
Therapeutic Targeting of Cancer Stem Cells in Lung, Head and Neck, and Bladder Cancers

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. CSC Signaling Pathways Implicated in Therapeutic Resistance
3. Unique Properties of CSCs by Cancer Type
3.1. Features of CSCs in Lung Cancer
3.1.1. Therapeutic Targeting of CSCs and CSC-Regulating Pathways in NSCLC
EGFR TKIs
Other Small Molecule Inhibitors
Other Agents with Unknown CSC Activity
3.2. Features of CSCs in Head and Neck Cancers
3.2.1. Therapy Targeting CSCs in Head and Neck Cancers
EGFR TKIs
Other Agents with Unknown CSC Activity
3.3. Features of CSCs in Bladder Cancer
Evaluation of Targeted Therapy Activity against CSCs in Bladder Cancer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890, discussion 1895–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin Jinesh, G.; Willis, D.L.; Kamat, A.M. Bladder cancer stem cells: Biological and therapeutic perspectives. Curr. Stem Cell Res. Ther. 2014, 9, 89–101. [Google Scholar] [PubMed]
- Li, Y.; Lin, K.; Yang, Z.; Han, N.; Quan, X.; Guo, X.; Li, C. Bladder cancer stem cells: Clonal origin and therapeutic perspectives. Oncotarget 2017, 8, 66668–66679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooki, A.; VandenBussche, C.J.; Kates, M.; Hahn, N.M.; Matoso, A.; McConkey, D.J.; Bivalacqua, T.J.; Hoque, M.O. CD24 regulates cancer stem cell (CSC)-like traits and a panel of CSC-related molecules serves as a non-invasive urinary biomarker for the detection of bladder cancer. Br. J. Cancer 2018, 119, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Mondala, P.K.; Vora, A.A.; Zhou, T.; Lazzari, E.; Ladel, L.; Luo, X.; Kim, Y.; Costello, C.; MacLeod, A.R.; Jamieson, C.H.M.; et al. Selective antisense oligonucleotide inhibition of human IRF4 prevents malignant myeloma regeneration via cell cycle disruption. Cell Stem Cell 2021, 28, 623–636.e9. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Torres-Ruiz, R.; Puig-Serra, P.; Moreno-Gaona, P.; Martin, M.C.; Moya, F.J.; Quintana-Bustamante, O.; Garcia-Silva, S.; Carcaboso, A.M.; Petazzi, P.; et al. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat. Commun. 2020, 11, 5060. [Google Scholar] [CrossRef]
- Kijima, H.; Scanlon, K.J. Ribozyme as an approach for growth suppression of human pancreatic cancer. Mol. Biotechnol. 2000, 14, 59–72. [Google Scholar] [CrossRef]
- Sharma, A.; Kansara, S.; Mahajan, M.; Yadav, B.; Garg, M.; Pandey, A.K. Long non-coding RNAs orchestrate various molecular and cellular processes by modulating epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166240. [Google Scholar] [CrossRef]
- Sourisseau, T.; Hassan, K.A.; Wistuba, I.; Penault-Llorca, F.; Adam, J.; Deutsch, E.; Soria, J.C. Lung cancer stem cell: Fancy conceptual model of tumor biology or cornerstone of a forthcoming therapeutic breakthrough? J. Thorac. Oncol. 2014, 9, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Brooks, M.; Wicha, M.S. Epithelial-mesenchymal plasticity of breast cancer stem cells: Implications for metastasis and therapeutic resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Shim, J.S. Existing drugs and their application in drug discovery targeting cancer stem cells. Arch. Pharm. Res. 2015, 38, 1617–1626. [Google Scholar] [CrossRef]
- Leon, G.; MacDonagh, L.; Finn, S.P.; Cuffe, S.; Barr, M.P. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signaling pathways. Pharmacol. Ther. 2016, 158, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradov, S.; Wei, X. Cancer stem cells and drug resistance: The potential of nanomedicine. Nanomedicine (Lond.) 2012, 7, 597–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F. CD10+ GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef] [PubMed]
- Sadhukhan, P.; Saha, S.; Dutta, S.; Sil, P.C. Mangiferin Ameliorates Cisplatin Induced Acute Kidney Injury by Upregulating Nrf-2 via the Activation of PI3K and Exhibits Synergistic Anticancer Activity With Cisplatin. Front. Pharmacol. 2018, 9, 638. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Hoque, M.O. Targeting cancer stem cells: A strategy for effective eradication of cancer. Cancers 2019, 11, 732. [Google Scholar] [CrossRef] [Green Version]
