
Employing Hybrid Lennard-Jones and Axilrod-Teller Potentials to Parametrize Force Fields for the Simulation of Materials’ Properties


Abstract
:1. Introduction
2. Materials and Methods
2.1. First Principles Calculations
2.2. Parametrization Using Molecular Dynamics
2.3. Parameters Set Validation through Molecular Dynamics Properties Determination
3. Results
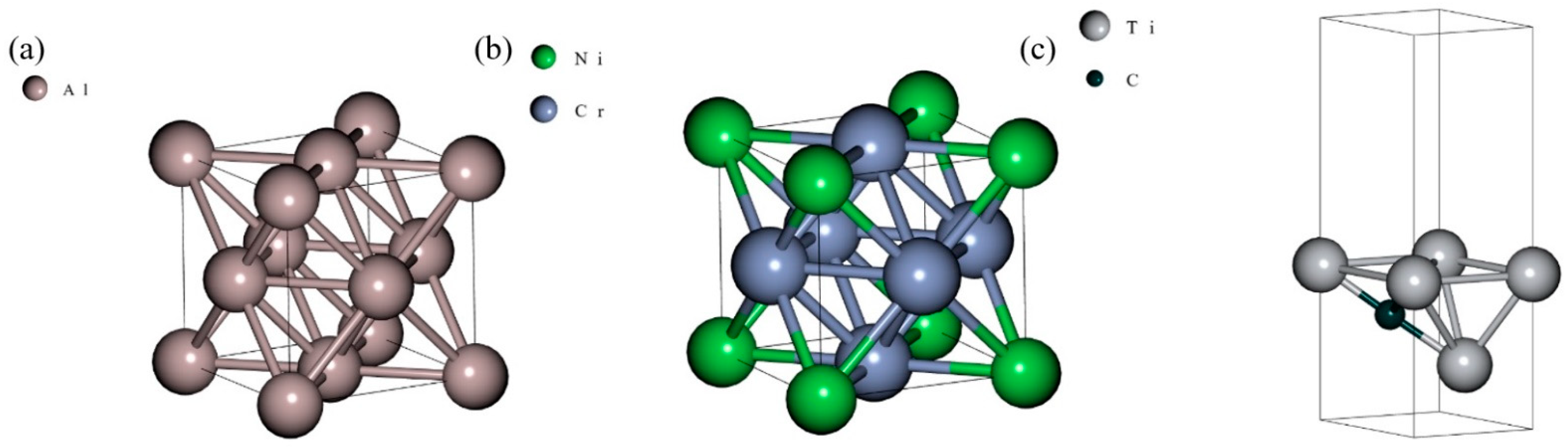
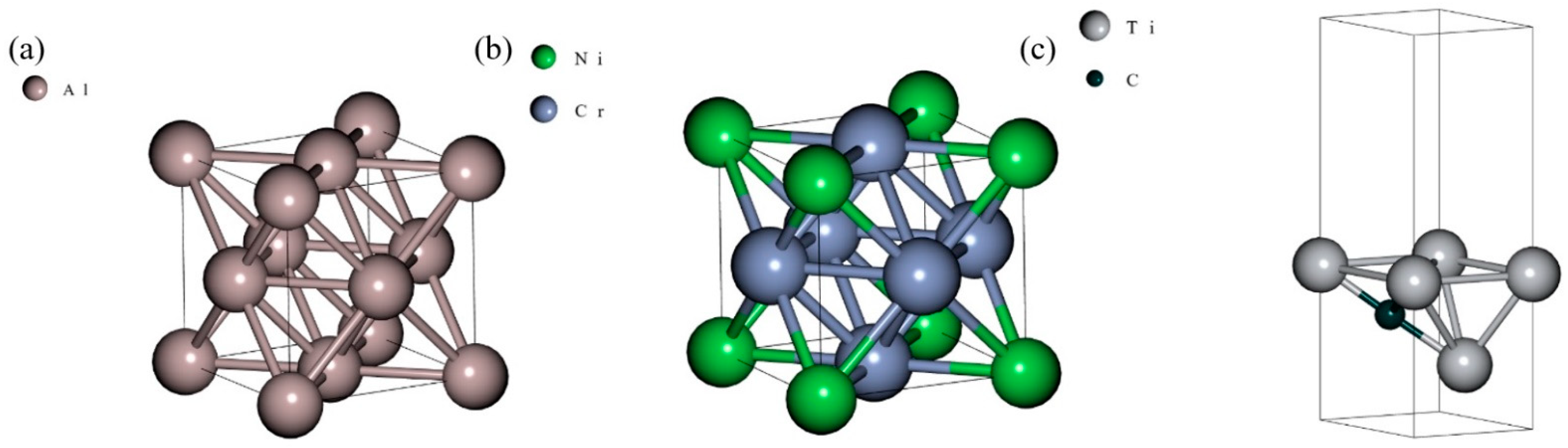
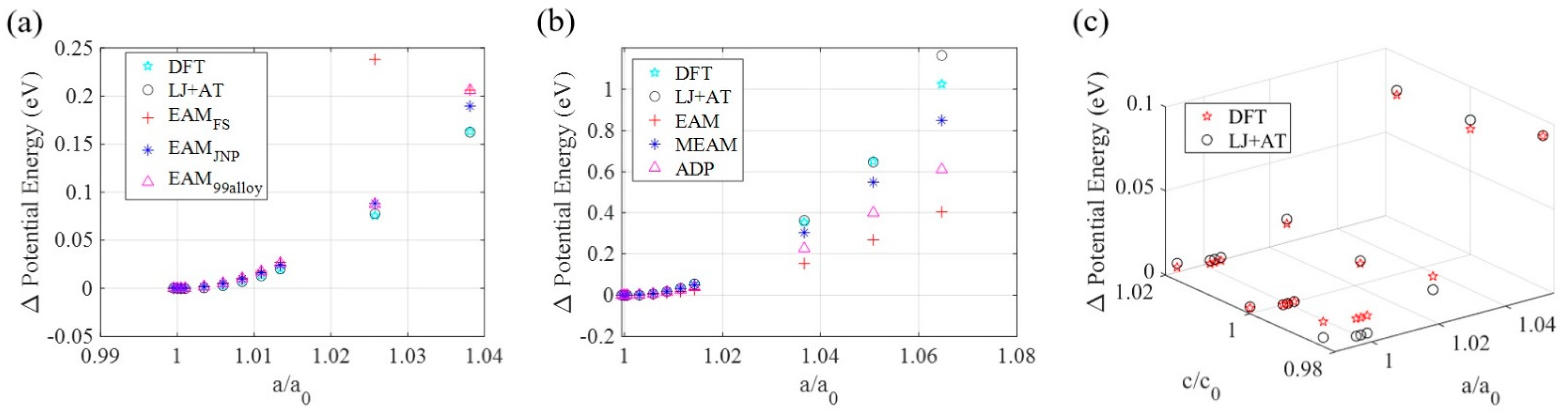
3.1. Ground State Determination
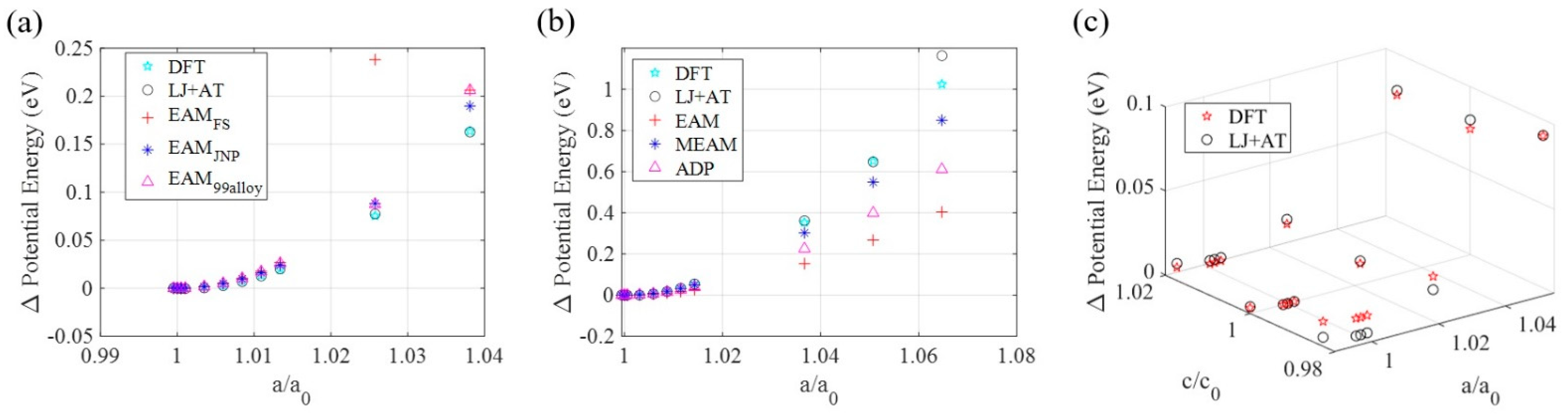
