Towards a Synthesis of Naphthalene Derived Natural Products

Abstract
:Introduction
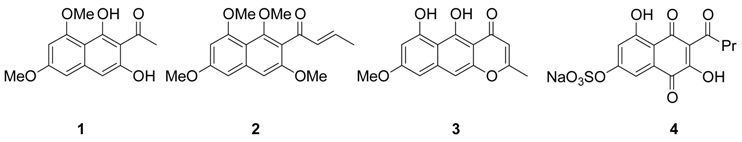
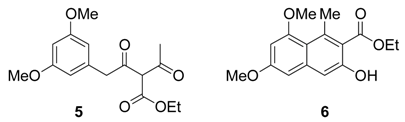
Results and Discussion
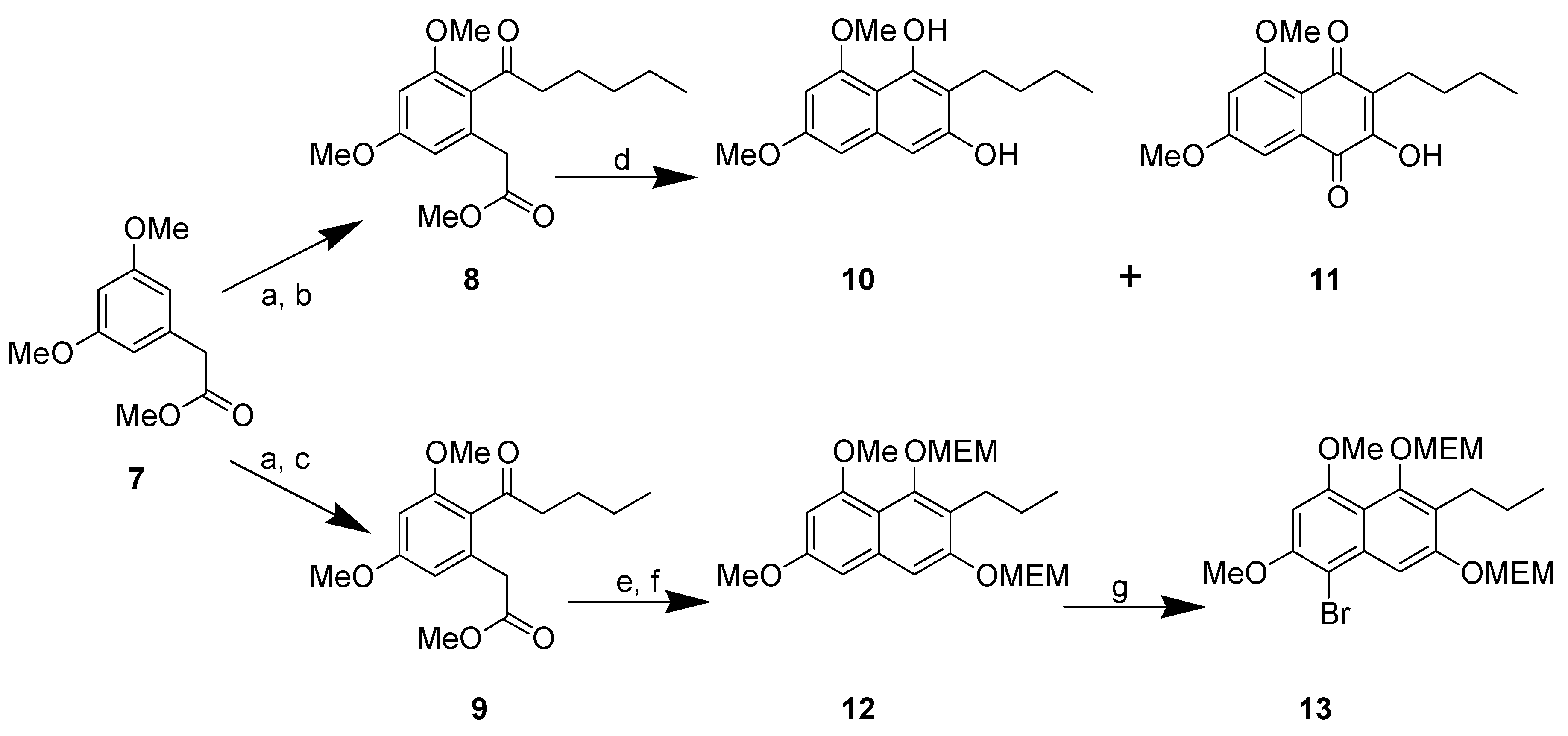

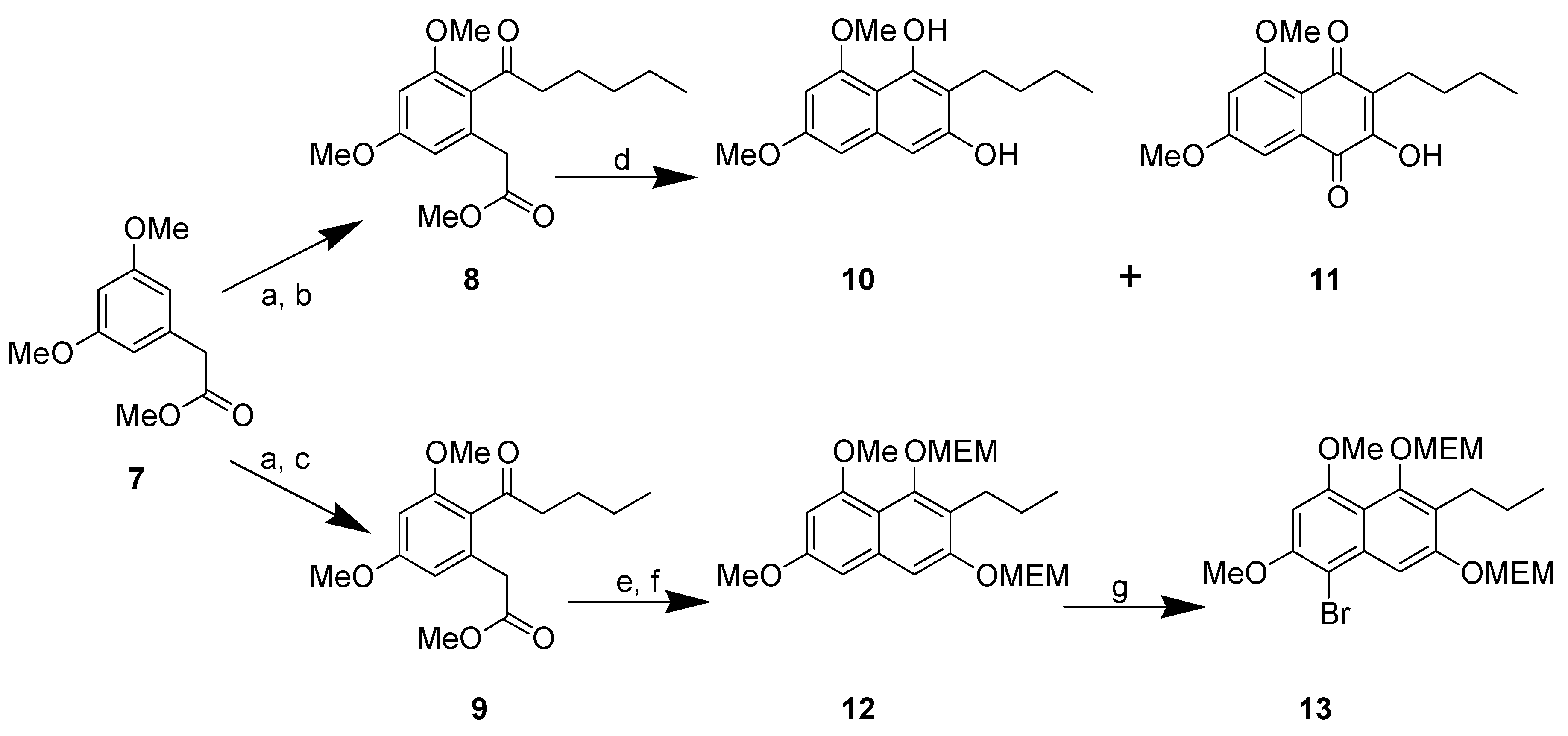{kind=link}
Experimental
General
General procedure to prepare the ketones \8 and 9
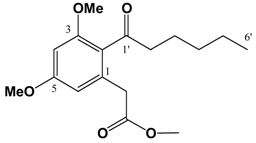 (v/v)]. IR: ν (cm-1): 2935 s, 2858 m, 1738 s, 1682 m, 1605 s, 1583 m, 1455 m, 1428 m, 1318 s, 1293 w, 1257 w, 1205 m, 1156 s, 1086 m, 1058 m, 1012 m, 954 w, 835 m. EIMS m/z: 308 (M+⋅, 26%), 277 (M-OCH3, 17%), 237 (M-(CH2)4CH3, 100%), 209 (M-C(O)(CH2)4CH3, 88%). HREI-MS m/z: found 308.1624 (C17H24O5 requires 308.1624). UV (MeOH) λmax (log ε): 201 (3.46), 266 (2.93). 1H-NMR (CDCl3): 6.39 (1H, d, 2.2 Hz, 4-H), 6.37 (1H, d, 2.2Hz, 6-H), 3.81 (3H, s, 3-OCH3)*, 3.80 (3H, s, 5-OCH3)*, 3.67 (3H, s, -CO2CH3), 3.62 (2H, s, -CH2CO2CH3), 2.81 (2H, t, 7.2 Hz, 2'-H), 1.64 (2H, m, 3'-H), 1.31 (4H, m, 4'&5'-H), 0.89 (3H, t, 6.9 Hz, 6'-H). 13C APT-NMR (CDCl3): 206.8 (C-1'), 171.6 (-CO2CH3), 161.2 (C-3)*, 158.8 (C-5)*, 134.3 (C-1), 123.9 (C-2), 107.8 (C-6), 97.4 (C-4), 55.5 (C-3-OCH3)∞, 55.3 (C-5-OCH3)∞, 51.9 (-CO2CH3), 44.3 ( C-2'), 38.7 (-CH2CO2CH3), 31.4 (C-4'), 23.7 (C-3'), 22.4 (C-5'), 13.9 (C-6'). Shifts with identical superscripts (*,∞) within a data set are interchangeable.