- Sadhukhan, P.; Sil, P.C. The regulation of intracellular redox homeostasis in cancer progression and its therapy. In Pathology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 105–114. [Google Scholar]
- Ooki, A.; Pena, M.D.C.R.; Marchionni, L.; Dinalankara, W.; Begum, A.; Hahn, N.M.; VandenBussche, C.J.; Rasheed, Z.A.; Mao, S.; Netto, G.J. YAP1 and COX2 coordinately regulate urothelial cancer stem-like cells. Cancer Res. 2018, 78, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, M.; Ham, K.; Hoque, M.O. A time for YAP1: Tumorigenesis, immunosuppression and targeted therapy. Int. J. Cancer 2018, 143, 2133–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, M.; Ooki, A.; Inokawa, Y.; Sadhukhan, P.; Ugurlu, M.T.; Izumchenko, E.; Munari, E.; Bogina, G.; Rudin, C.M.; Gabrielson, E. Concurrent Targeting of Potential Cancer Stem Cells Regulating Pathways Sensitizes Lung Adenocarcinoma to Standard Chemotherapy. Mol. Cancer Ther. 2020, 2020. 9, 2175–2185. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Spinola, M.; Dodge, M.; Raso, M.G.; Behrens, C.; Gao, B.; Schuster, K.; Shao, C.; Larsen, J.E.; Sullivan, L.A.; et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010, 70, 9937–9948. [Google Scholar] [CrossRef] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Giangreco, A.; Lu, L.; Vickers, C.; Teixeira, V.H.; Groot, K.R.; Butler, C.R.; Ilieva, E.V.; George, P.J.; Nicholson, A.G.; Sage, E.K.; et al. beta-Catenin determines upper airway progenitor cell fate and preinvasive squamous lung cancer progression by modulating epithelial-mesenchymal transition. J. Pathol. 2012, 226, 575–587. [Google Scholar] [CrossRef]
- Iderzorig, T.; Kellen, J.; Osude, C.; Singh, S.; Woodman, J.A.; Garcia, C.; Puri, N. Comparison of EMT mediated tyrosine kinase inhibitor resistance in NSCLC. Biochem. Biophys. Res. Commun. 2018, 496, 770–777. [Google Scholar] [CrossRef]
- Levina, V.; Marrangoni, A.; Wang, T.; Parikh, S.; Su, Y.; Herberman, R.; Lokshin, A.; Gorelik, E. Elimination of human lung cancer stem cells through targeting of the stem cell factor-c-kit autocrine signaling loop. Cancer Res. 2010, 70, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Wang, X.; Wang, Y.; Ma, D. Wnt/β-catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem. Biophys. Res. Commun. 2010, 392, 373–379. [Google Scholar] [CrossRef]
- Ghosh, G.; Lian, X.; Kron, S.J.; Palecek, S.P. Properties of resistant cells generated from lung cancer cell lines treated with EGFR inhibitors. BMC Cancer 2012, 12, 95. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.J.; Rho, J.K.; Kim, Y.M.; Jung, J.E.; Jin, Y.B.; Ko, Y.G.; Lee, J.S.; Lee, S.J.; Lee, J.C.; Park, M.J. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene 2013, 32, 209–221. [Google Scholar] [CrossRef]
- Nian, W.Q.; Chen, F.L.; Ao, X.J.; Chen, Z.T. CXCR4 positive cells from Lewis lung carcinoma cell line have cancer metastatic stem cell characteristics. Mol. Cell. Biochem. 2011, 355, 241–248. [Google Scholar] [CrossRef]
- Leung, E.L.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.; Cheng, L.C.; Sihoe, A.D.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Luo, H.; Zhou, X.; Zhu, B.; Wang, Y.; Bian, X. Identification of CD90 as a marker for lung cancer stem cells in A549 and H446 cell lines. Oncol. Rep. 2013, 30, 2733–2740. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutova, M.; Najbauer, J.; Gevorgyan, A.; Metz, M.Z.; Weng, Y.; Shih, C.-C.; Aboody, K.S. Identification of uPAR-positive Chemoresistant Cells in Small Cell Lung Cancer. PLoS ONE 2007, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Haas, T.L.; De Maria, R. Lung cancer stem cells: Tools and targets to fight lung cancer. Oncogene 2010, 29, 4625–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Sugano, T.; Seike, M.; Noro, R.; Soeno, C.; Chiba, M.; Zou, F.; Nakamichi, S.; Nishijima, N.; Matsumoto, M.; Miyanaga, A.; et al. Inhibition of ABCB1 Overcomes Cancer Stem Cell-like Properties and Acquired Resistance to MET Inhibitors in Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2015, 14, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Liu, S.; Breiter, D.R.; Wang, F.; Tang, Y.; Sun, S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res. 2008, 68, 6533–6540. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Hsu, H.-S.; Chen, Y.-W.; Tsai, T.-H.; How, C.