3.2. LJ + AT Parameters Determination
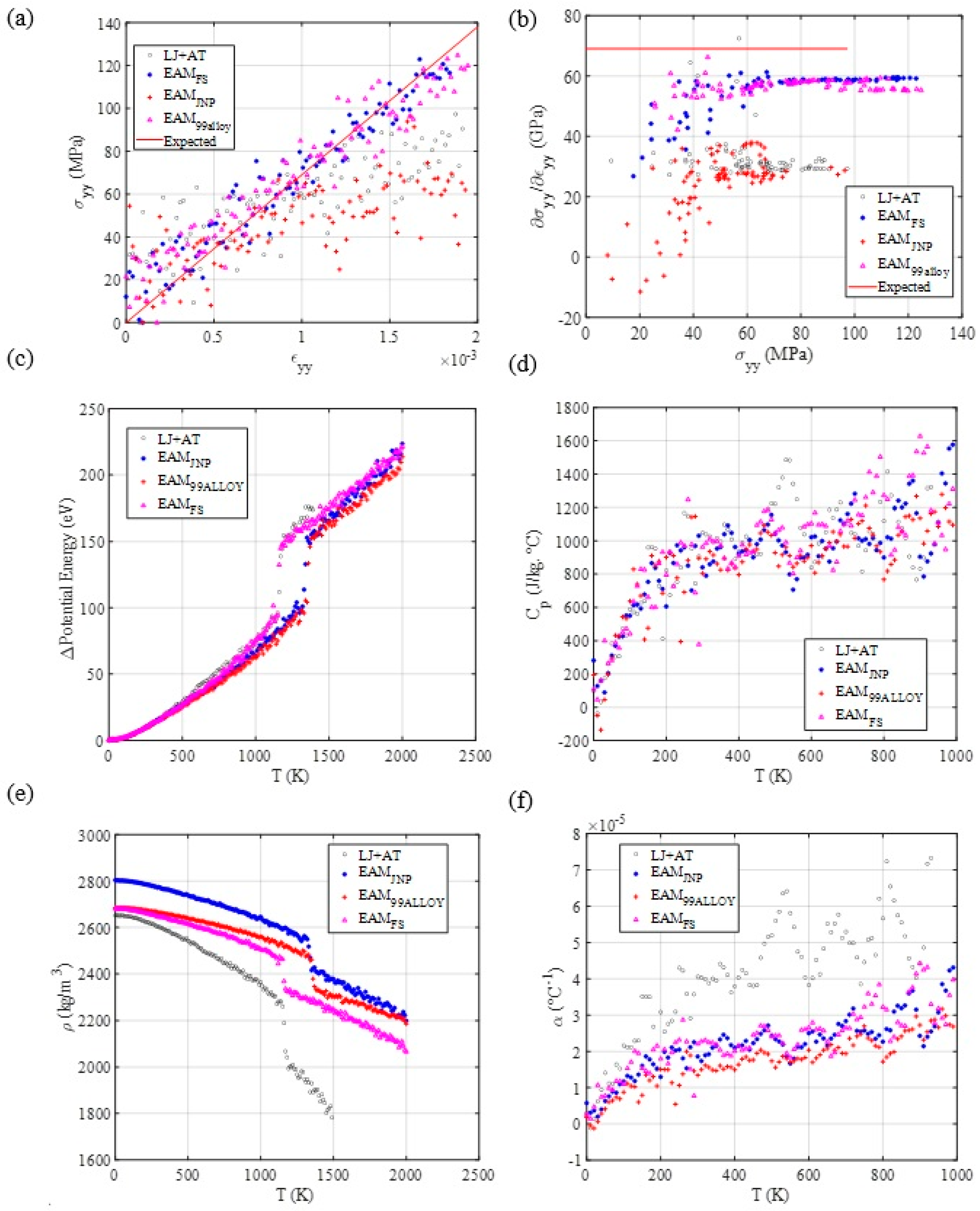
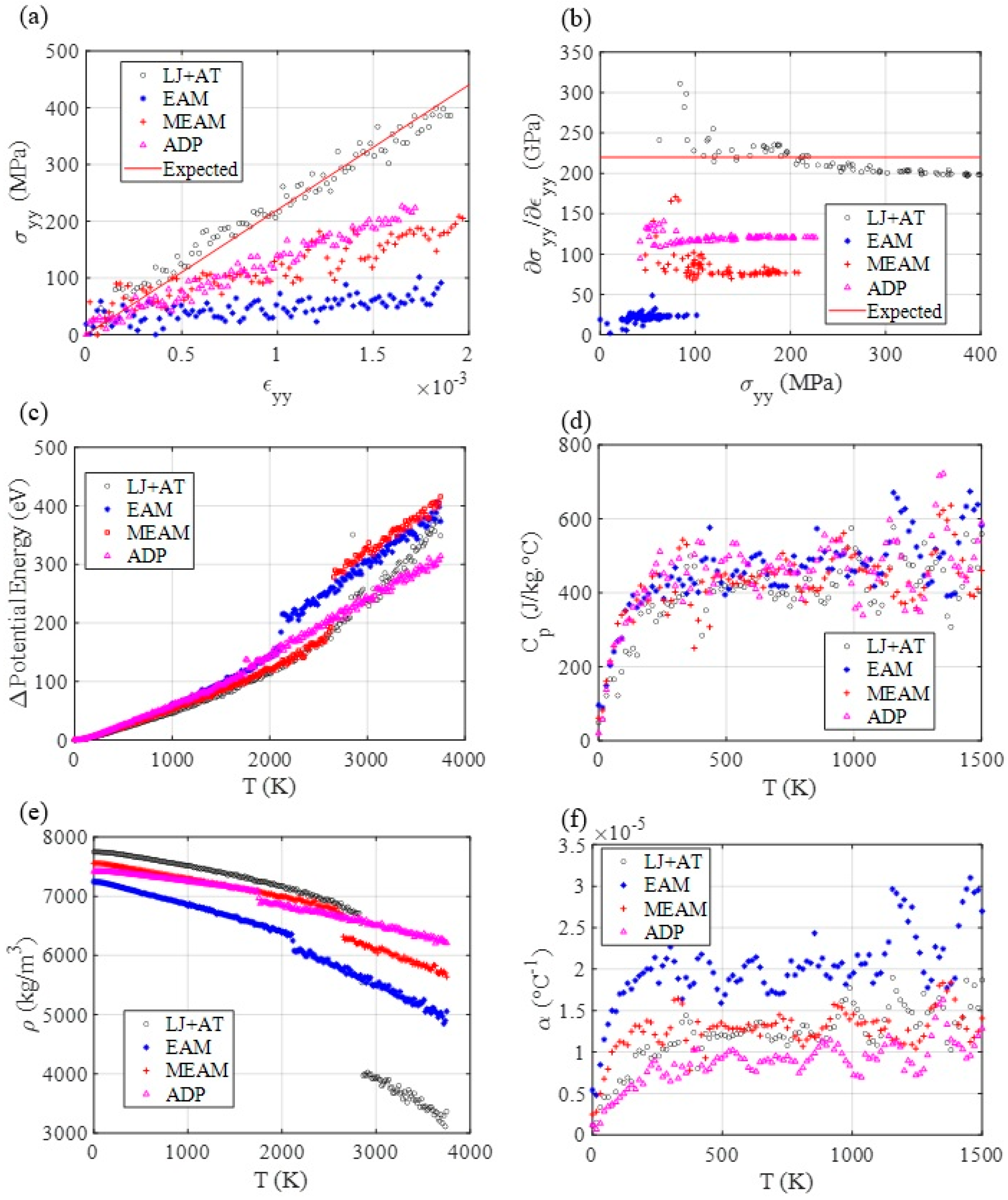
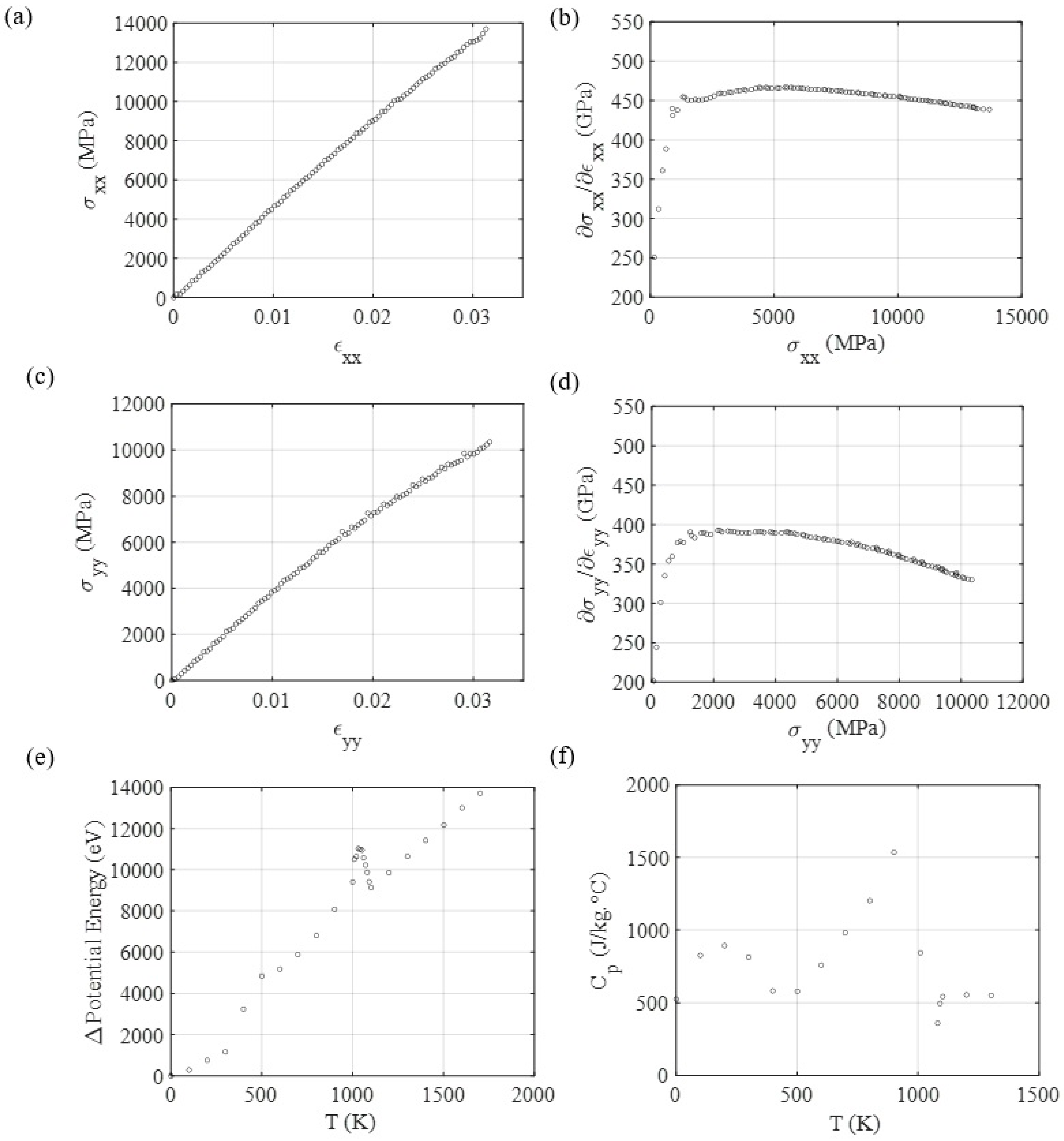
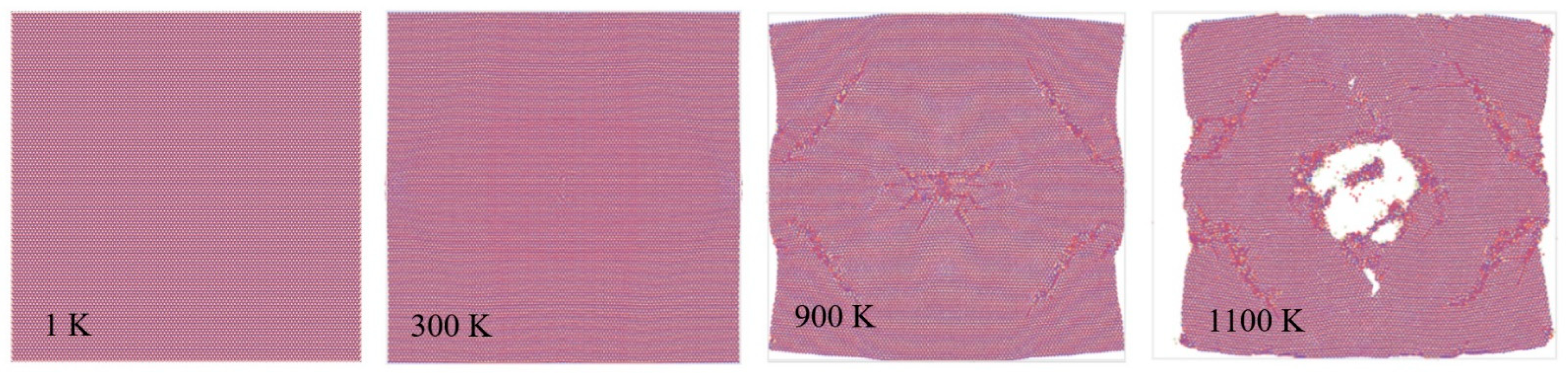
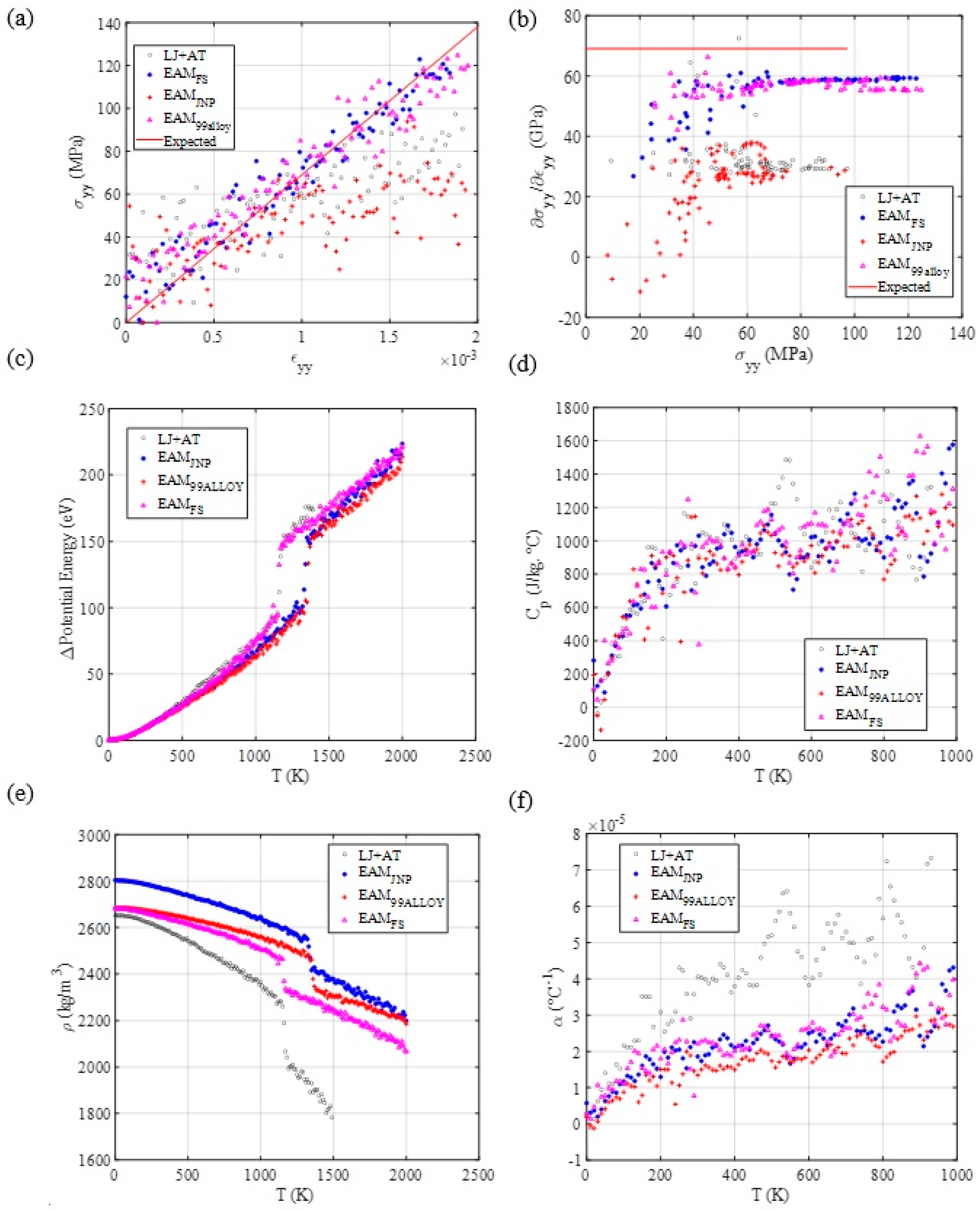
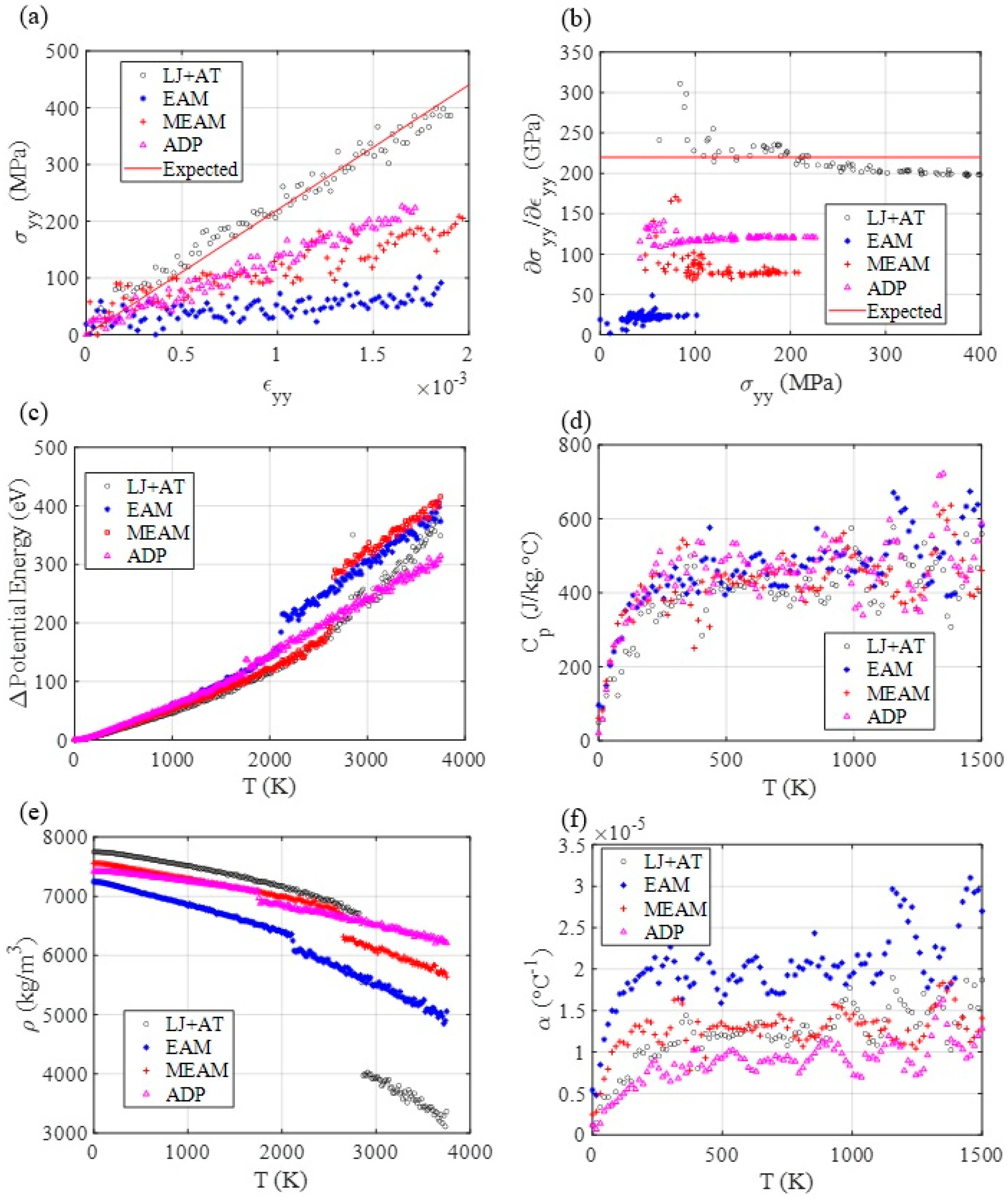