(v/v)]. IR: ν (cm-1): 2935 s, 2858 m, 1738 s, 1682 m, 1605 s, 1583 m, 1455 m, 1428 m, 1318 s, 1293 w, 1257 w, 1205 m, 1156 s, 1086 m, 1058 m, 1012 m, 954 w, 835 m. EIMS m/z: 308 (M+⋅, 26%), 277 (M-OCH3, 17%), 237 (M-(CH2)4CH3, 100%), 209 (M-C(O)(CH2)4CH3, 88%). HREI-MS m/z: found 308.1624 (C17H24O5 requires 308.1624). UV (MeOH) λmax (log ε): 201 (3.46), 266 (2.93). 1H-NMR (CDCl3): 6.39 (1H, d, 2.2 Hz, 4-H), 6.37 (1H, d, 2.2Hz, 6-H), 3.81 (3H, s, 3-OCH3)*, 3.80 (3H, s, 5-OCH3)*, 3.67 (3H, s, -CO2CH3), 3.62 (2H, s, -CH2CO2CH3), 2.81 (2H, t, 7.2 Hz, 2'-H), 1.64 (2H, m, 3'-H), 1.31 (4H, m, 4'&5'-H), 0.89 (3H, t, 6.9 Hz, 6'-H). 13C APT-NMR (CDCl3): 206.8 (C-1'), 171.6 (-CO2CH3), 161.2 (C-3)*, 158.8 (C-5)*, 134.3 (C-1), 123.9 (C-2), 107.8 (C-6), 97.4 (C-4), 55.5 (C-3-OCH3)∞, 55.3 (C-5-OCH3)∞, 51.9 (-CO2CH3), 44.3 ( C-2'), 38.7 (-CH2CO2CH3), 31.4 (C-4'), 23.7 (C-3'), 22.4 (C-5'), 13.9 (C-6'). Shifts with identical superscripts (*,∞) within a data set are interchangeable.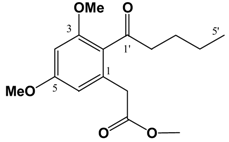 (v/v)]. IR: ν (cm-1): 2957 s, 2871 m, 1740 s, 1686 m, 1603 s, 1460 m, 1433 m, 1319 s, 1294 w, 1263 w, 1205m, 1157 s, 1099 m, 1082 m, 1013 m, 948 w, 835 m. EIMS m/z: 294 (M+⋅, 15%), 293 (52%), 275 (14%), 261 (35%), 250 (24%), 234 (100%), 206 (71%), 202 (33%), 176 (20%), 162 (28%), 148 (21%). HREI-MS m/z: found 294.1467 (C16H22O5 requires 294.1467). UV (MeOH) λmax (log ε): 202 (3.45), 263 (2.90). 