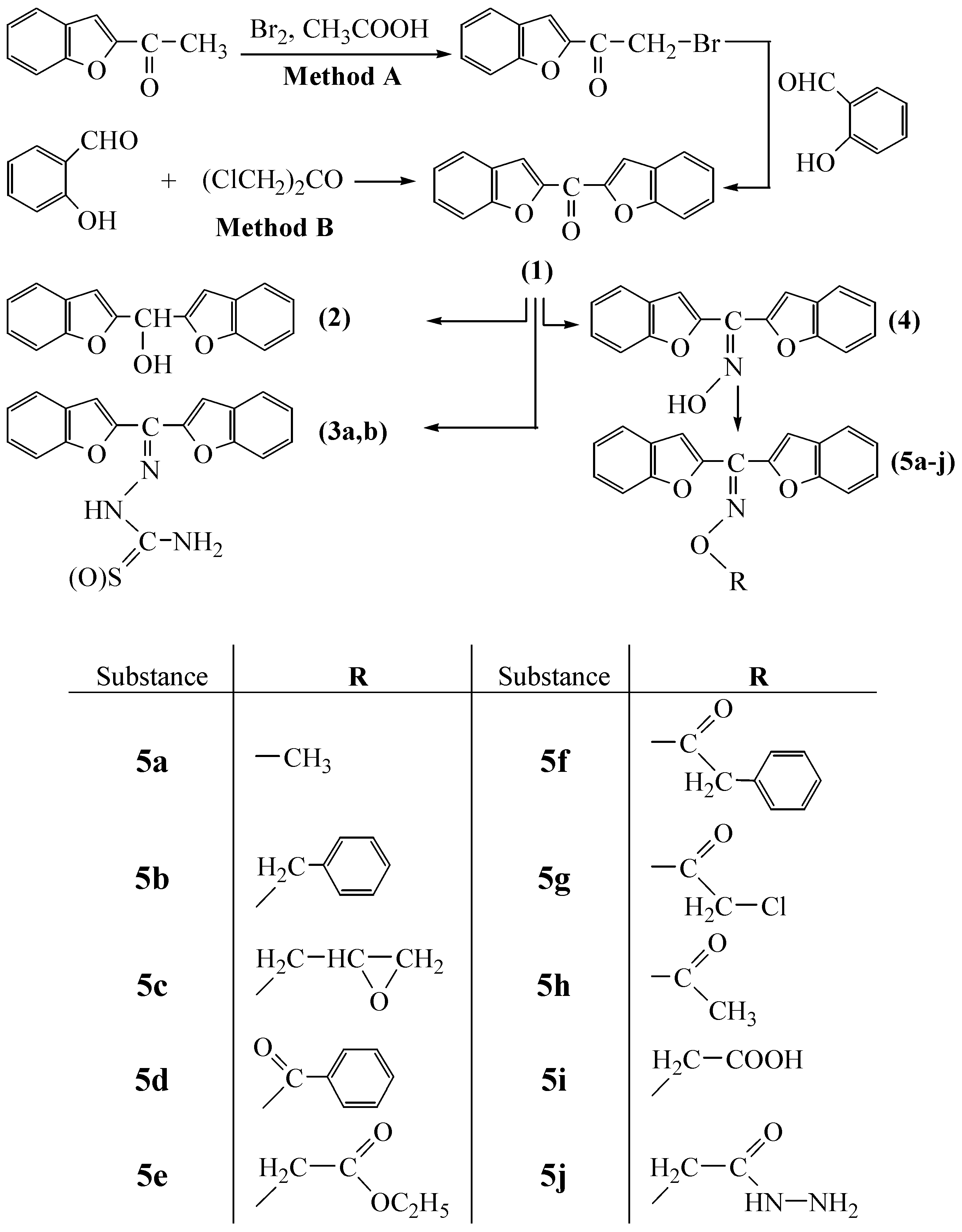
Bisbenzofuran-2-yl-methanone (
1) was synthesized in two different ways. The synthetic routes followed are shown in
Scheme 1. Overall yields of both routes are good and more or less similar. A is a new route, while B is the literature method [
14]. 1-(Benzofuran-2-yl)-2-bromoethanone, which is the starting material for method A, was synthesized from benzaldehyde and 1‑bromoacetone, followed by subsequent bromination with bromine in acetic acid, according to the general procedure described in [
14] with slight modifications. Treatment of this compound with salicylaldehyde in acetone solution gave bisbenzofuran-2-yl-methanone (
1) in the presence of NaH as neutralizing base. Since the synthesis of only one compound can result in the synthesis of bis-benzofuran-2-yl-methanone (
1) by route A, time consuming separation processes, for example chromatography, are not necessary. On the other hand, this new route is also provided slightly higher yield of compound (
1). For method B, the literature procedure was followed.
