
The Synthesis of Poly(Vinyl Alcohol) Grafted with Fluorinated Protic Ionic Liquids Containing Sulfo Functional Groups


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Characterization Methods
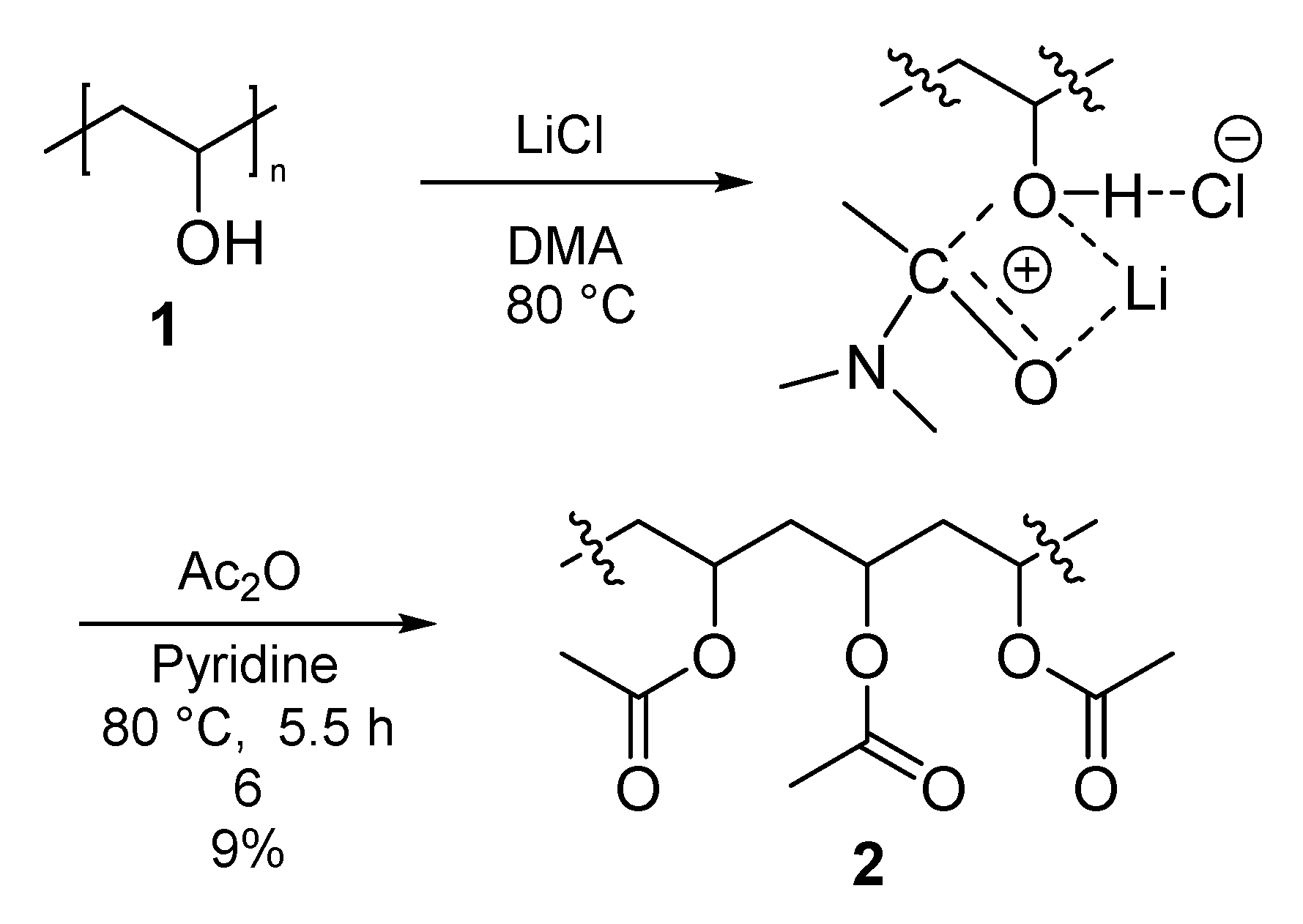