1H-NMR (CDCl3): 6.39 (1H, d, 2.2 Hz, 4-H), 6.36 (1H, d, 2.2Hz, 6-H), 3.80 (3H, s, 3-OCH3)*, 3.79 (3H, s, 5-OCH3)*, 3.67 (3H, s, -CO2CH3), 3.62 (2H, s, -CH2CO2CH3), 2.82 (2H, t, 7.3 Hz, 2'-H), 1.61 (2H, m, 3'-H), 1.36 (2H, m, 4'-H), 0.91 (3H, t, 7.3 Hz, 5'-H). 13C APT-NMR (CDCl3): 206.5 (C-1'), 171.4 (-CO2CH3), 161.1 (C-3)*, 158.6 (C-5)*, 134.2 (C-1), 123.7 (C-2), 107.8 (C-6), 97.2 (C-4), 55.4 (C-3-OCH3)∞, 55.3 (C-5-OCH3)∞, 51.7 (-CO2CH3), 43.9 ( C-2'), 38.5 (-CH2CO2CH3), 26.0 (C-3'), 22.2 (C-4'), 13.7 (C-5'). Shifts with identical superscripts (*,∞) within a data set are interchangeable.
(v/v)]. IR: ν (cm-1): 2957 s, 2871 m, 1740 s, 1686 m, 1603 s, 1460 m, 1433 m, 1319 s, 1294 w, 1263 w, 1205m, 1157 s, 1099 m, 1082 m, 1013 m, 948 w, 835 m. EIMS m/z: 294 (M+⋅, 15%), 293 (52%), 275 (14%), 261 (35%), 250 (24%), 234 (100%), 206 (71%), 202 (33%), 176 (20%), 162 (28%), 148 (21%). HREI-MS m/z: found 294.1467 (C16H22O5 requires 294.1467). UV (MeOH) λmax (log ε): 202 (3.45), 263 (2.90). 1H-NMR (CDCl3): 6.39 (1H, d, 2.2 Hz, 4-H), 6.36 (1H, d, 2.2Hz, 6-H), 3.80 (3H, s, 3-OCH3)*, 3.79 (3H, s, 5-OCH3)*, 3.67 (3H, s, -CO2CH3), 3.62 (2H, s, -CH2CO2CH3), 2.82 (2H, t, 7.3 Hz, 2'-H), 1.61 (2H, m, 3'-H), 1.36 (2H, m, 4'-H), 0.91 (3H, t, 7.3 Hz, 5'-H). 