-K.; Wang, C.-Y.; Hung, S.-C.; Chang, Y.-L.; Tsai, M.-L.; Lee, Y.-Y.; et al. Oct-4 Expression Maintained Cancer Stem-Like Properties in Lung Cancer-Derived CD133-Positive Cells. PLoS ONE 2008, 3, e2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Epstein, D.; Haley, J.D. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin. Exp. Metastasis 2008, 25, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.F.; Chang, Y.W.; Kuo, K.T.; Shen, Y.S.; Liu, C.Y.; Yu, Y.H.; Cheng, C.C.; Lee, K.Y.; Chen, F.C.; Hsu, M.K.; et al. NF-kappaB-driven suppression of FOXO3a contributes to EGFR mutation-independent gefitinib resistance. Proc. Natl. Acad. Sci. USA 2016, 113, E2526–E2535. [Google Scholar] [CrossRef] [Green Version]
- Murakami, A.; Takahashi, F.; Nurwidya, F.; Kobayashi, I.; Minakata, K.; Hashimoto, M.; Nara, T.; Kato, M.; Tajima, K.; Shimada, N.; et al. Hypoxia Increases Gefitinib-Resistant Lung Cancer Stem Cells through the Activation of Insulin-Like Growth Factor 1 Receptor. PLoS ONE 2014, 9, e86459. [Google Scholar] [CrossRef]
- Arasada, R.R.; Amann, J.M.; Rahman, M.A.; Huppert, S.S.; Carbone, D.P. EGFR blockade enriches for lung cancer stem-like cells through Notch3-dependent signaling. Cancer Res. 2014, 74, 5572–5584. [Google Scholar] [CrossRef] [Green Version]
- Codony-Servat, C.; Codony-Servat, J.; Karachaliou, N.; Molina, M.A.; Chaib, I.; Ramirez, J.L.; Gil, M.D.L.L.; Solca, F.; Bivona, T.G.; Rosell, R. Activation of signal transducer and activator of transcription 3 (STAT3) signaling in EGFR mutant non-small-cell lung cancer (NSCLC). Oncotarget 2017, 8, 47305. [Google Scholar] [CrossRef] [Green Version]
- Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyas, E.; Segura-Carretero, A.; Joven, J.; Martin-Castillo, B.; Barrajon-Catalan, E.; Micol, V.; Bosch-Barrera, J.; Menendez, J.A. Stem cell-like ALDH(bright) cellular states in EGFR-mutant non-small cell lung cancer: A novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibinin. Cell Cycle 2013, 12, 3390–3404. [Google Scholar] [CrossRef] [Green Version]
- Codony-Servat, J.; Codony-Servat, C.; Cardona, A.F.; Gimenez-Capitan, A.; Drozdowskyj, A.; Berenguer, O.; Bracht, M.; Ito, M.; Karachaliou, N.; Rosell, R. Cancer Stem Cell Biomarkers in EGFR-Mutation-Positive Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2019, 20, 167–177. [Google Scholar] [CrossRef]
- Du, W.; Ni, L.; Liu, B.; Wei, Y.; Lv, Y.; Qiang, S.; Dong, J.; Liu, X. Upregulation of SALL4 by EGFR activation regulates the stemness of CD44-positive lung cancer. Oncogenesis 2018, 7, 36. [Google Scholar] [CrossRef]
- Yu, C.C.; Hu, F.W.; Yu, C.H.; Chou, M.Y. Targeting CD133 in the enhancement of chemosensitivity in oral squamous cell carcinoma-derived side population cancer stem cells. Head Neck 2016, 38, E231–E238. [Google Scholar] [CrossRef]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Peitzsch, C.; Nathansen, J.; Schniewind, S.I.; Schwarz, F.; Dubrovska, A. Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma: Identification, Characterization and Clinical Implications. Cancers (Basel) 2019, 11, 616. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.W.; Wu, J.H.; Jiang, C.P. ABCG2: A potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci. 2010, 86, 631–637. [Google Scholar] [CrossRef]
- Galbiatti-Dias, A.L.S.; Fernandes, G.M.M.; Castanhole-Nunes, M.M.U.; Hidalgo, L.F.; Nascimento, C.H.; Kawasaki-Oyama, R.S.; Ferreira, L.A.M.; Biselli-Chicote, P.M.; Pavarino, E.C.; Goloni-Bertollo, E.M. Relationship between CD44(high)/CD133(high)/CD117(high) cancer stem cells phenotype and Cetuximab and Paclitaxel treatment response in head and neck cancer cell lines. Am. J. Cancer Res. 2018, 8, 1633–1641. [Google Scholar]