The reactions involve a nucleophilic substitution reaction in acetone between 1‑bromoacetone or 1,3-dichloroacetone and the sodium salts of 2-hydroxy-benzaldehyde (
Scheme 1). As a result of: (i) the strong nucleophilicity of the sodium salts of aldehydes, (ii) the absence of steric hindrance by dichloroacetone or 1-bromoacetylbenzofuran and (iii) the increased positive charge of the positive center of the carbocation formed by the elimination of chloride or bromide from the latter, due to the inductive effect of chlorine or bromine atoms, the acetone solvent provides a relatively mild temperature conditions for the formation of the expected substances with excellent yields.
The first compound, bisbenzofuran-2-yl-methanone (1), was prepared in the presence of potassium carbonate in excellent yields (81.9% by method B and 84.2% by method A). The disappearance of ‑CH2‑Cl or ‑CH2‑Br, (C=O, aldehyde) and -OH stretching bands of starting substances at about 732, 748, 1667 and 3319 cm–1, respectively, are evidence for the formation of the desired substance. Besides, the 1H-NMR spectra of the compound showed only aromatic protons. In the second step, 1 was reduced to its alcohol derivative 2 in the presence of NaBH4 in dioxane. The disappearance of the ketone C=O band and detection of a strong -OH band at about 3330 cm–1, are evidence for the formation of the alcohol derivative 2. In the 1H-NMR spectra of this compound, the –OH proton appears at 6.11 ppm. Treatment of 1 with semicarbazide or thiosemicarbazide in ethanol resulted in the formation of the semicarbazone and thiosemicarbazone derivatives 3a and 3b, respectively. Reaction of 1 with NH2OH·HCl in pyridine gave its oxime derivative 4. The absence of the C=O band of 1 and the existence of a broad =N-OH band ceneterd around 3166 cm-1 in the IR spectra of 4, confirm the formation of the compound. The oxime –OH proton appears as a broad singlet at 13.01 ppm in the corresponding 1H-NMR spectrum.
Reactions of 4 in acetone with appropriate alkyl iodides or bromides, or acyl chlorides in the presence of K2CO3, gave the oxime ether type compounds 5a-j. The use of acetone as solvent provides relatively mild temperature conditions for the formation of the expected substances in excellent yields. The analytical and spectroscopic characterization data of the compounds 5a-j given in the Experimental are in good agreement with the expected structures’ characteristics, as are the elemental analysis results. All the compounds synthesized here are solid substances, stable at room temperature and not affected by atmospheric conditions over at least in two weeks. On the other hand, as these compounds contain benzofuran, epoxide, ester, alkyl chloride, carbazone and hydrazide functions in their molecules, they seem to be suitable candidates for further chemical modifications and may be pharmacologically active and useful as ligands in coordination chemistry.
The compounds, except
5i, were tested against two gram–positive (
Staphylococcus aureus COWAN I,
Bacillus megaterium DSM 32) and two gram–negative bacteria (
Klebsiella pneumonia FMC 5,
Escherichia coli ATCC 25922). The antifungal activiti
es of compounds were evaluated
in vitro against a yeast–like fungus such as
Candida albicans CCM 314. The test results obtained are listed in
Table 1. Antifungal and antibacterial data for streptomycin and nystatin are also included in this Table for comparison purposes. The data show that these compounds generally exhibited moderate activity at the higher concentrations towards many of the bacteria and the fungus tested. The most active compound is
1. This compound exhibited activity towards all the microorganisms studied. Unexpectedly, six of the substances tested showed no toxicity any of the bacteria and the yeast under the test conditions. Studies of the biological activity of the substances towards some other different bacteria and yeasts are in progress.