13C APT-NMR (CDCl3): 206.5 (C-1'), 171.4 (-CO2CH3), 161.1 (C-3)*, 158.6 (C-5)*, 134.2 (C-1), 123.7 (C-2), 107.8 (C-6), 97.2 (C-4), 55.4 (C-3-OCH3)∞, 55.3 (C-5-OCH3)∞, 51.7 (-CO2CH3), 43.9 ( C-2'), 38.5 (-CH2CO2CH3), 26.0 (C-3'), 22.2 (C-4'), 13.7 (C-5'). Shifts with identical superscripts (*,∞) within a data set are interchangeable.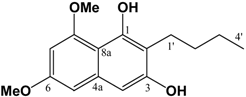 (cm-1): 3402 (s, br), 2932 s, 2858 m, 2362 w, 1636 s, 1597 s, 1448 m, 1404 m, 1371 s, 1338 w, 1246 w, 1209 m, 1150 m, 1107 m, 1041 m. ESI-MS (+ve ion, cv 50V) m/z: 277 (M+H, 100%), 263 (M-CH3+H, 10%), 220 (2%), 205 (2%). UV (MeOH) λmax (log ε): 245 (4.90), 292 (3.91). 1H-NMR (CDCl3): 9.43 (1H, s, 1-OH), 6.55 (1H, s, 4-H), 6.50 (1H, d, 2.1 Hz, 5-H), 6.28 (1H, d, 2.1 Hz, 7-H), 3.98 (3H, s, 8-OCH3), 3.85 (3H, s, 6-OCH3), 2.74 (2H, t, 7.5 Hz, 1'-H), 1.58 (2H, m, 2'-H), 1.44 (2H, m, 3'-H), 0.94 (3H, t, 7.2 Hz, 4'-H). 13C APT-NMR (CDCl3): 157.5 (C-6)*, 157.0 (C-8) *, 154.6 (C-3)∞, 152.9 (C-1)∞, 135.9 (C-4a), 112.1 (C-2), 106.2 (C-8a), 100.8 (C-4) 98.0 (C-5), 95.4 (C-7), 56.0 (C-8-OCH3), 55.3 (C-6-OCH3), 31.5 (C-2'), 22.9 (C-1')Δ, 22.8 (C-3')Δ, 14.1 (C-4'). Shifts with identical superscripts (*,∞,Δ) are interchangeable.
(cm-1): 3402 (s, br), 2932 s, 2858 m, 2362 w, 1636 s, 1597 s, 1448 m, 1404 m, 1371 s, 1338 w, 1246 w, 1209 m, 1150 m, 1107 m, 1041 m. ESI-MS (+ve ion, cv 50V) m/z: 277 (M+H, 100%), 263 (M-CH3+H, 10%), 220 (2%), 205 (2%). UV (MeOH) λmax (log ε): 245 (4.90), 292 (3.91). 