- Yao, W.; Wang, L.; Huang, H.; Li, X.; Wang, P.; Mi, K.; Cheng, J.; Liu, H.; Gu, C.; Huang, L.; et al. All-trans retinoic acid reduces cancer stem cell-like cell-mediated resistance to gefitinib in NSCLC adenocarcinoma cells. BMC Cancer 2020, 20, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, T.; Ozasa, H.; Aoki, W.; Aburaya, S.; Yamamoto Funazo, T.; Furugaki, K.; Yoshimura, Y.; Yamazoe, M.; Ajimizu, H.; Yasuda, Y.; et al. YAP1 mediates survival of ALK-rearranged lung cancer cells treated with alectinib via pro-apoptotic protein regulation. Nat. Commun. 2020, 11, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, J.S.; Herrmann, A.C.; Bernatchez, C.; Haymaker, C.; Molldrem, J.J.; Hong, W.K.; Perez-Soler, R. Immune-Modulation by Epidermal Growth Factor Receptor Inhibitors: Implication on Anti-Tumor Immunity in Lung Cancer. PLoS ONE 2016, 11, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sette, G.; Salvati, V.; Mottolese, M.; Visca, P.; Gallo, E.; Fecchi, K.; Pilozzi, E.; Duranti, E.; Policicchio, E.; Tartaglia, M.; et al. Tyr1068-phosphorylated epidermal growth factor receptor (EGFR) predicts cancer stem cell targeting by erlotinib in preclinical models of wild-type EGFR lung cancer. Cell Death Dis. 2015, 6, e1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, A.C.; Sham, D.; Hristova, M.; Danyal, K.; Heppner, D.E.; Bauer, R.A.; Sipsey, L.M.; Habibovic, A.; van der Vliet, A. DUOX1 silencing in lung cancer promotes EMT, cancer stem cell characteristics and invasive properties. Oncogenesis 2016, 5, e261. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C.; Lin, H.C.; Tsai, K.J.; Chiang, Y.W.; Lim, K.H.; Chen, C.G.S.; Su, Y.W.; Peng, C.L.; Ho, A.S.; Huang, L.; et al. Epidermal growth factor induces STAT1 expression to exacerbate the IFNr-mediated PD-L1 axis in epidermal growth factor receptor-positive cancers. Mol. Carcinog. 2018, 57, 1588–1598. [Google Scholar] [CrossRef]
- Han, J.; Zhao, F.; Zhang, J.; Zhu, H.; Ma, H.; Li, X.; Peng, L.; Sun, J.; Chen, Z. miR-223 reverses the resistance of EGFR-TKIs through IGF1R/PI3K/Akt signaling pathway. Int. J. Oncol. 2016, 48, 1855–1867. [Google Scholar] [CrossRef] [Green Version]
- Hashida, S.; Yamamoto, H.; Shien, K.; Miyoshi, Y.; Ohtsuka, T.; Suzawa, K.; Watanabe, M.; Maki, Y.; Soh, J.; Asano, H.; et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015, 106, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zhou, X.; Li, S.; Qin, Y.; Chen, Y.; Liu, H. Inhibition of miR-23a increases the sensitivity of lung cancer stem cells to erlotinib through PTEN/PI3K/Akt pathway. Oncol. Rep. 2017, 38, 3064–3070. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.C.; Chang, J.; Huang, S.C.; Lin, H.C.; Ho, A.S.; Lim, K.H.; Chang, C.C.; Huang, L.; Chang, Y.C.; Chang, Y.F.; et al. YM155 as an inhibitor of cancer stemness simultaneously inhibits autophosphorylation of epidermal growth factor receptor and G9a-mediated stemness in lung cancer cells. PLoS ONE 2017, 12, e0182149. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Lv, H.; Zhong, D.S.; Zhou, Q.H. EGFR-TKI resistance and MAP17 are associated with cancer stem cell like properties. Oncol. Lett. 2018, 15, 6655–6665. [Google Scholar] [CrossRef] [PubMed]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non-Small Cell Lung Cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Huang, S.T.; Zhang, L.; Liu, Z.G.; Liang, R.X.; Jiang, S.W.; Jiang, Y.N.; Yu, X.J.; Jiang, Y.C.; Li, X.Z.; et al. Combined prognostic value of the cancer stem cell markers CD47 and CD133 in esophageal squamous cell carcinoma. Cancer Med. 2019, 8, 1315–1325. [Google Scholar] [CrossRef]
- Setúbal Destro Rodrigues, M.F.; Gammon, L.; Rahman, M.M.; Biddle, A.; Nunes, F.D.; Mackenzie, I.C. Effects of Cetuximab and Erlotinib on the behaviour of cancer stem cells in head and neck squamous cell carcinoma. Oncotarget 2018, 9, 13488–13500. [Google Scholar] [CrossRef] [Green Version]
- Nigro, A.; Ricciardi, L.; Salvato, I.; Sabbatino, F.; Vitale, M.; Crescenzi, M.A.; Montico, B.; Triggiani, M.; Pepe, S.; Stellato, C.; et al. Enhanced Expression of CD47 Is Associated With Off-Target Resistance to Tyrosine Kinase Inhibitor Gefitinib in NSCLC. Front. Immunol. 2019, 10, 3135. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.H.; Rho, Y.S.; Lee, S.H.; Koo, B.S.; Lee, H.J.; Do, S.I.; Cho, J.H.; Eun, Y.G.; Park, M.W.; Shin, H.A.; et al. Role of integrin β1 as a biomarker of stemness in head and neck squamous cell carcinoma. Oral Oncol. 2019, 96, 34–41. [Google Scholar] [CrossRef]
- Kobayashi, I.; Takahashi, F.; Nurwidya, F.; Nara, T.; Hashimoto, M.; Murakami, A.; Yagishita, S.; Tajima, K.; Hidayat, M.; Shimada, N.; et al. Oct4 plays a crucial role in the maintenance of gefitinib-resistant lung cancer stem cells. Biochem. Biophys. Res. Commun. 2016, 473, 125–132. [Google Scholar] [CrossRef]