3.3. Process of PVA Acetylation
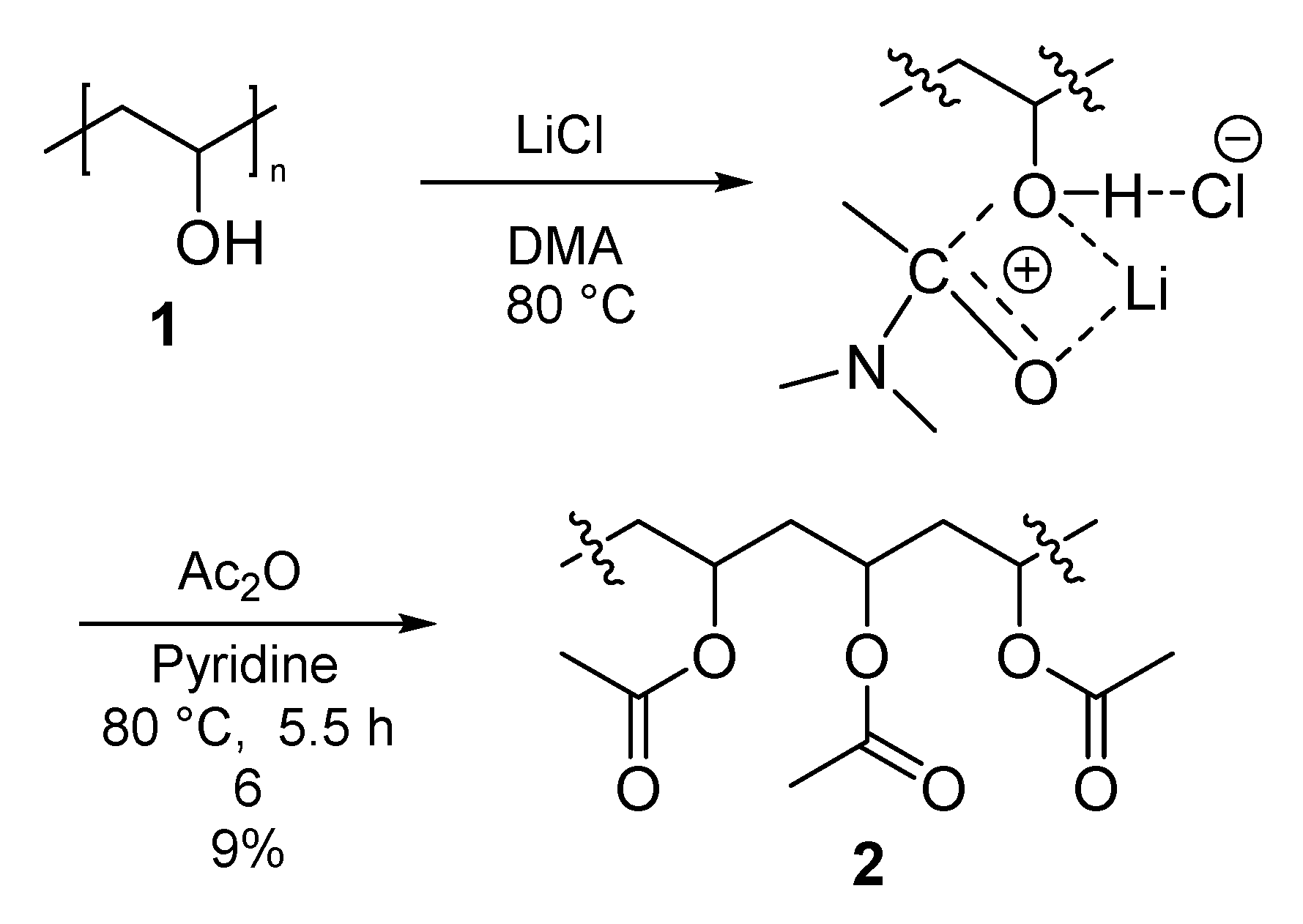
3.3.1. Polymer I (PVA-Ac) (2)