1H-NMR (CDCl3): 9.43 (1H, s, 1-OH), 6.55 (1H, s, 4-H), 6.50 (1H, d, 2.1 Hz, 5-H), 6.28 (1H, d, 2.1 Hz, 7-H), 3.98 (3H, s, 8-OCH3), 3.85 (3H, s, 6-OCH3), 2.74 (2H, t, 7.5 Hz, 1'-H), 1.58 (2H, m, 2'-H), 1.44 (2H, m, 3'-H), 0.94 (3H, t, 7.2 Hz, 4'-H). 13C APT-NMR (CDCl3): 157.5 (C-6)*, 157.0 (C-8) *, 154.6 (C-3)∞, 152.9 (C-1)∞, 135.9 (C-4a), 112.1 (C-2), 106.2 (C-8a), 100.8 (C-4) 98.0 (C-5), 95.4 (C-7), 56.0 (C-8-OCH3), 55.3 (C-6-OCH3), 31.5 (C-2'), 22.9 (C-1')Δ, 22.8 (C-3')Δ, 14.1 (C-4'). Shifts with identical superscripts (*,∞,Δ) are interchangeable.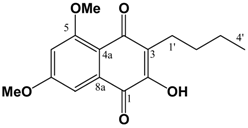 (v/v)]. mp: 160∞C. IR: ν (cm-1): 3209 m, 2932 m, 2359 m, 1655 m, 1638 s, 1595 m, 1460 m, 1321 s, 1209 s, 1157 m, 1126 w, 1034 w. EIMS m/z: 290 (M+∞, 100%), 248 (M-C3H6, 43%), 233 (M-(CH2)3CH3, 13%), 219 (35%), 165 (22%). HREI-MS m/z: found 290.1163 (C16H18O5 requires 290.1154). UV (MeOH) λmax (log ε): 212 (4.68), 261 (4.51), 304 (4.29). 1H-NMR (CDCl3): 7.25 (1H, d, 2.4 Hz, 8-H), 6.75 (1H, d, 2.4 Hz, 5-H), 3.96 (3H, s, 5-OCH3)*, 3.93 (3H, s, 7-OCH3)*, 2.56 (2H, t, 7.5 Hz, 1'-H), 1.50 (2H, m, 2'-H), 1.40 (2H, m, 3'-H), 0.92 (3H, t, 7.2 Hz, 4'-H). 13C APT-NMR (CDCl3): 183.6 (C-4)*, 181.7 (C-1)*, 163.7 (C-7)∞, 161.7 (C-5)∞, 150.9 (C-2), 133.2 (C-8a), 126.2 (C-3), 114.2 (C-4a), 105.2 (C-6), 103.0 (C-8), 56.4 (C-5-OCH3)Δ, 55.8 (C-7-OCH3)Δ, 30.5 (C-2'), 23.2 (C-1'), 22.9 (C-3'), 13.8(C-4'). Shifts with identical superscripts (*,∞,Δ) within a data set are interchangeable.