- Shien, K.; Toyooka, S.; Yamamoto, H.; Soh, J.; Jida, M.; Thu, K.L.; Hashida, S.; Maki, Y.; Ichihara, E.; Asano, H.; et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013, 73, 3051–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurwidya, F.; Takahashi, F.; Winardi, W.; Tajima, K.; Mitsuishi, Y.; Murakami, A.; Kobayashi, I.; Nara, T.; Hashimoto, M.; Kato, M.; et al. Zinc-finger E-box-binding homeobox 1 (ZEB1) plays a crucial role in the maintenance of lung cancer stem cells resistant to gefitinib. Thorac. Cancer 2021, 12, 1536–1548. [Google Scholar] [CrossRef]
- Cheng, H.; Ge, X.; Zhuo, S.; Gao, Y.; Zhu, B.; Zhang, J.; Shang, W.; Xu, D.; Ge, W.; Shi, L. β-Elemene Synergizes With Gefitinib to Inhibit Stem-Like Phenotypes and Progression of Lung Cancer via Down-Regulating EZH2. Front. Pharmacol. 2018, 9, 1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.K.; To, K.K.W.; Huang, L.Y.; Xu, J.H.; Yang, K.; Wang, F.; Huang, Z.C.; Ye, S.; Fu, L.W. Afatinib circumvents multidrug resistance via dually inhibiting ATP binding cassette subfamily G member 2 in vitro and in vivo. Oncotarget 2014, 5, 11971–11985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, K.K.; Stewart, R.; Napier, D.; Claudio, P.P.; Craven, R.J. PGRMC1 Elevation in Multiple Cancers and Essential Role in Stem Cell Survival. Adv. Lung Cancer (Irvine) 2015, 4, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Song, H.Y.; Sun, B.B.; Liao, Y.L.; Xu, D.L.; Guo, W.Z.; Wang, T.; Jing, B.; Hu, M.; Li, K.M.; Yao, F.; et al. GPRC5A deficiency leads to dysregulated MDM2 via activated EGFR signaling for lung tumor development. Int. J. Cancer 2019, 144, 777–787. [Google Scholar] [CrossRef]
- Si, J.; Ma, Y.; Bi, J.W.; Xiong, Y.; Lv, C.; Li, S.; Wu, N.; Yang, Y. Shisa3 brakes resistance to EGFR-TKIs in lung adenocarcinoma by suppressing cancer stem cell properties. J. Exp. Clin. Cancer Res. 2019, 38, 481. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, J.; Xie, N.; Huang, H.; Xu, S.; Cai, J.; Qi, S. lincROR influences the stemness and crizotinib resistance in EML-ALK(+) non-small-cell lung cancer cells. OncoTargets Ther. 2018, 11, 3649–3657. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.J.; Noh, K.H.; Lee, Y.H.; Hong, S.O.; Song, K.H.; Lee, H.J.; Kim, S.; Kim, T.M.; Jeon, J.H.; Seo, J.H.; et al. Targeting stemness is an effective strategy to control EML4-ALK+ non-small cell lung cancer cells. Oncotarget 2015, 6, 40255–40267. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Tian, X.; Zhang, Y.; Huang, X.; Li, Q.; Li, W.; Li, S. LINC00319 promotes cancer stem cell-like properties in laryngeal squamous cell carcinoma via E2F1-mediated upregulation of HMGB3. Exp. Mol. Med. 2021. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Yasui, H.; Kakudo, K.; Nozaki, M. Cetuximab-resistant oral squamous cell carcinoma cells become sensitive in anchorage-independent culture conditions through the activation of the EGFR/AKT pathway. Int. J. Oncol. 2015, 47, 2165–2172. [Google Scholar] [CrossRef] [Green Version]
- Leong, H.S.; Chong, F.T.; Sew, P.H.; Lau, D.P.; Wong, B.H.; Teh, B.-T.; Tan, D.S.W.; Iyer, N.G. Targeting Cancer Stem Cell Plasticity Through Modulation of Epidermal Growth Factor and Insulin-Like Growth Factor Receptor Signaling in Head and Neck Squamous Cell Cancer. Stem Cells Transl. Med. 2014, 3, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wu, M.; Li, Y.; Chang, I.; Yuan, Q.; Ekimyan-Salvo, M.; Deng, P.; Yu, B.; Yu, Y.; Dong, J.; et al. Targeting BMI1(+) Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell 2017, 20, 621–634.e626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, S.C.; Rodriguez-Ramirez, C.; McDermott, S.P.; Wicha, M.S.; Nör, J.E. FGFR signaling regulates resistance of head and neck cancer stem cells to cisplatin. Oncotarget 2018, 9, 25148–25165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Shin, J.H.; Chen, C.H.; Cruz, L.; Farnebo, L.; Yang, J.; Borges, P.; Kang, G.; Mochly-Rosen, D.; Sunwoo, J.B. Targeting aldehyde dehydrogenase activity in head and neck squamous cell carcinoma with a novel small molecule inhibitor. Oncotarget 2017, 8, 52345–52356. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Liu, S.; Duan, S.Z.; Zhang, L.; Zhou, H.; Hu, Y.; Zhou, X.; Shi, C.; Zhou, R.; Zhang, Z. Targeting the c-Met/FZD8 signaling axis eliminates patient-derived cancer stem-like cells in head and neck squamous carcinomas. Cancer Res. 2014, 74, 7546–7559. [Google Scholar] [CrossRef] [Green Version]