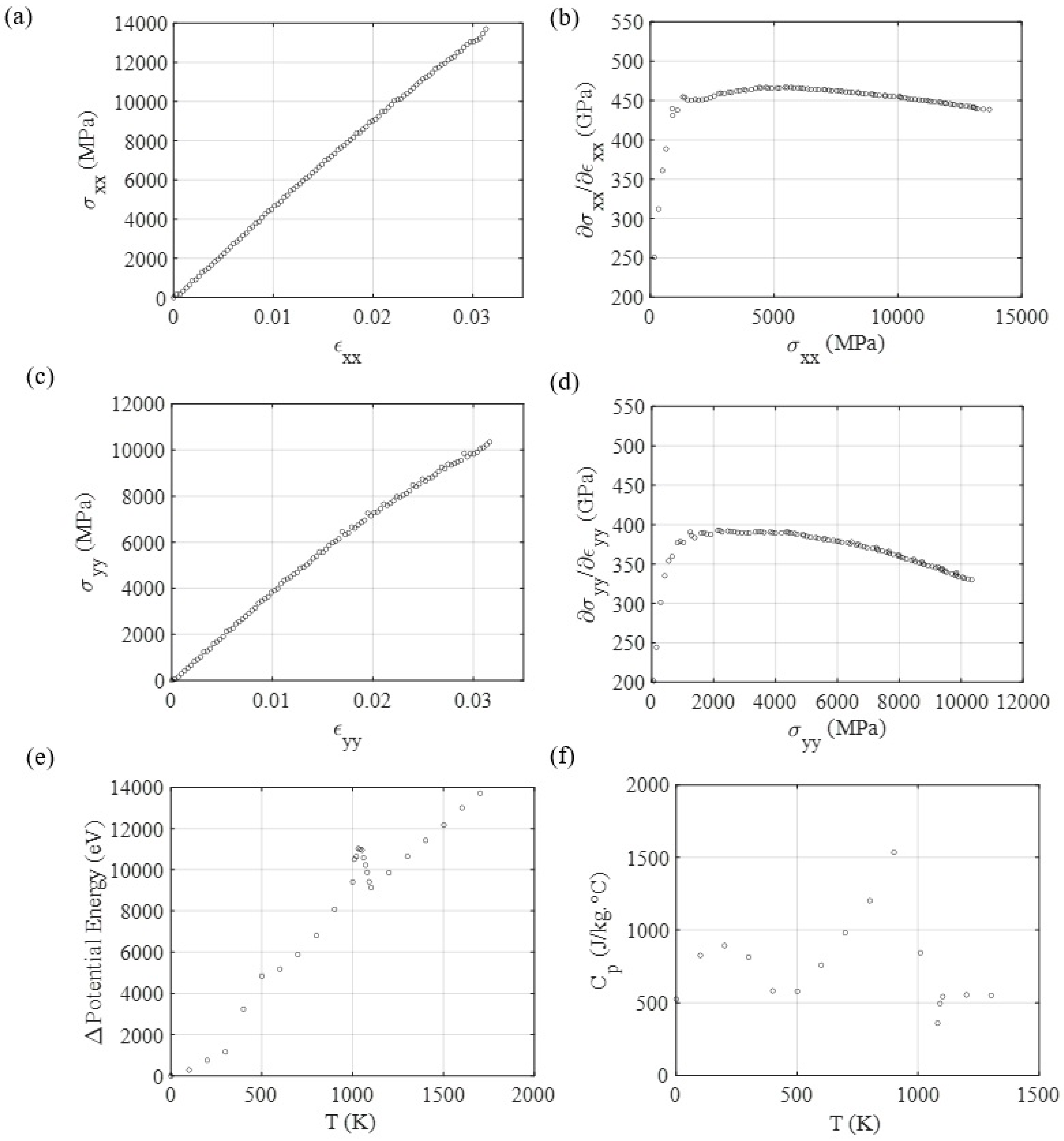
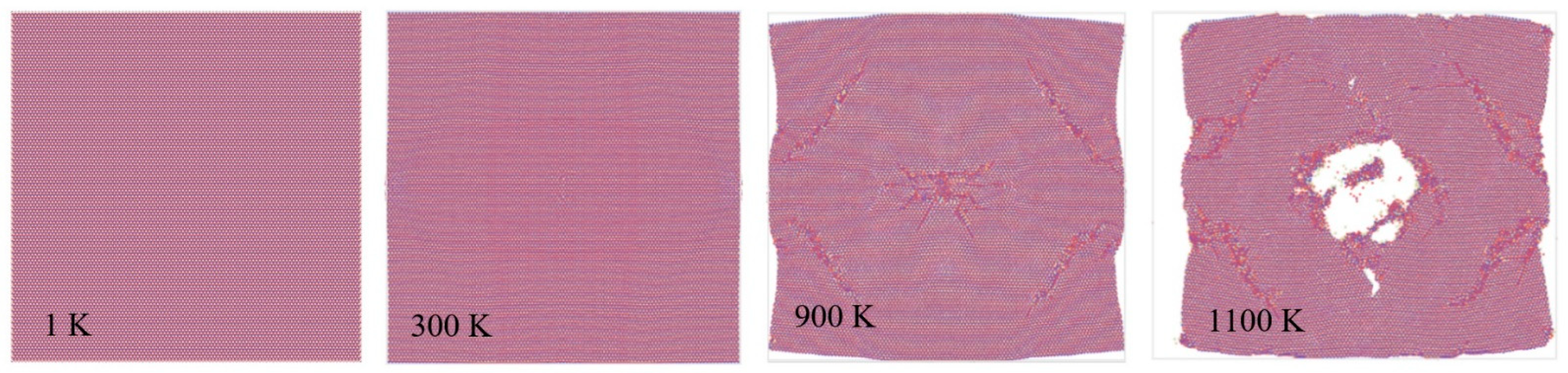
3.3. Mechanical and Thermal Properties Determination
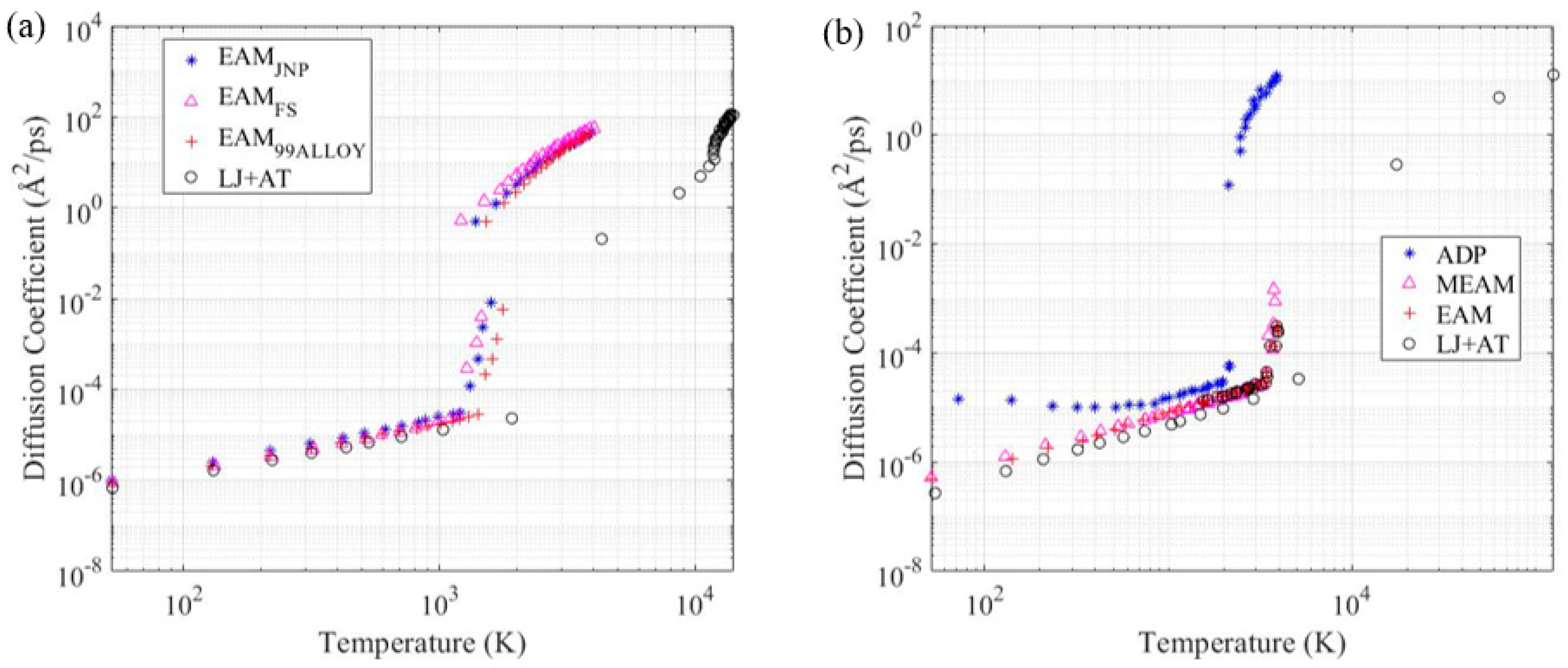
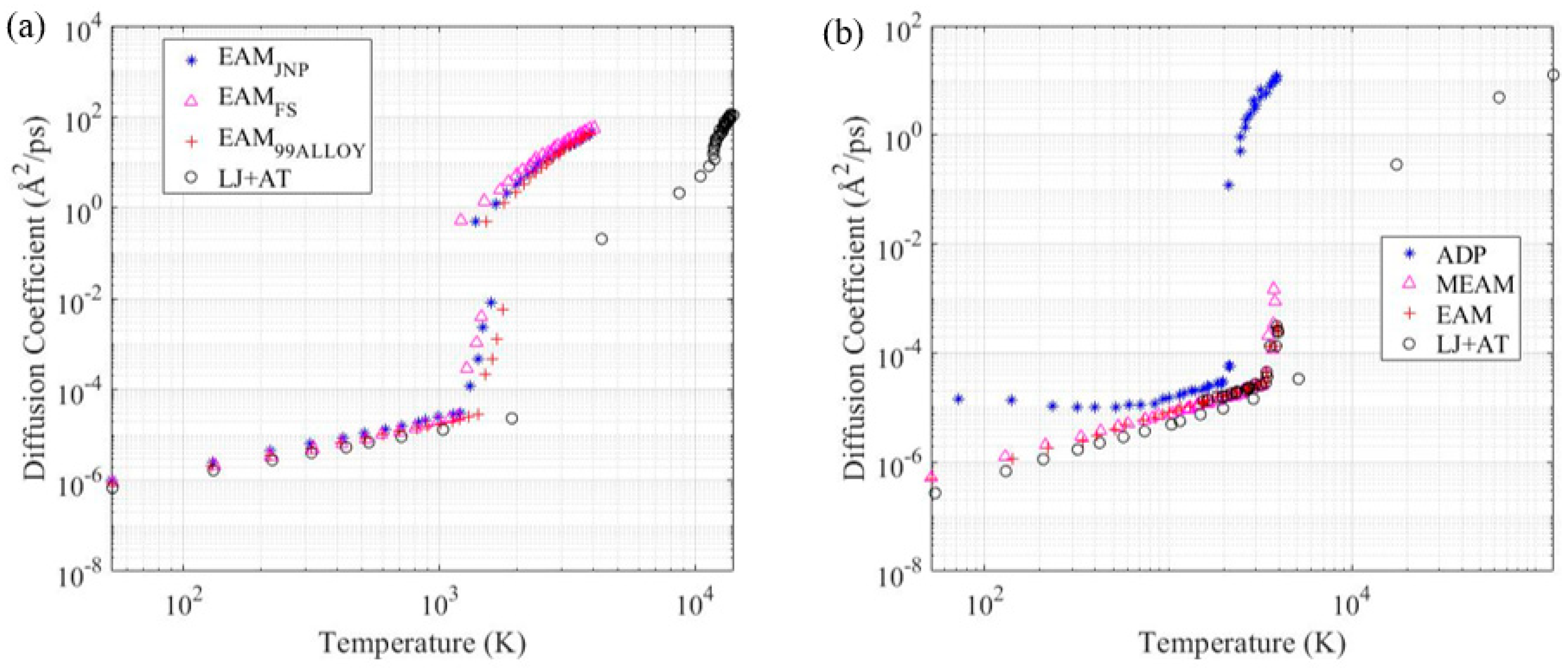
3.4. Diffusion Coefficient
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rutkai, G.; Thol, M.; Span, R.; Vrabec, J. How well does the Lennard-Jones potential represent the thermodynamic properties of noble gases? Mol. Phys. 2017, 115, 1104–1121. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Semiempirical quantum mechanical calculation of hydrogen embrittlement in metals. Phys. Rev. Lett. 1983, 50, 1285–1288. [Google Scholar] [CrossRef]
- Pair_Style Eam Command—LAMMPS Documentation (Sandia.Gov). Available online: https://lammps.sandia.gov/doc/pair_eam.html (accessed on 27 May 2021).