(v/v)]. mp: 160∞C. IR: ν (cm-1): 3209 m, 2932 m, 2359 m, 1655 m, 1638 s, 1595 m, 1460 m, 1321 s, 1209 s, 1157 m, 1126 w, 1034 w. EIMS m/z: 290 (M+∞, 100%), 248 (M-C3H6, 43%), 233 (M-(CH2)3CH3, 13%), 219 (35%), 165 (22%). HREI-MS m/z: found 290.1163 (C16H18O5 requires 290.1154). UV (MeOH) λmax (log ε): 212 (4.68), 261 (4.51), 304 (4.29). 1H-NMR (CDCl3): 7.25 (1H, d, 2.4 Hz, 8-H), 6.75 (1H, d, 2.4 Hz, 5-H), 3.96 (3H, s, 5-OCH3)*, 3.93 (3H, s, 7-OCH3)*, 2.56 (2H, t, 7.5 Hz, 1'-H), 1.50 (2H, m, 2'-H), 1.40 (2H, m, 3'-H), 0.92 (3H, t, 7.2 Hz, 4'-H). 13C APT-NMR (CDCl3): 183.6 (C-4)*, 181.7 (C-1)*, 163.7 (C-7)∞, 161.7 (C-5)∞, 150.9 (C-2), 133.2 (C-8a), 126.2 (C-3), 114.2 (C-4a), 105.2 (C-6), 103.0 (C-8), 56.4 (C-5-OCH3)Δ, 55.8 (C-7-OCH3)Δ, 30.5 (C-2'), 23.2 (C-1'), 22.9 (C-3'), 13.8(C-4'). Shifts with identical superscripts (*,∞,Δ) within a data set are interchangeable.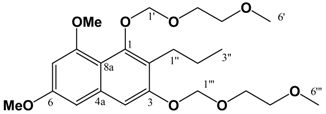 Pet. Sp, 30:70 (v/v)]. IR: ν (cm-1): 2932 s, 1622 s, 1580m, 1458 m, 1389 m, 1337 m, 1252 w, 1204 m, 1155 s, 1119 m, 1097 m, 1036 s, 847 w. ESI-MS (+ve ion, cv 50V) m/z: 461 (M+Na, 100%), 239 (M+H, 11%), 373 ([M-MEM]+Na), 28%), 221 (22%). HREI-MS m/z: found 438.2258 (C23H34O8 requires 438.2254). UV (MeOH) λmax (log ε): 216 (4.25), 242 (4.46), 288 (3.74). 1H-NMR (CDCl3): 7.14 (1H, s, 4-H), 6.65 (1H, d, 2.2 Hz, 5-H), 6.37 (1H, d, 2.2 Hz, 7-H), 5.38 (2H, s, 1'''-H), 5.12 (2H, s, 1'-H), 4.00-3.57 (8H, m, {3'-H, 4'-H, 3'''-H, 4'''-H}), 3.90 (3H, s, 8-OCH3)*, 3.86 (3H, s, 6-OCH3)*, 3.40 (3H, s, 6'-H)∞, 3.39 (3H, s, 6'''-H)∞, 2.80 (2H, t, 7.5 Hz, 1''-H), 1.60 (2H, m, 2''-H), 0.97 (3H, t, 7.3 Hz, 3''-H). 13C APT-NMR (CDCl3): 157.6 (C-8), 156.4 (C-6), 154.9 (C-3), 151.5 (C-1), 136.6 (C-4a), 123.4 (C-2), 111.3 (C-8a), 105.2 (C-4), 100.2 (C-1')∗, 98.5 (C-5), 97.0 (C-7), 93.3 (C-1''')∗, 71.7 (C-4')Δ, 71.6 (C-4''')Δ, 69.2 (C-3'), 67.6 (C-3'''), 59.1 (C-6')Ψ, 59.0 (C-6''')Ψ, 55.8 (C-8-OCH3)Ω, 55.2 (C-6-OCH3)Ω, 26.5 (C-2''), 23.2 (C-1''), 14.5 (C-3''). Shifts with identical superscripts (*,∞,Δ,Ψ,Ω) within a data set are interchangeable.