- Ohishi, T.; Koga, F.; Migita, T. Bladder Cancer Stem-Like Cells: Their Origin and Therapeutic Perspectives. Int. J. Mol. Sci. 2015, 17, 43. [Google Scholar] [CrossRef] [Green Version]
- Hofner, T.; Macher-Goeppinger, S.; Klein, C.; Schillert, A.; Eisen, C.; Wagner, S.; Rigo-Watermeier, T.; Baccelli, I.; Vogel, V.; Trumpp, A.; et al. Expression and prognostic significance of cancer stem cell markers CD24 and CD44 in urothelial bladder cancer xenografts and patients undergoing radical cystectomy. Urol. Oncol. 2014, 32, 678–686. [Google Scholar] [CrossRef]
- Hayashi, M.; Guida, E.; Inokawa, Y.; Goldberg, R.; Reis, L.O.; Ooki, A.; Pilli, M.; Sadhukhan, P.; Woo, J.; Choi, W. GULP1 regulates the NRF2-KEAP1 signaling axis in urothelial carcinoma. Sci. Signal. 2020, 13, eaba0443. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Pan, X.; Zhao, L.; Li, Z.; Dai, K.; Yan, F.; Liu, S.; Ma, H.; Lai, Y. LncRNA as a diagnostic and prognostic biomarker in bladder cancer: A systematic review and meta-analysis. OncoTargets Ther. 2018, 11, 6415–6424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, Y.; Kong, J.; Kim, E.; Choi, J.H.; Yuk, H.D.; Lee, H.; Kim, H.R.; Lee, K.H.; Kang, M.; et al. Epigenetic regulation of mammalian Hedgehog signaling to the stroma determines the molecular subtype of bladder cancer. Elife 2019, 8, e43024. [Google Scholar] [CrossRef]
- Shi, M.J.; Meng, X.Y.; Wu, Q.J.; Zhou, X.H. High CD3D/CD4 ratio predicts better survival in muscle-invasive bladder cancer. Cancer Manag. Res. 2019, 11, 2987–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirtonia, A.; Gala, K.; Fernandes, S.G.; Pandya, G.; Pandey, A.K.; Sethi, G.; Khattar, E.; Garg, M. Repurposing of drugs: An attractive pharmacological strategy for cancer therapeutics. Semin. Cancer Biol. 2021, 68, 258–278. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.; Kumari, P. Anti-cancer potential of some commonly used drugs. Curr. Pharm. Des. 2021. [Google Scholar] [CrossRef] [PubMed]
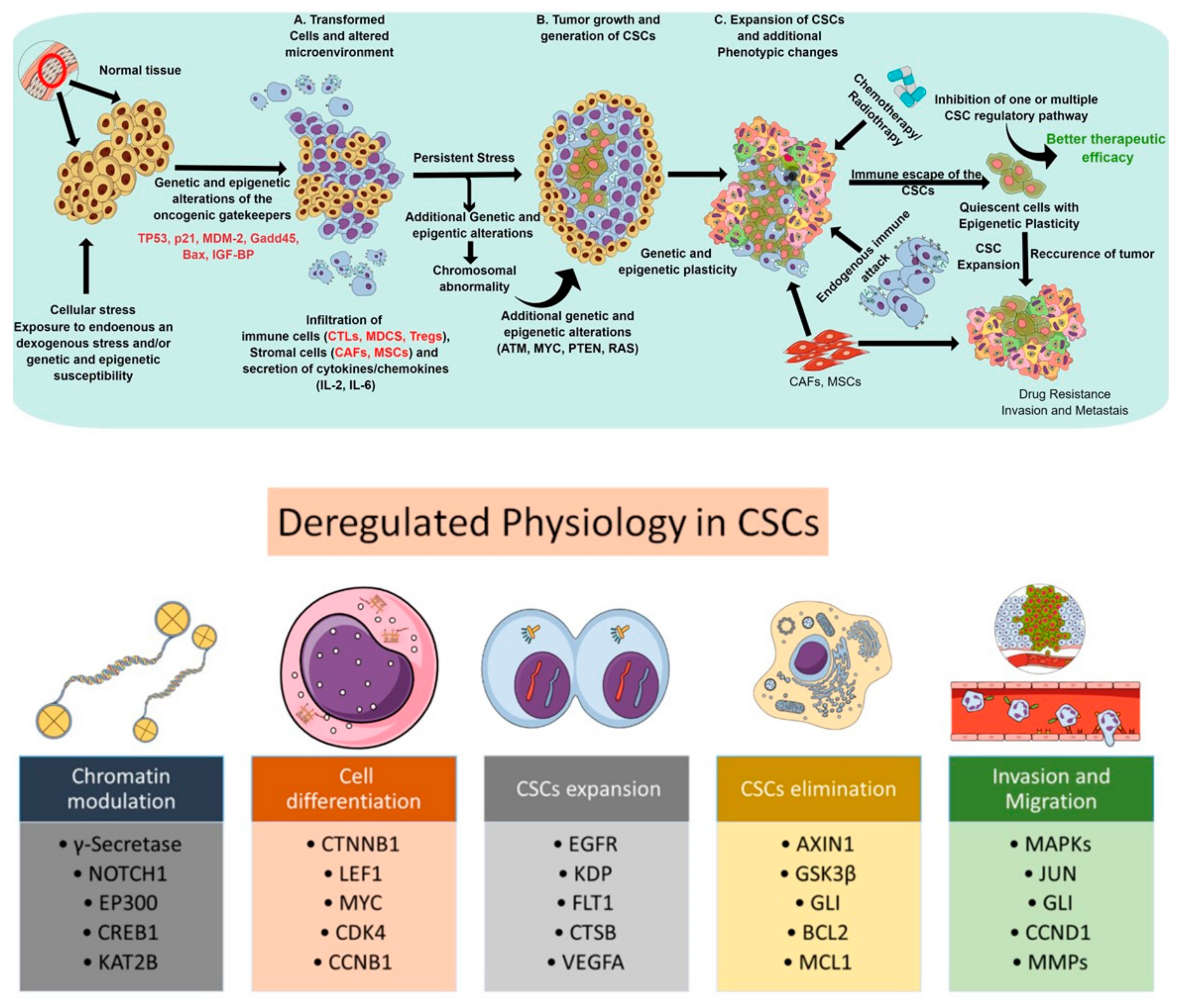

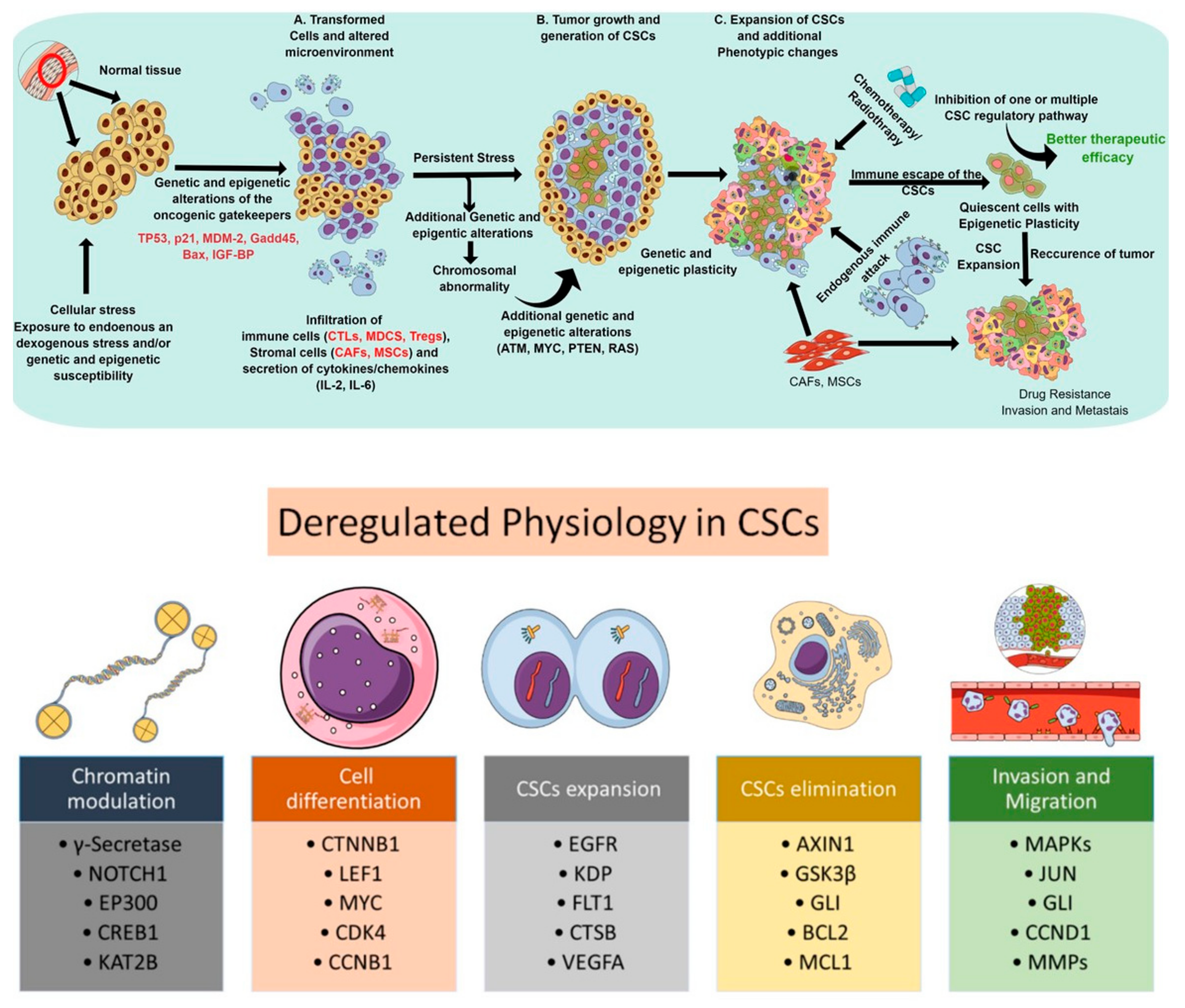{kind=link}
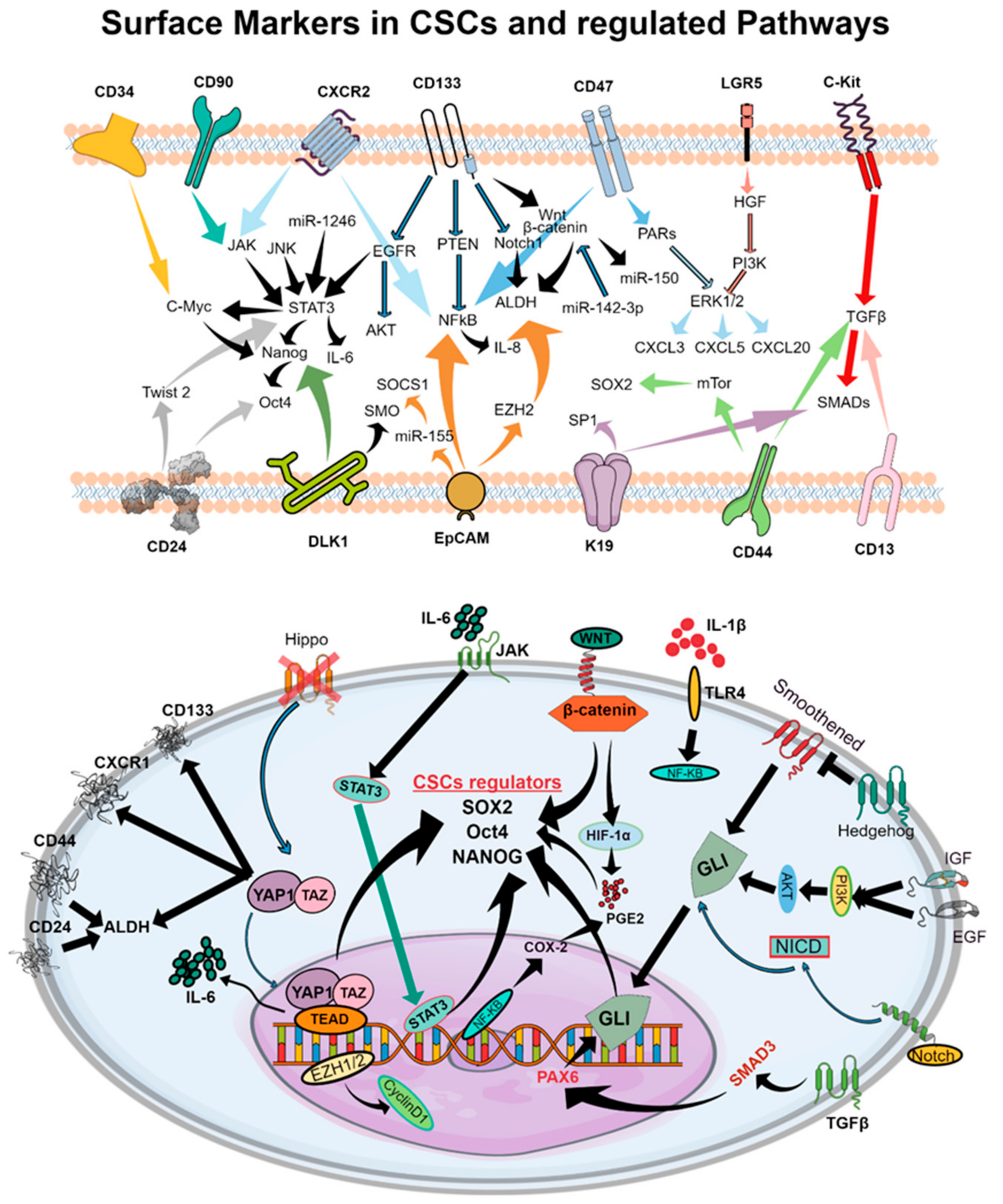
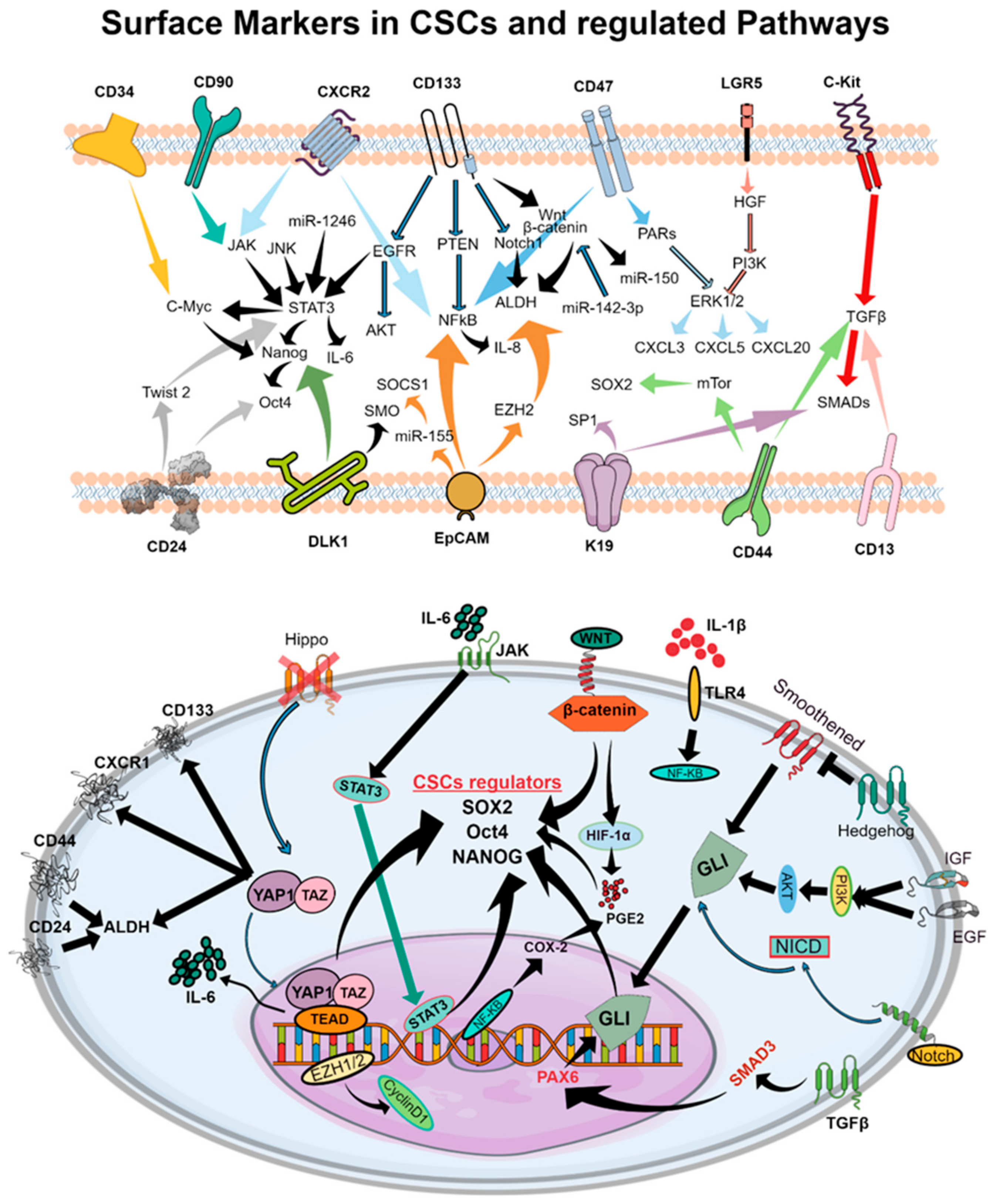{kind=link}
| Intracellular Factors | Common CSC Markers | LC | HNC | BC |
|---|---|---|---|---|
| Cell Surface Receptors | CD133 | [21] | [39] | [52] |
| CD44 | [19] | [34] | [53] | |
| ALDH1A | [19] | [49] | [54] | |
| Transcription Factors | OCT4 | [19] | [41] | [54] |
| BMI1 | [19] | [42] | [54] | |
| SOX2 | [5] | [19] | [54] | |
| NANOG | [39] | [54] | [5] |
| Cancer Type | Potential CSC Markers | References |
|---|---|---|
| Lung Cancer | ||
| Cell Surface Receptors | CXCR4 | [32] |
| CD90 | [35] | |
| CD166 | [36] | |
| uPAR | [37] | |
| CSC Molecules | CEA | [39] |
| EpCAM | ||
| NCAM | ||
| Growth Factor | IGF-1 | [27] |
| Membrane Transporter | ABCB1 | [40] |
| ABCG2 | [55] | |
| Head and Neck Cancer | ||
| Cell Surface Receptors | CD166 | [54] |
| CD98 | [54] | |
| CD117 | [56] | |
| Bladder Cancer | ||
| Cell Surface Receptors | AR (Androgen receptor) | [19] |
| CD24 | [5] | |
| CD90 | [21] | |
| Transcription Factors | SOX4 | [21] |
| Membrane Transporter | ABCG2 | [21] |
| CSC-Related Proteins, Molecules | Nestin | [21] |
| CK14 | [21] | |
| CK5 | [21] | |
| P-cadherin | [21] | |
| COX2/PGE2 | [21] | |
| YAP1/STAT3 | [24] | |
| Wnt/β-catenin | [19] |
| FDA Approved Drugs | Pathway Blocked | Evidence of Generation and Expansion of CSCs |
|---|---|---|
| Lung | ||
| Afatinib | EGFR | EGFR inhibition via NOTCH enriches cancer stem cell populations. Suggestion that combination therapy with STAT3 and Src inhibitors may improve blockade of CSCs [48,50]. In vivo, afatinib’s blockade of EGFR significantly suppresses PD-L1 expression via the inhibition of STAT1 and IRF1 [57]. |
| Afinitor (Everolimus) | mTOR | Afinitor can deregulate the PI3K/AKT/mTOR pathway and dampen the CSC phenotype in the cancer cells. Clinical data also indicate its potential anticancer effects with paclitaxel in lung cancer patients. |