Table 1.
Antimicrobial effect of the compounds*
Table 1.
Antimicrobial effect of the compounds*
| Compound | S. aureus | B. megaterium | K. pneumonia | E. coli | C. albicans |
|---|
| 1 | 13 | 18 | 15 | 18 | 17 |
| 2 | - | 15 | 13 | - | - |
| 3a | - | - | - | - | - |
| 3b | - | - | - | - | - |
| 4 | 8 | 13 | 11 | 14 | 12 |
| 5a | 9 | 11 | 11 | - | 15 |
| 5b | - | - | - | - | - |
| 5c | - | - | - | - | - |
| 5d | - | - | - | - | - |
| 5e | - | 8 | - | - | - |
| 5f | 13 | 15 | 13 | 11 | - |
| 5g | 15 | 18 | 18 | 13 | 11 |
| 5h | - | 11 | - | - | - |
| 5j | - | - | - | - | - |
| S.10 | 17 | 17 | 16 | - | - |
| N.30 | - | - | - | - | 18 |
Experimental
General
All the chemicals were purchased from Merck and used as received, except dichloroacteone and acetylbenzofuran, which were synthesized in our laboratory. Melting points were determined on a Thomas Hoover melting point apparatus and are uncorrected. The IR spectra were measured with Mattson 1000 FT-IR spectrophotometer. The 1H-NMR spectra were recorded on a Varian-Gemini 200 MHz spectrometer and are reported in ppm (δ) relative to tetramethylsilane (TMS) as the internal standard and 13C-NMR spectra (50.34 MHz) are referenced to deuterochloroform (CDCl3). Elemental analyses were determined on a LECO CHNSO-932 auto elemental analysis apparatus. Analyses (TLC) were performed on precoated 0.2 mm Merck Kieselgel 60 F254 plates, visualizing with a 254 nm UV lamp. Column chromatography was performed using Merck silica gel, 70-230 mesh. Solvents were dried and purified by known conventional methods prior to use.
General procedure for the synthesis of bisbenzofuran-2-yl-methanone (1), method A.
In a two necked 250 mL flask equipped with a reflux condenser, 2-acetylbenzofuran (16 g, 100 mmol) in acetic acid (150 mL) was heated to 50 ˚C. To this, bromine (16 g, 100 mmol) in acetic acid (50 mL) was added dropwise under nitrogen atmosphere and stirred at same temperature for 2 h while monitoring the course of the reaction by TLC. After cooling to room temperature, water was added. The solid, bromoacetylbenzofuran, thus separated was filtered off, washed with water several times and crystallized from ethanol. In the second stage, a mixture of salicylaldehyde (0.8 g, 3.34 mmol) and sodium hydride (0.08 g, 3.34 mmol) in acetone (50 mL) was stirred for 1h at room temperature. To this solution, a solution of bromoacetylbenzofuran (0.8 g, 3.34 mmol) in acetone (20 mL) was added dropwise and stirred for 18 h at room temperature. The solid 1 thus separated was filtered off, washed with water several times and crystallized from ethanol.
General procedure for the synthesis of bisbenzofuran-2-yl-methanone (1), method B.
In a two necked 500 mL flask, a mixture of salicylaldehyde (61.73 g, 0.51 mol) and K2CO3 (104 g, 0.76 mol) in acetone (200 mL) was stirred so that a bulky salt was obtained. To this mixture, a solution of dichloroactone (32.1 g, 0.25 mol) in acetone was added at room temperature and the mixture was stirred for 2 h. while the course of reaction was monitored by TLC. After cooling to room temperature, the mixture was poured into water and the precipitate of bisbenzofuran-2-yl-methanone (1) formed was filtered off, washed with water several times and crystallized from ethanol. Yield: 54.3 g (81.93 %); m.p.: 174.2 ˚C; IR (KBr): 1631 cm-1 (C=O); 1H‑NMR (CDCl3) δ: 7.35 (t, 2H, jH3-H2 = 7.33 Hz, jH3-H4 = 7.33 Hz, H3 aromatics ), 7.52 (t, 2H, H4 aromatics which has same signals with H3, aromatics), 7.66 (d, 2H, jH5-H4 = 7.11 Hz, H5 aromatics), 7.78 (d, 2H, jH2-H3 = 7.33 Hz, H2 aromatics ), 8.02 (d, 2H, jH1-H2 = 1.1 Hz, may be jH1-H5 = 1.1 Hz, H1 aromatics); 13C‑NMR (CDCl3) δ: 114.4, 118.2, 125.5, 126.11, 129.1, 130.6, 153.6, 157,8, 173.5 (C=O). Anal. Calcd. For C17H10O: C 77.85, H 3.84. Found: C 77.63, H 4.02.