Pet. Sp, 30:70 (v/v)]. IR: ν (cm-1): 2932 s, 1622 s, 1580m, 1458 m, 1389 m, 1337 m, 1252 w, 1204 m, 1155 s, 1119 m, 1097 m, 1036 s, 847 w. ESI-MS (+ve ion, cv 50V) m/z: 461 (M+Na, 100%), 239 (M+H, 11%), 373 ([M-MEM]+Na), 28%), 221 (22%). HREI-MS m/z: found 438.2258 (C23H34O8 requires 438.2254). UV (MeOH) λmax (log ε): 216 (4.25), 242 (4.46), 288 (3.74). 1H-NMR (CDCl3): 7.14 (1H, s, 4-H), 6.65 (1H, d, 2.2 Hz, 5-H), 6.37 (1H, d, 2.2 Hz, 7-H), 5.38 (2H, s, 1'''-H), 5.12 (2H, s, 1'-H), 4.00-3.57 (8H, m, {3'-H, 4'-H, 3'''-H, 4'''-H}), 3.90 (3H, s, 8-OCH3)*, 3.86 (3H, s, 6-OCH3)*, 3.40 (3H, s, 6'-H)∞, 3.39 (3H, s, 6'''-H)∞, 2.80 (2H, t, 7.5 Hz, 1''-H), 1.60 (2H, m, 2''-H), 0.97 (3H, t, 7.3 Hz, 3''-H). 13C APT-NMR (CDCl3): 157.6 (C-8), 156.4 (C-6), 154.9 (C-3), 151.5 (C-1), 136.6 (C-4a), 123.4 (C-2), 111.3 (C-8a), 105.2 (C-4), 100.2 (C-1')∗, 98.5 (C-5), 97.0 (C-7), 93.3 (C-1''')∗, 71.7 (C-4')Δ, 71.6 (C-4''')Δ, 69.2 (C-3'), 67.6 (C-3'''), 59.1 (C-6')Ψ, 59.0 (C-6''')Ψ, 55.8 (C-8-OCH3)Ω, 55.2 (C-6-OCH3)Ω, 26.5 (C-2''), 23.2 (C-1''), 14.5 (C-3''). Shifts with identical superscripts (*,∞,Δ,Ψ,Ω) within a data set are interchangeable.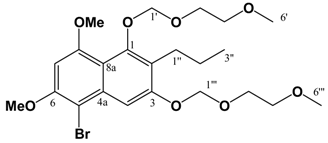 [MeOH/ DCM, 2:98 (v/v)]. ESI-EIMS (+ve ion, cv 20 V) m/z: 541 (M+Na, 81Br, 100%), 539 (M+Na, 79Br, 100%), 519 (M+H, 81Br, 55%), 517 (M+H, 79Br, 55%). HREI-MS m/z: found 516.1356 (C23H33O879Br1 requires 516.1359). 1H-NMR (CDCl3): 7.65 (1H, s, 7-H), 6.55 (1H, s, 4-H), 5.43 (2H, s, 1'''-H), 5.10 (2H, s, 1'-H), 4.00 (3H, s, 8-OCH3)*, 3.97 (3H, s, 6-OCH3)*, 3.87 (4H, m, (3'-H, 3'''-H))∞, 3.61 (4H, m, (4'-H, 4'''-H))∞, 3.40 (6H, s (coincident), 6'-H & 6'''-H), 2.82 (2H, t, 7.8 Hz, 1''-H), 1.58 (2H, m, 2''-H), 0.98 (3H, t, 7.4 Hz, 3''-H). Shifts with identical superscripts (*,∞) within a data set are interchangeable.