| Alectinib | ALK | Alectinib is an FDA-approved drug for ALK-positive lung cancer patients[58] showed its significant effect in combination with YAP1 inhibitors. |
| Bevacizumab | VEGF-A | Bevacizumab is approved by the FDA for the treatment of common primary brain tumors, and can prevent lung metastasis; it can attenuate tumor cell proliferation, and is simultaneously effective in relieving underlying disease symptoms. |
| Brigatinib | ALK, ROS1, IGF1, EGFR | Brigatinib is approved by the FDA for the treatment of metastatic non-small-cell lung cancer; it is capable of overcoming resistance against other ALK inhibitors. |
| Ceritinib | ALK | Ceritinib is approved by the FDA for lung cancer patients who have not been previously treated with ALK inhibitors; it also can dampen the CSC phenotype via the downregulation of PI3K-driven proliferative cascade. |
| Crizotinib | ALK, ROS1 | Expression of linc-ROR and crizotinib concentration are negatively correlated. Linc-ROR elevates the viability of EML–ALK+ NSCLC cells, while crizotinib suppresses cell viability and CSC features. Thus, linc-ROR is a potential target for therapy with crizotinib [59]. Crizotinib-treated cells show decreases in NANOG, OCT4, and ALDH+ expression (all dose-dependent). Cells treated with crizotinib gradually and consistently lose the ability to form spheres, in a dose-dependent manner. Thus, the antitumor effect of crizotinib is at least partially related to the loss of stemness in these NSCLC cells [60]. |
| Dabrafenib | BRAF | Dabrafenib has been shown to be effective against BRAF V600E-mutant non-small-cell lung cancer. The FDA has approved combinatorial use of this with trametinib for metastatic NSCLC patients. Dabrafenib mainly affects the CSC phenotype and cell proliferation by downregulating the MAPK cascade. |
| Dacomitinib | EGFR | Dacomitinib is effective against both EGFR-resistant and -sensitive advanced NSCLC patients. |
| Durvalumab | PD-L1 | Durvalumab can effectively bind with PD-L1, but not PD-L2; it shows poor response against EGFR- and ALK-positive patients. |
| Erlotinib | EGFR | In EGFR wild-type cells, increased erlotinib activity was observed for cells with tyr1068 phosphorylation [61] Since EGFR wild-type CSCs express EGFR, the presence of tyr1068 may indicate the possibility of erlotinib response in CSCs [61]. Continuous exposure to erlotinib is associated with increased CSC trait expression, including CSC behaviors (self-renewal) and CSC molecules (NANOG, Oct 4, etc.). Treatment with anti-CSC molecules in combination with EGFR inhibitors may reduce the efficacy of these resistance mechanisms [31]. Erlotinib did not suppress the phosphorylation of PI3K and AKT in CSCs (despite EGFR inhibition). Compared to non-CSCs, there was an observed upregulation of miR-23a and downregulation of PTEN in CSCs. Knockdown of miR-23a may act as a mechanism to enhance the antitumor effect of erlotinib and increase subsequent apoptosis—novel strategy to eliminate the erlotinib resistance of lung cancer stem cells [62]. Erlotinib significantly reduces MDM2 levels. MDM2 is an oncoprotein that regulates p53 by inhibiting its transcriptional activity. However, there was no information on CSC markers [63]. Knockdown of SALL4 increased erlotinib sensitivity and promoted erlotinib-induced apoptosis. SALL4 knockdown reduced spheroid formation in vitro, as well as spheroid formation in CD44+ cells (CD44 is a surface marker of CSCs) [51]. Erlotinib-resistant NSCLC cells express markers of CSCs (CD44+, CD24-), and are able to form spheres more efficiently [31]. Expression of ABCG2 and CD133 was significantly elevated following treatment with erlotinib. Upon knockdown of MAP17, sphere cells were less resistant to erlotinib. Moreover, CSC cells with MAP17 knockdown decreased the efficiency of sphere formation [64]. Erlotinib-resistant cells had increased p120-catenin and Kaiso factor levels, which led to upregulation of EMT transcription factors. Via knockdown of p120-catenin and PRMT-1, cells were re-sensitized [28]. Erlotinib increased pSTAT3 and ALDH activity in EGFR mutant cells [48]. Silencing of DUOX1 led to enhanced resistance to erlotinib and upregulated levels of the CSC markers CD133 and ALDH1. The loss of DUOX1 was associated with acquired resistance