Synthesis of bisbenzofuran-2-yl-methanol (2).
In a two necked 25 mL flask equipped with a reflux condenser, a mixture of bisbenzofuran-2-yl-methanone (1, 2 g, 7.625 mmol) and NaBH4 (0.29 g, 7.7 mmol) in dioxane (50 mL) was refluxed for 2 h. while the course of the reaction was monitored by IR. After cooling to room temperature, the mixture was poured into water and formed precipitate was filtered off by suction, dried in air and crystallized from ethanol. Yield: 1.83 g (91.5 %); m.p.: 91.7 ˚C; IR (KBr): 3230 cm-1 (-OH); 1H‑NMR (CDCl3) δ: 3.02 (s, 1H, >CH-), 6.11 (s, 1H, -OH), 6.79 (s, 2H, aromatics), 7.21-7.60 (m, 8H, aromatics); 13C‑NMR (CDCl3) δ: 157.2, 157.1, 129.9, 128.7, 125.1, 123.4, 113.5, 106.8, 67.1. Anal. Calcd. For C17H12O3: C 77.26, H 4.58. Found: C 76.89, H 4.61.
General procedure for the synthesis of semicarbazones/thiosemicarbazones 3a and 3b.
The preparation of 3b is given as an example. A mixture of 1 (1 g, 4 mmol), thiosemicarbazide (0.365 g, 4 mmol) and p-toluenesulphonic acid (0.005 g, as catalyst) in absolute ethanol (50 mL) was refluxed. The course of the reaction was monitored by TLC. After the solvent was removed under reduced pressure, the residue was treated with water and solid product 3b thus obtained was recrystallized from ethanol.
Bisbenzofuran-2-yl-methanone semicarbazone (3a). Yield: 1.09 g (89.64 %); m.p.: 189.4 ˚C; IR (KBr): 3467 cm-1 (-NH2), 3286 cm-1 (-NH-), 1738 cm-1 (C=O), 1587 cm-1 (C=N); 1H‑NMR (CDCl3) δ: 1.67 (s, 1H, -NH-), 6.57 (s, 1H, -NH-), 7.21-7.75 (m, 10H, aromatics), 10.73 (s, 1H, -NH- ), 13C‑NMR (CDCl3) δ: 181.4, 157.3, 156.9, 148.3, 130.8, 126.5, 125.6, 123.8, 114.1, 111.7. Anal. Calcd. For C18H13N3O3: C 67.71, H 4.10, N 13.16. Found: C 68.04, H 4.04, N 13.08.
Bisbenzofuran-2-yl-methanone thiosemicarbazone (3b). Yield: 0.53 g (41.73 %); m.p.: 194 ˚C; IR (KBr): 3399 and 3351 cm-1 (-NH2), 3249 cm-1 (-NH-), 1592 cm-1 (C=N), 1077 (C=S); 1H‑NMR (CDCl3) δ: 7.1 (s, 1H, -NH-), 7.2-7.7 (m, 11H, aromatics plus -NH-), 9.8 (s, 1H, -NH-); 13C‑NMR (CDCl3) δ: 178.4, 158.4, 157.1, 156.7, 131.5, 126.3, 125.7, 123.9, 113.7, 111.4. Anal. Calcd. For C18H13N3O2S: C 64.46, H 3.91, N 12.53, S 9.56. Found: C 64.27, H 4.03, N 12.66, S 9.91.
General procedure for the synthesis of ketoxime 4.