- Gullett, M.P.; Wagner, G.; Slepoy, A. Numerical Tools for Atomistic Simulations; Sandia National Laboratories: Albuquerque, NM, USA, 2004; SAND2003-8782.
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef] [Green Version]
- Chenoweth, K.; Van Duin, A.C.T.; Goddard, W.A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, D.R.; Morrissey, L.S.; Nakhla, S. Molecular dynamics simulations of nanoindentation–the importance of force field choice on the predicted elastic modulus of FCC aluminum. Mol. Simul. 2020, 46, 923–931. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Asahi, R.; Freeman, C.M.; Saxe, P.; Wimmer, E. Thermal expansion, diffusion and melting of Li2O using a compact forcefield derived from ab initio molecular dynamics. Model. Simul. Mater. Sci. Eng. 2014, 22, 075009. [Google Scholar] [CrossRef]
- Pedone, A.; Malavasi, G.; Menziani, M.C.; Cormack, A.N.; Segre, U. A new self-consistent empirical interatomic potential model for oxides, silicates, and silicas-based glasses. J. Phys. Chem. B 2006, 110, 11780–11795. [Google Scholar] [CrossRef]
- Daksha, C.M.; Yeon, J.; Chowdhury, S.C.; Gillespie, J.W. Automated ReaxFF parametrization using machine learning. Comput. Mater. Sci. 2021, 187, 110107. [Google Scholar] [CrossRef]
- Wang, L.P.; Martinez, T.J.; Pande, V.S. Building force fields: An automatic, systematic, and reproducible approach. J. Phys. Chem. Lett. 2014, 5, 1885–1891. [Google Scholar] [CrossRef]
- van der Spoel, D. Systematic design of biomolecular force fields. Curr. Opin. Struct. Biol. 2021, 67, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Anagnostopoulos, A.; Alexiadis, A.; Ding, Y. Simplified force field for molecular dynamics simulations of amorphous SiO2 for solar applications. Int. J. Therm. Sci. 2021, 160, 106647. [Google Scholar] [CrossRef]
- Filippova, V.P.; Blinova, E.N.; Shurygina, N.A. Constructing the pair interaction potentials of iron atoms with other metals. Inorg. Mater. Appl. Res. 2015, 6, 402–406. [Google Scholar] [CrossRef]
- Chen, K. Bonding Characteristics of TiC and TiN. Model. Numer. Simul. Mater. Sci. 2013, 3, 7–11. [Google Scholar] [CrossRef]
- Osti, N.C.; Naguib, M.; Ostadhossein, A.; Xie, Y.; Kent, P.R.C.; Dyatkin, B.; Rother, G.; Heller, W.T.; Van Duin, A.C.T.; Gogotsi, Y.; et al. Effect of Metal Ion Intercalation on the Structure of MXene and Water Dynamics on its Internal Surfaces. ACS Appl. Mater. Interfaces 2016, 8, 8859–8863. [Google Scholar] [CrossRef] [PubMed]
- Plummer, G.; Anasori, B.; Gogotsi, Y.; Tucker, G.J. Nanoindentation of monolayer Tin+1CnTx MXenes via atomistic simulations: The role of composition and defects on strength. Comput. Mater. Sci. 2019, 157, 168–174. [Google Scholar] [CrossRef]
- Borysiuk, V.N.; Mochalin, V.N.; Gogotsi, Y. Molecular dynamic study of the mechanical properties of two-dimensional titanium carbides Tin+1Cn (MXenes). Nanotechnology 2015, 26, 265705. [Google Scholar] [CrossRef] [Green Version]
- Borysiuk, V.N.; Mochalin, V.N.; Gogotsi, Y. Bending rigidity of two-dimensional titanium carbide (MXene) nanoribbons: A molecular dynamics study. Comput. Mater. Sci. 2018, 143, 418–424. [Google Scholar] [CrossRef]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum Expresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009, 21, 395502. [Google Scholar] [CrossRef]
- Gaillac, R.; Pullumbi, P.; Coudert, F.X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, P.; Wang, M.; Liu, J. Molecular Dynamics Simulations of Melting Iron Nanoparticles with/without Defects Using a Reaxff Reactive Force Field. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Jacobsen, K.W.; Norskov, J.K.; Puska, M.J. Interatomic interactions in the effective-medium theory. Phys. Rev. B 1987, 35, 7423–7442. [Google Scholar] [CrossRef] [Green Version]
- Mishin, Y.; Farkas, D.; Mehl, M.J.; Papaconstantopoulos, D.A. Interatomic potentials for monoatomic metals from experimental data and ab initio calculations. Phys. Rev. B-Condens. Matter Mater. Phys. 1999, 59, 3393–3407. [Google Scholar] [CrossRef] [Green Version]
- Mendelev, M.I.; Rodin, A.O.; Bokstein, B.S. Computer simulation of diffusion in dilute Al-Fe alloys. Defect Diffus. Forum 2009, 289, 733–740. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Srolovitz, D.J.; Ackland, G.J.; Han, S. Effect of Fe segregation on the migration of a non-symmetric ∑5 tilt grain boundary in Al. J. Mater. Res. 2005, 20, 208–218. [Google Scholar] [CrossRef]
- Bonny, G.; Castin, N.; Terentyev, D. Interatomic potential for studying ageing under irradiation in stainless steels: The FeNiCr model alloy. Model. Simul. Mater. Sci. Eng. 2013, 21, 085004. [Google Scholar] [CrossRef]
- Howells, C.A.; Mishin, Y. Angular-dependent interatomic potential for the binary Ni-Cr system. Model. Simul. Mater. Sci. Eng. 2018, 26, 085008. [Google Scholar] [CrossRef] [Green Version]
- Reade Advanced Materials-Nickel-Chromium Alloys (NiCr). Available online: https://www.reade.com/products/nickel-chromium-alloys-nicr (accessed on 27 May 2021).