to erlotinib and enhanced EMT and CSC features [65]. In lung CSCs, downregulation of miR-223 has been implicated in erlotinib resistance. Inhibition of miR-233 was observed in stem-like cells, and led to increased expression of IGF1R. The downregulation of miR-223 induces activation of the PI3k/AKT pathway in lung CSCs, and may also be responsible for the resistance of stem-like cells [66]. Elevated PGRMC1 levels were seen in lung tumor CSCs. However, no increased cancer stem cell death was noted with PGRMC1 inhibitor (AG-205) treatment as opposed to erlotinib treatment [67]. When coupled with Hedgehog pathway inhibitors, cells treated with erlotinib or gefitinib had decreased sphere formation [68]. |
| Gefitinib | EGFR | EGFR inhibition through NOTCH enriches cancer stem cell populations. Cells that survived EGFR inhibition have elevated CSC marker expression. Moreover, single blockade of EGFR increases the population of CSCs. It has been suggested that combination therapy with STAT3 and Src inhibitors may improve blockade of CSCs [48]. Levels of FOXO3a (transcription factor that triggers apoptosis) are correlated with sensitivity to EGFR-TKI (gefitinib, erlotinib). Suppression of FOXO3a increases gefitinib resistance and enhances the stem-like properties of lung cancer cells. Moreover, miR-155 transcriptionally regulates NF-kB, leading to repressed FOXO3a, increased gefitinib resistance, and enhanced cancer stemness in vitro and in vivo [45]. Gefitinib acts upon CSC regulator SALL4 in CD44+ CSCs. SALL4 is associated with increased CSC characteristics, and is a potential target whose inhibition could decrease Gefitinib resistance [51]. |
| Imatinib | SCF-c-kit | Imatinib inhibits c-kit, or CD177, signaling. The c-kit pathway is implicated in lung cancer CSC characteristics. Coupling with chemotherapy may inhibit CSC and bulk tumor cell growth [29]. |
| Osimertinib | EGFR | EGFR inhibition through NOTCH enriches cancer stem cell populations. Cells that survive EGFR inhibition have elevated CSC marker expression. Single blockade of EGFR increases the population of CSCs. It has been suggested that combination therapy with STAT3 and Src may improve blockade of CSCs [48]. |
| Head and Neck | ||
| Cetuximab | EGFR | Cetuximab is ineffective in the CSC subpopulation of head and neck cancer cell lines, likely due to migratory behavior from EGFR expression [56]. Cetuximab also failed to consistently inhibit migratory behavior through different cell lines [69]. Cetuximab inhibits sphere formation [69] and decreases CD44 expression [70]. This is indicative of some chemical effects on CSCs in head and neck cancers. |
| Afatinib | EGFR, PD-L1 | Afatinib can reduce SP cell count and CSC characteristics and increase CSC chemotherapy sensitivity via the inhibition of ABCG2 activity and expression. Activity on SP cells is likely independent of its EGFR activity. Thus, coupling with chemotherapy could lead to effective growth [71]. |
| Erlotinib | EGFR | Erlotinib treatment is correlated with elevated CD133 and ABCG2 expression, possibly due to proliferation of TKI-resistant CSCs [67]. ALD (aldehyde dehydrogenase) enables CSCs to metabolize chemotherapy and oxidants. ALD can be targeted with the use of erlotinib in head and neck cancers [72]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mudra, S.E.; Sadhukhan, P.; Ugurlu, M.T.; Alam, S.; Hoque, M.O. Therapeutic Targeting of Cancer Stem Cells in Lung, Head and Neck, and Bladder Cancers. Cancers 2021, 13, 5098. https://doi.org/10.3390/cancers13205098
Mudra SE, Sadhukhan P, Ugurlu MT, Alam S, Hoque MO. Therapeutic Targeting of Cancer Stem Cells in Lung, Head and Neck, and Bladder Cancers. Cancers. 2021; 13(20):5098. https://doi.org/10.3390/cancers13205098
Chicago/Turabian StyleMudra, Sarah E., Pritam Sadhukhan, M. Talha Ugurlu, Shorna Alam, and Mohammad O. Hoque. 2021. "Therapeutic Targeting of Cancer Stem Cells in Lung, Head and Neck, and Bladder Cancers" Cancers 13, no. 20: 5098. https://doi.org/10.3390/cancers13205098
APA StyleMudra, S. E., Sadhukhan, P., Ugurlu, M. T., Alam, S., & Hoque, M. O. (2021). Therapeutic Targeting of Cancer Stem Cells in Lung, Head and Neck, and Bladder Cancers. Cancers, 13(20), 5098. https://doi.org/10.3390/cancers13205098