Compound 1 (20 g, 76 mmol), hydroxylamine hydrochloride (5.56 g, 80 mmol) and pyridine (100 mL) were mixed. The mixture was refluxed to complete the reaction while monitoring its course by IR. After cooling to room temperature, the mixture was poured into water. The solid bis-benzofuran-2-yl-methanone oxime (4) thus separated was filtered off, washed with water several times and crystallized from ethanol. Yield: 20.75 g (98.15 %); m.p.: 230.9 ˚C; IR (KBr): 3166 cm-1 (-OH oxime), 1581 cm-1 (C=N), 1036 (N-O); 1H‑NMR (CDCl3) δ: 13.01 (br s, 1H, -OH), 7.25-7.94 (m, 10H, aromatics), 13C‑NMR (CDCl3) δ: 155.9, 154.8, 145.9, 135.8, 128.6, 127.4, 125.1, 123.6, 110.4. Anal. Calcd. For C17H11NO3: C 73.64, H 4.00, N 5.05. Found: C 73.55, H 4.06, N 5.11.
General procedure for the synthesis of oxime-ethers 5a-h.
Compound 4 (1.0 g, 3.6 mmol), K2CO3 (0.55 g, 4 mmol) and an appropriate alkyl halogenide or acid chloride (3.6 mmol) were mixed in absolute acetone. The mixture was refluxed to complete the reaction while monitoring its course by IR. After cooling to room temperature, the mixture was poured into water. The solid thus separated was filtered off, washed with copious water and recrystallized from ethanol.
Bisbenzofuran-2-yl-methanone O-methyl oxime (5a). Yield: 0.43 g (41.03 %); m.p.: 131.0 ˚C; IR (KBr): 2937 cm-1 (-CH3 aliphatics), 1613 cm-1 (C=N); 1H‑NMR (CDCl3) δ: 4.30 (s, 3H, -CH3), 7.18-7.77 (m, 10H, aromatics). 13C‑NMR (CDCl3) δ: 156.9, 155.7, 148.3, 141.0, 130.2, 128.5, 127.8, 123.9, 113.8, 54.4. Anal. Calcd. For C18H13NO3: C 74.22, H 4.50, N 4.81. Found: C 73.89, H 4.61, N 5.02.
Bisbenzofuran-2-yl-methanone O-benzyl oxime (5b). Yield: 1.87 g (70.56 %); m.p.: 127.6 ˚C; IR (KBr): 2936 cm-1 (-CH3, aliphatics), 1614 cm-1 (C=N), 1004 (N-O); 1H‑NMR (CDCl3) δ: 5.56 (s, 2H, -CH2-), 7.26-7.76 (m, 10H, aromatics). 13C‑NMR (CDCl3) δ: 156.9, 155.7, 150.9, 148.4, 138.8, 130.3, 129.8, 127.7, 125.2, 123.7, 113.6, 80.5. Anal. Calcd. For C24H17NO3: C 78.46, H 4.66, N 3.81. Found: C 78.44, H 4.75, N 3.94.
Bisbenzofuran-2-yl-methanone O-oxiranylmethyl oxime (5c). Yield: 0.52 g (21.66 %); m.p.: 88.4 ˚C; IR (KBr): 2930 cm-1 (-CH2-, aliphatics), 1613 cm-1 (C=N), 1259 cm-1 (C-O, on benzofuran ring), 1046 cm-1 (C-O, epoxide), 1014 (N-O); 1H‑NMR (CDCl3): δHD, 2.76 (dd, 1H, jDC = 5.3 Hz, jDB = 2.6 Hz), δHC, 2.85 (dd, 1H, jCB = 4.8 Hz, jCD = 5.3 Hz), δB, 3.47 (m, 1H, HB), δHE, 4.41 (dd, 1H, jAE = 12.2 Hz, jBE = 6.4 Hz), δHA, 4.77 (dd, 1H, jAB = 2.8 Hz, jAE = 12.2 Hz), ), 7.25-7.83 (m, 10H, aromatics). 13C‑NMR (CDCl3) δ: 156.9, 155.8, 146.1, 130.1, 127.8, 125.2, 123.7, 113.7, 78.4, 52.1, 46.8. Anal. Calcd. For C20H15NO4: C 72.06, H 4.54, N 4.20. Found: C 71.96, H 4.49, N 4.17.