[MeOH/ DCM, 2:98 (v/v)]. ESI-EIMS (+ve ion, cv 20 V) m/z: 541 (M+Na, 81Br, 100%), 539 (M+Na, 79Br, 100%), 519 (M+H, 81Br, 55%), 517 (M+H, 79Br, 55%). HREI-MS m/z: found 516.1356 (C23H33O879Br1 requires 516.1359). 1H-NMR (CDCl3): 7.65 (1H, s, 7-H), 6.55 (1H, s, 4-H), 5.43 (2H, s, 1'''-H), 5.10 (2H, s, 1'-H), 4.00 (3H, s, 8-OCH3)*, 3.97 (3H, s, 6-OCH3)*, 3.87 (4H, m, (3'-H, 3'''-H))∞, 3.61 (4H, m, (4'-H, 4'''-H))∞, 3.40 (6H, s (coincident), 6'-H & 6'''-H), 2.82 (2H, t, 7.8 Hz, 1''-H), 1.58 (2H, m, 2''-H), 0.98 (3H, t, 7.4 Hz, 3''-H). Shifts with identical superscripts (*,∞) within a data set are interchangeable.References
- Silva, O.; Gomes, E.T. J. Nat. Prod. 2003, 66, 447–449. [CrossRef] [PubMed]
- Ashley, J.N.; Hobbs, B.C.; Raistrick, H. Biochem. J. 1937, 31, 385–397.
- Stout, G.; Jensen, L. Acta Crystallogr 1962, 15, 451–457.
- Takahashi, D.; Maoka, T.; Tsushima, M.; Fujitani, K.; Kozuka, M.; Matsuno, T.; Shingu, T. Chem. Pharm. Bull. 2002, 12, 1609–1612. [CrossRef]
- Shibata, S.; Morishita, E. Chem. Pharm. Bull. 1963, 11, 821–823. [CrossRef]
- Bycroft, B.W.; Roberts, J.C. Chem. Soc. J. 1963, 4868–4872.
- Abell, C.; Bush, B.D.; Staunton, J. J. Chem. Soc., Chem. Commun. 1986, 15–17. [CrossRef]
- Rideout, J.A.; Smith, I.R.; Sutherland, M.D. Aust. J. Chem. 1976, 29, 1087–1098.
- Pang, Y.P.; Kozikowski, A.P. J. Org. Chem. 1991, 56, 4499–508.
- Bhatt, M.V.; Perumal, P.T. Tetrahedron Lett. 1981, 22, 2605–2608.
- Noureldin, N.A.; Zhao, D.; Lee, D.G. J. Org. Chem. 1997, 62, 8767–8772. [CrossRef]
- Sample Availability: Not available
© 2005 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
McCulloch, M.; Barrow, R. Towards a Synthesis of Naphthalene Derived Natural Products. Molecules 2005, 10, 1272-1278. https://doi.org/10.3390/10101272
McCulloch M, Barrow R. Towards a Synthesis of Naphthalene Derived Natural Products. Molecules. 2005; 10(10):1272-1278. https://doi.org/10.3390/10101272
Chicago/Turabian StyleMcCulloch, M., and R. Barrow. 2005. "Towards a Synthesis of Naphthalene Derived Natural Products" Molecules 10, no. 10: 1272-1278. https://doi.org/10.3390/10101272
APA StyleMcCulloch, M., & Barrow, R. (2005). Towards a Synthesis of Naphthalene Derived Natural Products. Molecules, 10(10), 1272-1278. https://doi.org/10.3390/10101272