Synthesis and Molecular Structure of Methyl 4-O-methyl-α-D-glucopyranuronate

Abstract
:Introduction
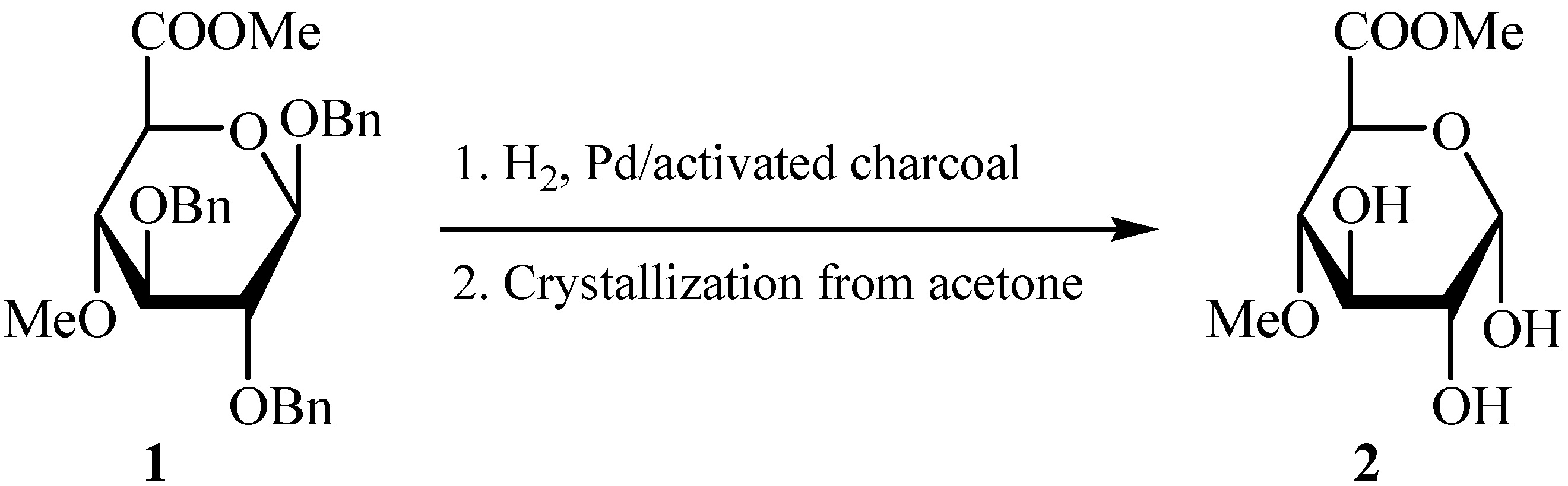
Results and Discussion
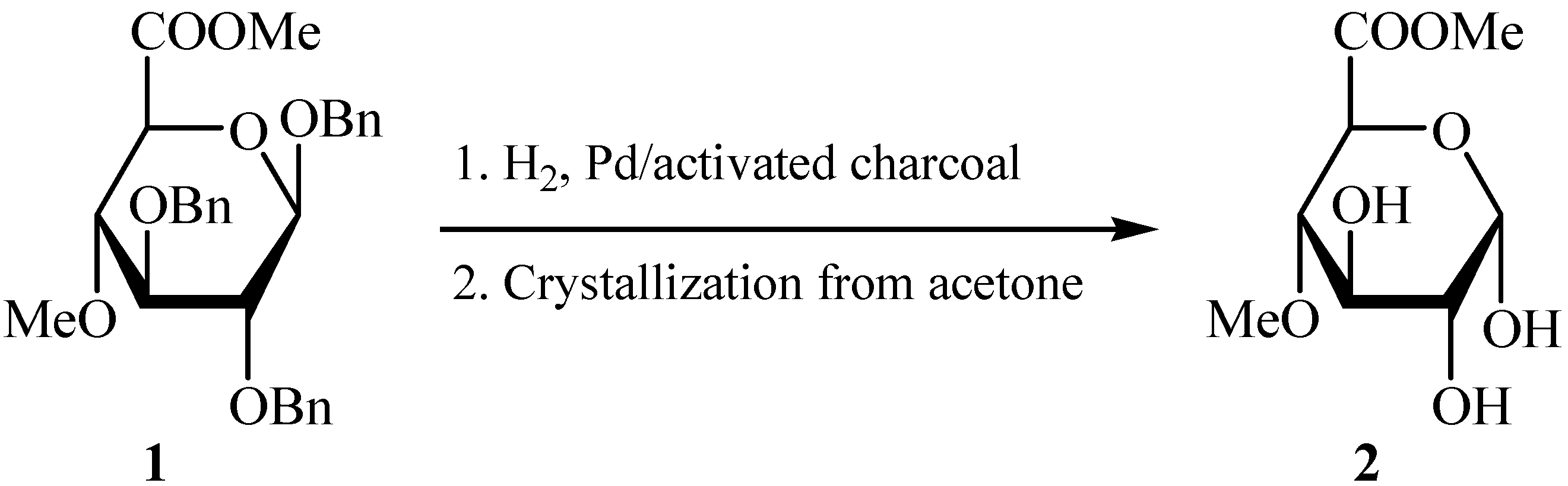