Bisbenzofuran-2-yl-methanone O-benzoyl oxime (5d). Yield: 1.29 g (94.16 %); m.p.: 154.9 ˚C; IR (KBr): 1747 cm-1 (C=O), 1612 cm-1 (C=N), 1057 (N-O); 1H‑NMR (CDCl3) δ: 7.28-7.79 (m, 13H, aromatics), 8.18-21 (m, 2H, aromatics); 13C‑NMR (CDCl3) δ: 165.4, 157.7, 156.9, 149.8, 146.1, 132.0, 130.8, 129.4, 128.5, 125.9, 125.5, 124.6, 124.1, 113.9. Anal. Calcd. For C24H15NO4: C 75.58, H 3.96, N 3.67. Found: C 75.43, H 4.02, N 3.88.
(Bis-benzofuran-2-yl-methyleneaminooxy)-acetic acetic acid ethyl ester (5e). Yield: 5.87 g (98.62 %); m.p.: 116.7 ˚C; IR (KBr): 2977 cm-1 (aliphatics), 1755 cm-1 (C=O), 1614 cm-1 (C=N), 1033 cm‑1 (N-O), 1087 cm-1 (C-O); 1H‑NMR (CDCl3) δ: 1.34 (t, 3H, j=7.21. –CH3), 4.31 (q, j=7.05, 2H, ‑CH2-), 5.05 (s, 2H, -CH2-), 7.27-7.74 (m, 10H, aromatics); 13C‑NMR (CDCl3) δ: 171.0, 157.0, 155.9, 145.9, 130.0, 128.6, 125.2, 124.4, 123.8, 113.7, 74.4, 63.2, 16.2. Anal. Calcd. For C21H17NO5: C 69.41, H 4.72, N 3.85. Found: C 69.55, H 4.87, N 4.01.
Bis-benzofuran-2-yl-methanone O-(2-phenylacetyl) oxime (5f). Yield: 1.28 g (89.95 %); m.p.: 141.4 ˚C; IR (KBr): 2919 cm-1 (aliphatics), 1753 cm-1 (C=O), 1613 cm-1 (C=N), 1021 (N-O), 1260 (C-O, on furan ring); 1H‑NMR (CDCl3) δ: 3.99 (s, 2H, –CH2-), 7.05-7.70 (m, 15H, aromatics); 13C‑NMR (CDCl3) δ: 169.9, 157.4, 156.3, 147.4, 134.9, 130.9, 129.6, 128.9, 125.5, 124.0, 118.5, 114.5, 113.0, 42.6. Anal. Calcd. For C25H17NO4: C 75.94, H 4.33, N 3.54. Found: C 76.12, H 4.46, N 3.43.
Bis-benzofuran-2-yl-methanone O-(2-chloroacetyl) oxime (5g). Yield: 1.18 g (92.69 %); m.p.: 135 ˚C; IR (KBr): 2954 cm-1 (aliphatics), 1785 cm-1 (C=O), 1613 cm-1 (C=N), 1012 (N-O), 1221 (C-O, on furan ring), 747 cm-1 (-CH2-Cl); 1H‑NMR (CDCl3) δ: 4.43 (s, 2H, –CH2-), 7.26-7.81 (m, 10H, aromatics); 13C‑NMR (CDCl3) δ: 166.8, 157.6, 144.9, 129.7, 129.0, 128.8, 125.6, 124.7, 124.2, 113.9, 42.1. Anal. Calcd. For C19H12ClNO4: C 64.51, H 3.42, N 3.96. Found: C 64.68, H 3.37, N 3.74.
Bis-benzofuran-2-yl-methanone O-acetyl oxime (5h). Yield: 1.18 g (95.65%); m.p.: 95.2 ˚C; IR (KBr): 2931 cm-1 (aliphatics), 1780 cm-1 (C=O), 1613 cm-1 (C=N), 1256 cm-1 (C-O, on benzofuran ring), 1021 cm-1 (N-O),; 1H‑NMR (CDCl3) δ: 2.40 (s, 3H, –CH3), 7.29-7.77 (m, 10H, aromatics); 13C‑NMR (CDCl3) δ: 169.9, 157.5, 156.4, 145.4, 129.4, 128.3, 125.5, 124.4, 118.2, 113.9, 21.8. Anal. Calcd. For C18H13N3O2S: C 71.47, H 4.10, N 4.39. Found: C 71.57, H 4.03, N 4.25.