- Kurtoglu, M.; Naguib, M.; Gogotsi, Y.; Barsoum, M.W. First principles study of two-dimensional early transition metal carbides. MRS Commun. 2012, 2, 133–137. [Google Scholar] [CrossRef]
- Lipatov, A.; Lu, H.; Alhabeb, M.; Anasori, B.; Gruverman, A.; Gogotsi, Y.; Sinitskii, A. Elastic properties of 2D Ti3C2Tx MXene monolayers and bilayers. Sci. Adv. 2018, 4, eaat0491. [Google Scholar] [CrossRef] [Green Version]
- Borysiuk, V.; Mochalin, V.N. Thermal stability of two-dimensional titanium carbides Ti n+1 C n (MXenes) from classical molecular dynamics simulations. MRS Commun. 2019, 9, 203–208. [Google Scholar] [CrossRef]
- Wyatt, B.C.; Nemani, S.K.; Desai, K.; Kaur, H.; Nanosystems, I. High-Temperature Stability and Phase Transformations of Titanium Carbide (Ti 3 C 2 T x) MXene. J. Phys. Condens. Matter. 2021, 33, 224002. [Google Scholar] [CrossRef] [PubMed]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Ma, X.Y.; Lyu, H.Y.; Hao, K.R.; Zhao, Y.M.; Qian, X.; Yan, Q.B.; Su, G. Large family of two-dimensional ferroelectric metals discovered via machine learning. Sci. Bull. 2020, 66, 233–242. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Parameter | Value | Unit |
|---|---|---|---|
| Aluminum | 0.168 | eV | |
| 2.5487 | Å | ||
| 236 | eV | ||
| Nichrome | 0.36 | eV | |
| 3.1796 | Å | ||
| 0.12 | eV | ||
| 2.2483 | Å | ||
| 0.58 | eV | ||
| 2.2483 | Å | ||
| 540 | eV | ||
| 80 | eV | ||
| 240 | eV | ||
| 0.18 | eV | ||
| 2.7031 | Å | ||
| 0.6 | eV | ||
| 1.8693 | Å | ||
| 0.01 | eV | ||
| 2.5827 | Å | ||
| 0.5 | eV | ||
| 2.7031 | Å | ||
| 0.6 | eV | ||
| 1.8693 | Å | ||
| 0.18 | eV | ||
| 2.7031 | Å | ||
| 10 | eV | ||
| 10 | eV | ||
| 10 | eV | ||
| 10 | eV | ||
| 10 | eV | ||
| 10 | eV |
| Property | LJ + AT | EAM 99 | EAM FS | EAM JNP | ELATE | Experimental [26] |
|---|---|---|---|---|---|---|
| C11 (GPa) | 107.2695 | 113.7967 | 105.0917 | 111.3806 | 104 | 114 |
| C12 (GPa) | 70.8877 | 61.5546 | 59.4629 | 85.1381 | 73 | 61.9 |
| C44 (GPa) | 56.3227 | 31.5946 | 30.6588 | 45.9262 | 32 | 31.6 |
| Bulk Modulus (GPa) | 83.0150 | 78.9683 | 74.6725 | 93.8857 | 83 | 79 |
| Young (GPa) | 72.4258 | 66.1612 | 61.2460 | 38.0122 | 65.4847 | 70 |
| Poisson Ratio | 0.3979 | 0.3510 | 0.3614 | 0.4332 | 0.37 | 0.35 |
| Shear Modulus (GPa) | 37.2568 | 28.8576 | 26.7366 | 29.5237 | 23.6667 | 26 |
| Latent Heat of Fusion (kJ/kg) | 405.86 | 345.43 | 360.81 | 371.18 | --- | 396 |
| Melting point (K) | 934.16 | 1096.6 | 934.16 | 1072.3 | --- | 933 |
| ) | 2601 | 2663 | 2645 | 2774 | --- | 2700 |
| ) | 1049 | 804.3 | 962.6 | 841.3 | --- | 921 |
| ) | 40.6 | 13.4 | 22.1 | 18.6 | --- | 23 |
| Property | LJ + AT | EAM | MEAM | ADP | Experimental [31] |
|---|---|---|---|---|---|
| C11 (GPa) | 437.3272 | 111.15091 | 315.5455 | 143.4087 | --- |
| C12 (GPa) | 216.3067 | 98.4087 | 129.8099 | 144.2840 | --- |
| C44 (GPa) | 161.6199 | 83.1493 | 99.1138 | 91.4083 | --- |
| Bulk Modulus (GPa) | 289.9802 | 102.6561 | 191.7217 | 143.9923 | 110–205 |
| Young (GPa) | 311.0619 | 48.7332 | 170.8231 | 140.5154 | 150–245 |
| Poisson Ratio | 0.3309 | 0.4696 | 0.2915 | 0.4990 | 0.26–0.325 |
| Shear Modulus (GPa) | 136.0651 | 44.7602 | 95.9908 | 45.4853 | 55–100 |
| Latent Heat of Fusion (kJ/kg) | 328.62 | 225.79 | 303.46 | 90.96 | 275–320 |
| Melting point (K) | 2290.7 | 1705.9 | 2144.5 | 1413.4 | 1475–1710 |
| ) | 7704 | 7147 | 7493 | 7385 | 7750–8650 |
| ) | 376.9 | 467.7 | 401.9 | 419.2 | 380–500 |
| ) | 10.94 | 22.69 | 12.91 | 6.47 | 9–16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco, D.d.C.; Cheng, G.J. Employing Hybrid Lennard-Jones and Axilrod-Teller Potentials to Parametrize Force Fields for the Simulation of Materials’ Properties. Materials 2021, 14, 6352. https://doi.org/10.3390/ma14216352
Branco DdC, Cheng GJ. Employing Hybrid Lennard-Jones and Axilrod-Teller Potentials to Parametrize Force Fields for the Simulation of Materials’ Properties. Materials. 2021; 14(21):6352. https://doi.org/10.3390/ma14216352
Chicago/Turabian StyleBranco, Danilo de Camargo, and Gary J. Cheng. 2021. "Employing Hybrid Lennard-Jones and Axilrod-Teller Potentials to Parametrize Force Fields for the Simulation of Materials’ Properties" Materials 14, no. 21: 6352. https://doi.org/10.3390/ma14216352
APA StyleBranco, D. d. C., & Cheng, G. J. (2021). Employing Hybrid Lennard-Jones and Axilrod-Teller Potentials to Parametrize Force Fields for the Simulation of Materials’ Properties. Materials, 14(21), 6352. https://doi.org/10.3390/ma14216352