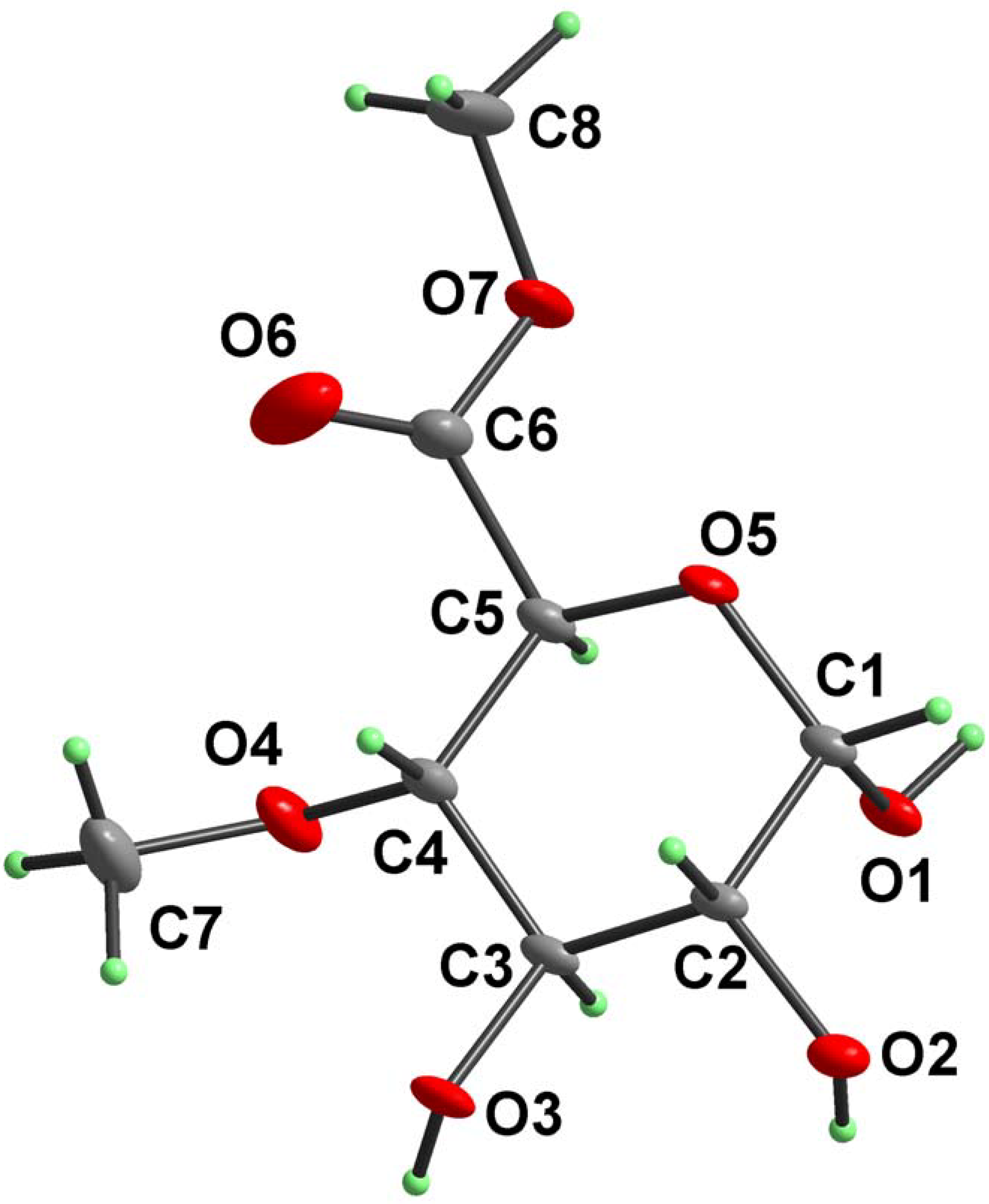

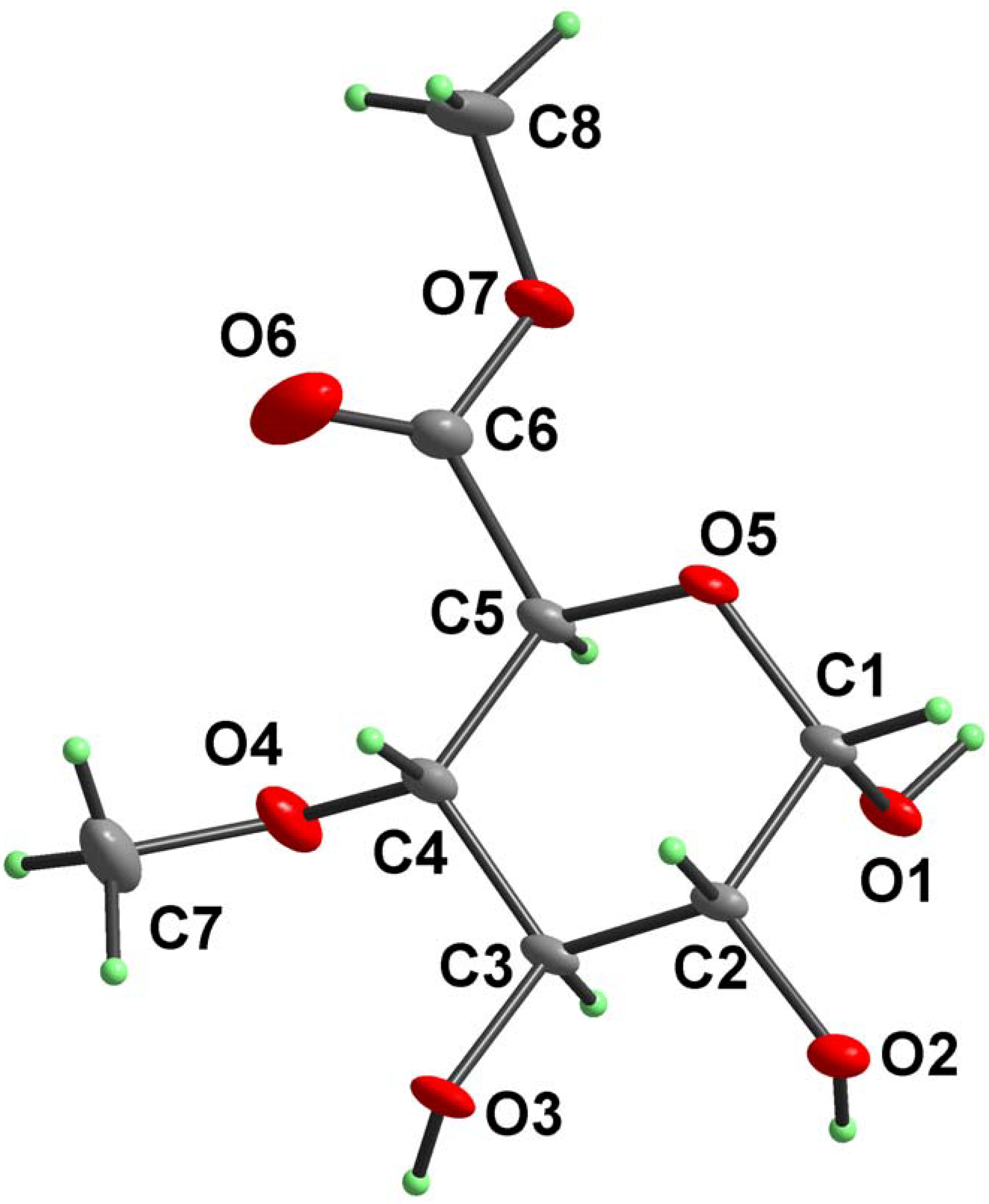{kind=link}
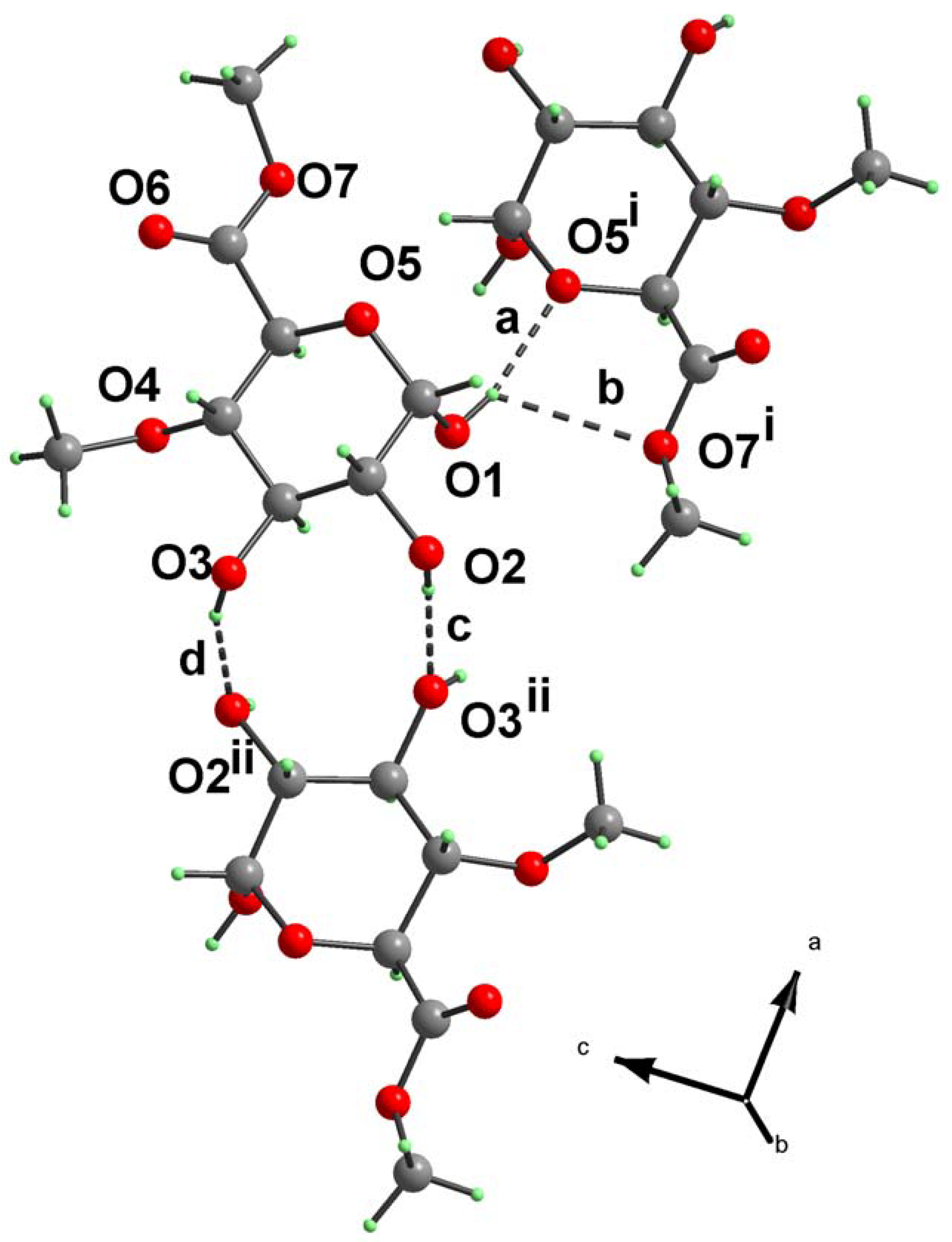{kind=link}
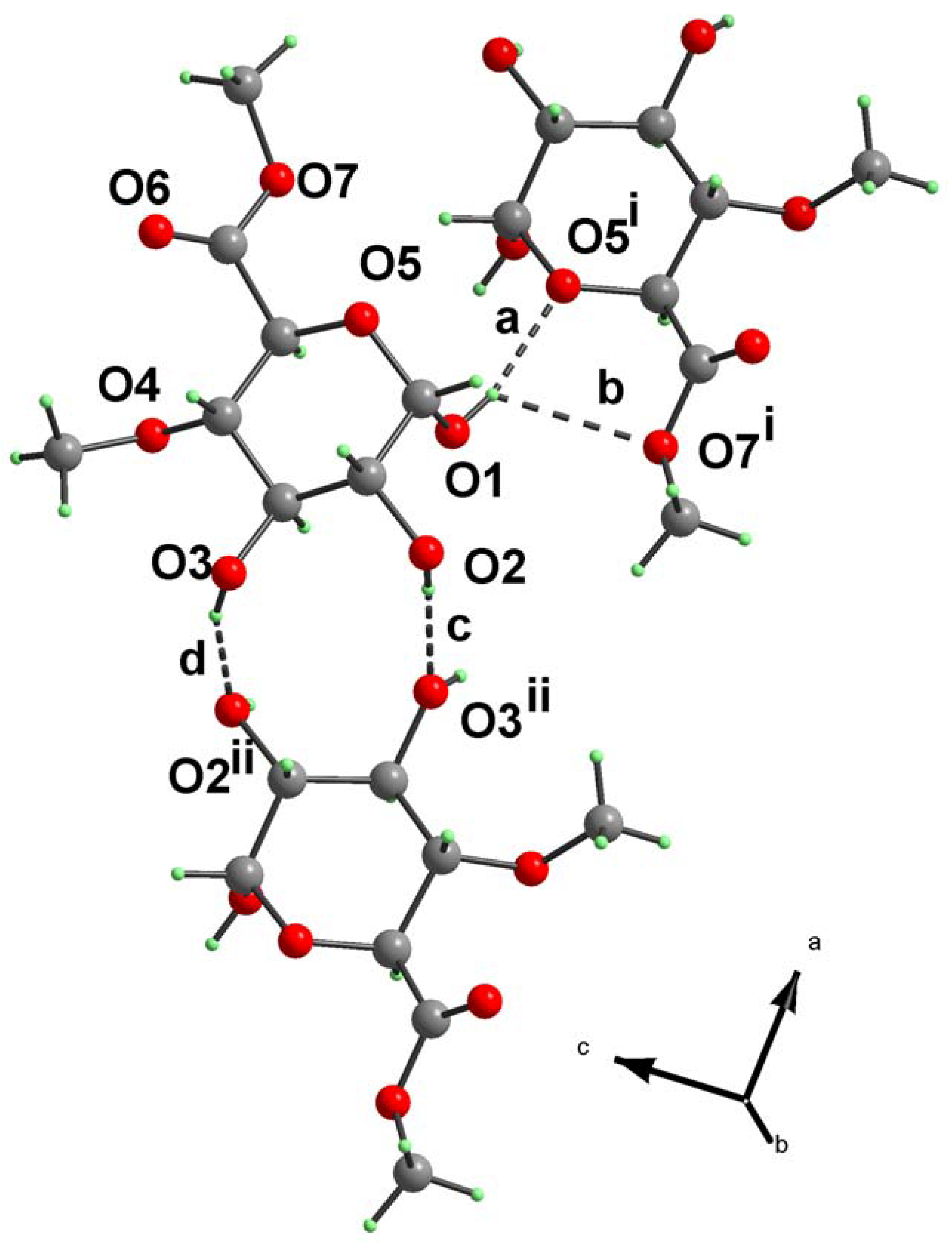
{kind=link}
| Bond | Distance | Bond angle | Angle |
|---|---|---|---|
| O1–C1 | 1.3922(17) | C1–O5–C5 | 113.76(10) |
| Torsion angle | Angle | Torsion angle | Angle |
|---|---|---|---|
| C5–O5–C1–O1 | 58.47(15) | O5–C1–C2–C3 | 60.05(13) |
| Notation | X–H…Y | Symmetry code | X–H (Å) | H…Y (Å) | X…Y (Å) | X–H…Y (°) |
|---|---|---|---|---|---|---|
| a | O1–H1…O5i | –x+2, y+1/2, –z+1 | 0.84 | 2.00 | 2.8219(13) | 166.9 |
Experimental
General
X-ray techniques
| Empirical formula | C8H14O7 |
Syntheses
Methyl 4-O-methyl-α-D-glucopyranuronate (2).