General procedure for the synthesis of 5i.
Compound 5e (1.5 g, 4.13 mmol) and NaOH (0.32 g, 8 mmol) in EtOH (50 mL) were mixed. The mixture was refluxed to complete the reaction while monitoring its course by IR. After cooling to room temperature, the mixture was poured into water. Acidified with 5% HCl (pH = 3-4). The solid (bis-benzofuran–2-yl-methyleneaminooxy) acetic acid (5i) thus separated was filtered off, washed with water several times and crystallized from ethanol. Yield: 1.23 g (89.13 %); m.p.: 176.2 ˚C; IR (KBr): 2924 cm-1 (aliphatics), 1701 cm-1 (C=O), 1614 cm-1 (C=N), 1039 cm-1 (N-O), 1006 cm-1 (C-O); 1H‑NMR (CDCl3) δ: 5.18 (s, 2H, –CH2-), 7.24-7.75 (m, 10H, aromatics), 8.43 (br s, 1H, -OH); 13C‑NMR (CDCl3) δ: 176.0, 157.0, 145.9, 142.9, 130.0, 128.7, 128.0, 125.3, 123.9, 113.7, 73.9. Anal. Calcd. For C19H13NO5: C 68.06, H 3.91, N 4.18. Found: C 68.17, H 4.02, N 4.22.
General procedure for the synthesis of 5j.
The mixture of compound 5e (1.5 g, 4.13 mmol) and NH2NH2·H2O (0.25 g, 5 mmol) in absolute EtOH (50 mL) was refluxed for 8 h. to complete the reaction while monitoring its course by IR. After cooling to room temperature, the mixture was poured into cold water. The solid (bis-benzofuran-2-yl-methleneaminooxy) acetic acid hydrazide (5j) thus separated was filtered off, washed with water several times and crystallized from ethanol. Yield: 1.08 g (75.00 %); m.p.: 198 ˚C; IR (KBr): 3334-3314 cm-1 (-NH2 and –NH-), 2940 cm-1 (aliphatics), 1660 cm-1 (C=N), 1030 cm-1 (N-O), 949 cm-1 (C-O); 1H‑NMR (CDCl3) δ: 4.40 (br s, 2H, –NH2), 4.85 (s, 2H, -CH2-), 7.34-7.88 (m, 10H, aromatics), 9.34 (s, 1H, -NH-); 13C‑NMR (CDCl3) δ: 168.7, 156.0, 155.1, 149.8, 145.2, 129.4, 128.8, 125.6, 123.9, 113.6, 75.2. Anal. Calcd. For C19H15N3O4: C 65.32, H 4.33, N 12.03. Found: C 65.47, H 4.42, N 12.16.
Preparation of microbial cultures and biological assays
Microorganisms were obtained from the culture collection of the Microbiology Laboratory of the Biological Sciences of Faculty of Science and Arts, Firat University, Turkey. In this work, Bacillus subtilis LM 622 (B.s.), Enterobacter aeruginosa CCM 2531 (E.a.), Eschericia coli ATCC 25922 (E.c.), Listeria monocytogenes SCOTTA (L.m.), Pseudomonas aeruginosa DSM 50071 (P.a.), Staphilococcus aureus COWAN I (S.a.) Candida albicans FMC 17 (C.a.) and Saccharamyces cerevisiae FMC 16 (S.c.) were used to investigate the bacteriological and antifungal activities of selected substances.
The bacteria and yeast strains were nourished in nutrient broth (Difco) and in malt extract broth (Difco), and incubated for 24 and 48 h, respectively. In the Disk Diffusion method, the sterile Mueller Hinton Agar (Oxoid) for bacteria and Saburoud Dextrose Agar for yeast were separately inoculated with the test microorganisms. The compounds, dissolved in DMSO as 500 µg/disc solutions, were placed in 6 mm wells in the agar media and the plates were incubated at 32 °C for bacteria (18-24 h) and at 25° C for yeast (72 h) [
15]. The resulting inhibition zones on the plates were measured (in mm) after 48 h (
Table 1). The control samples were only dissolved in DMSO. The data reported in
Table 1 are the average of three experiments.


{kind=link}