Acknowledgements
References
- Timell, T.E. Adv. Carbohydr. Chem. 1964, 19, 247.
- Timell, T.E. Can. J. Chem. 1959, 37, 827.
- Kováč, P.; Petráková, E.; Kočiš, P. Carbohydr. Res. 1981, 93, 144.
- Hirsch, J.; Koóš, M.; Kováč, P. Carbohydr. Res. 1998, 310, 145.
- Das, N.N.; Das, S.C.; Dutt, A.S.; Roy, A. Carbohydr. Res. 1981, 94, 73.
- Kanie, O.; Takeda, T.; Ogihara, Y. Carbohydr. Res. 1989, 190, 53.
- Kováč, P.; Palovčík, R. Chem. Zvesti 1978, 32, 501.
- CCDC 246572 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (Fax: +44-1223-336033; e-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
- Cremer, D.; Pople, J. A. J. Am. Chem. Soc. 1975, 97, 1354.
- Bernstein, J.; Davis, R. E.; Shimoni, L.; Chang, N.-L. Angew. Chem., Int. Ed. Engl. 1995, 34, 1555.
- Motherwell, W. D. S.; Shields, G. P.; Allen, F. H. Acta Crystallogr., Sect. B 1999, 55, 1044.
- Siemens AXS. In SMART & SAINT; Madison, WI, USA, 1995.
- Sheldrick, G. M. Program SADABS; University of Göttingen: Germany, 2001. [Google Scholar]
- Blessing, R. H. Acta Crystallogr., Sect. A. 1995, 51, 33.
- Bruker AXS Inc. SHELXTL Version 6.10; Madison, WI, USA, 2001. [Google Scholar]
- Brandenburg, K. DIAMOND: Visual Crystal Structure Information System, Version 2.1e; Crystal Impact GbR: Bonn, Germany, 2001. [Google Scholar]
- Klemer, A. Chem. Ber. 1959, 92, 218.
- Sample Availability: Samples of methyl 4-O-methyl-α-D-glucopyranuronate may be obtained from the authors.
© 2005 by MDPI (http://www.mdpi.org) Reproduction is permitted for noncommercial purposes.
Share and Cite
Hirsch, J.; Langer, V.; Koóš, M. Synthesis and Molecular Structure of Methyl 4-O-methyl-α-D-glucopyranuronate. Molecules 2005, 10, 251-258. https://doi.org/10.3390/10010251
Hirsch J, Langer V, Koóš M. Synthesis and Molecular Structure of Methyl 4-O-methyl-α-D-glucopyranuronate. Molecules. 2005; 10(1):251-258. https://doi.org/10.3390/10010251
Chicago/Turabian StyleHirsch, Ján, Vratislav Langer, and Miroslav Koóš. 2005. "Synthesis and Molecular Structure of Methyl 4-O-methyl-α-D-glucopyranuronate" Molecules 10, no. 1: 251-258. https://doi.org/10.3390/10010251
APA StyleHirsch, J., Langer, V., & Koóš, M. (2005). Synthesis and Molecular Structure of Methyl 4-O-methyl-α-D-glucopyranuronate. Molecules, 10(1), 251-258. https://doi.org/10.3390/10010251