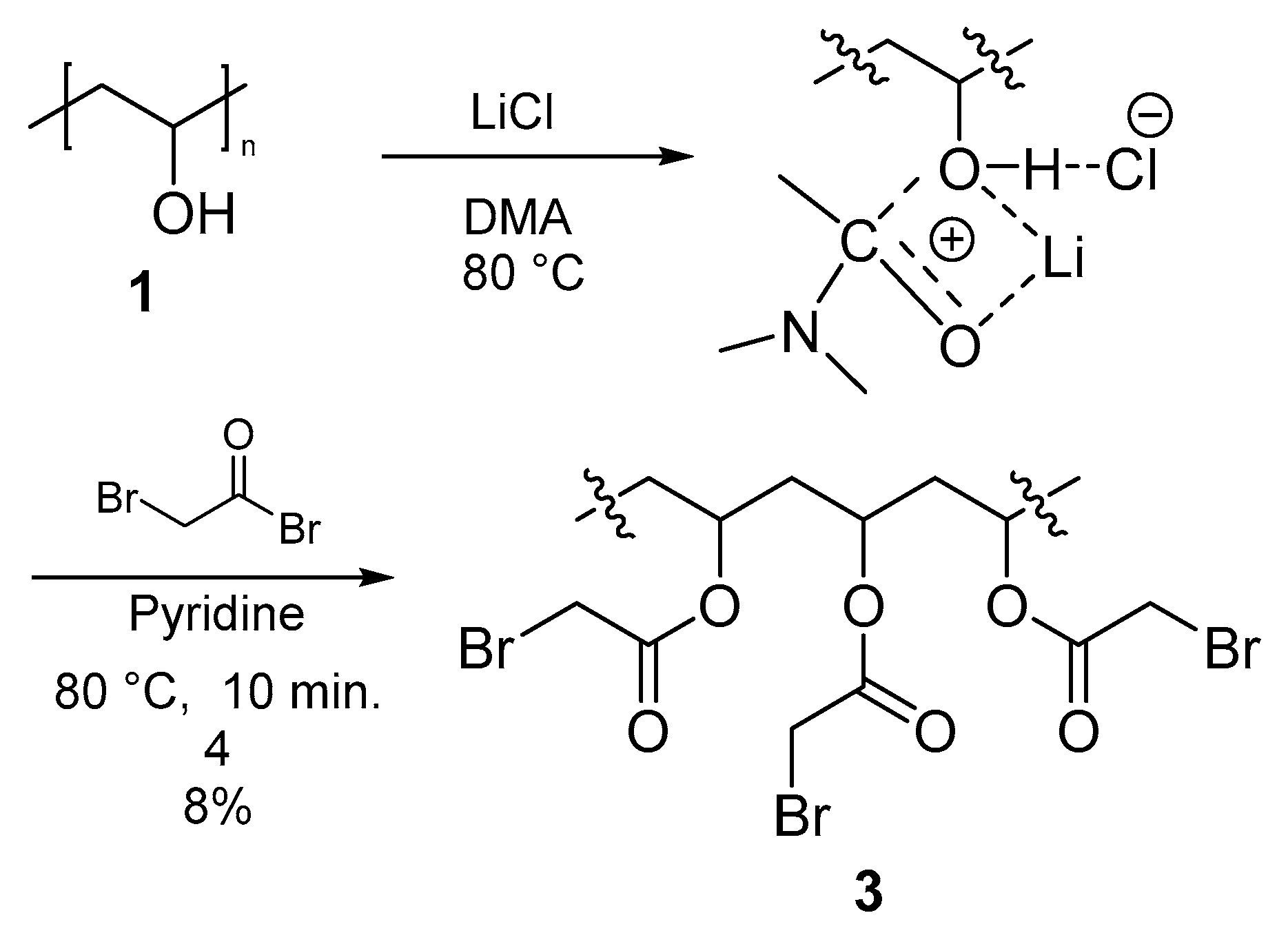
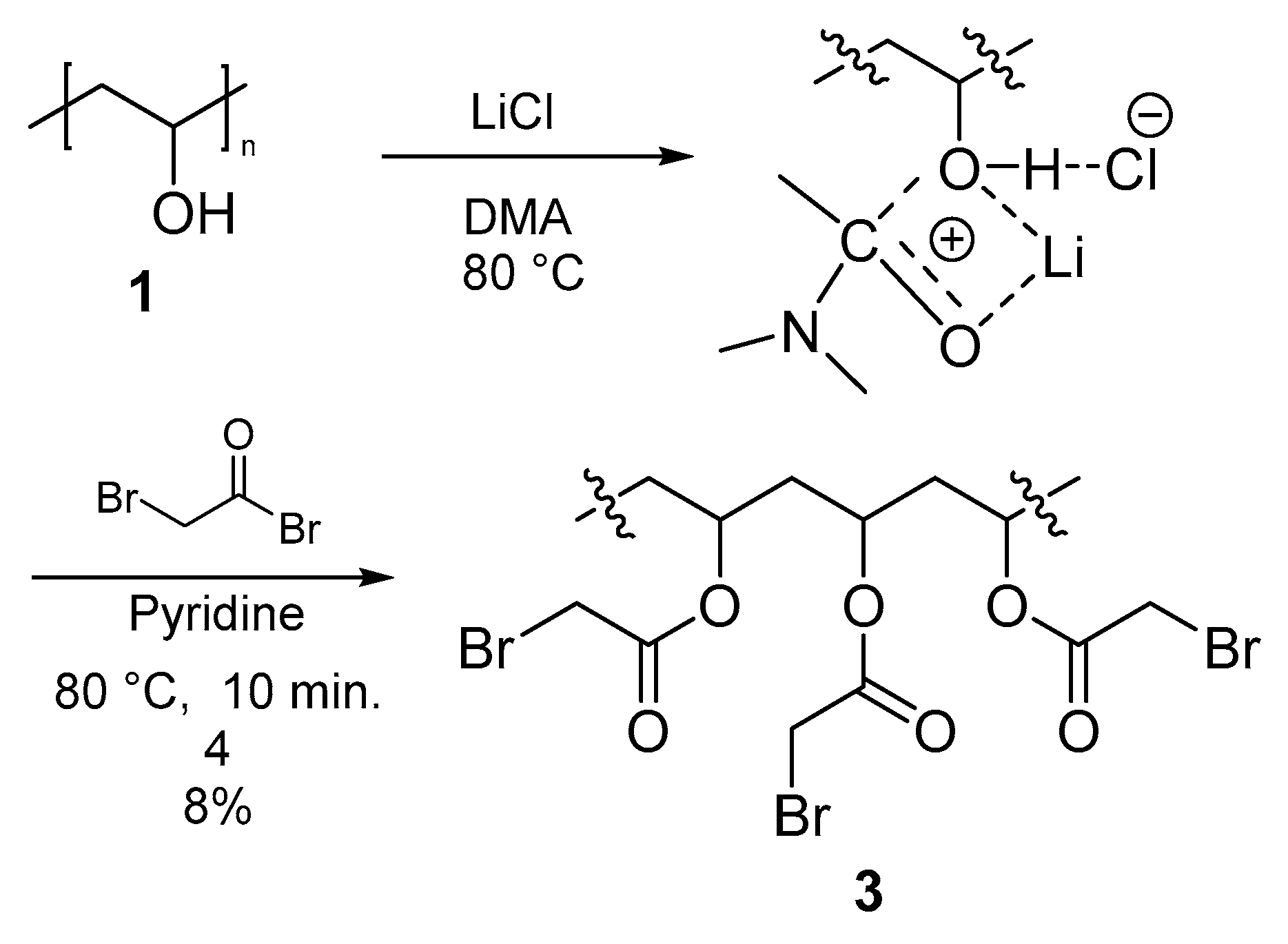
3.3.2. Polymer II (PVA-AcBr) (3)
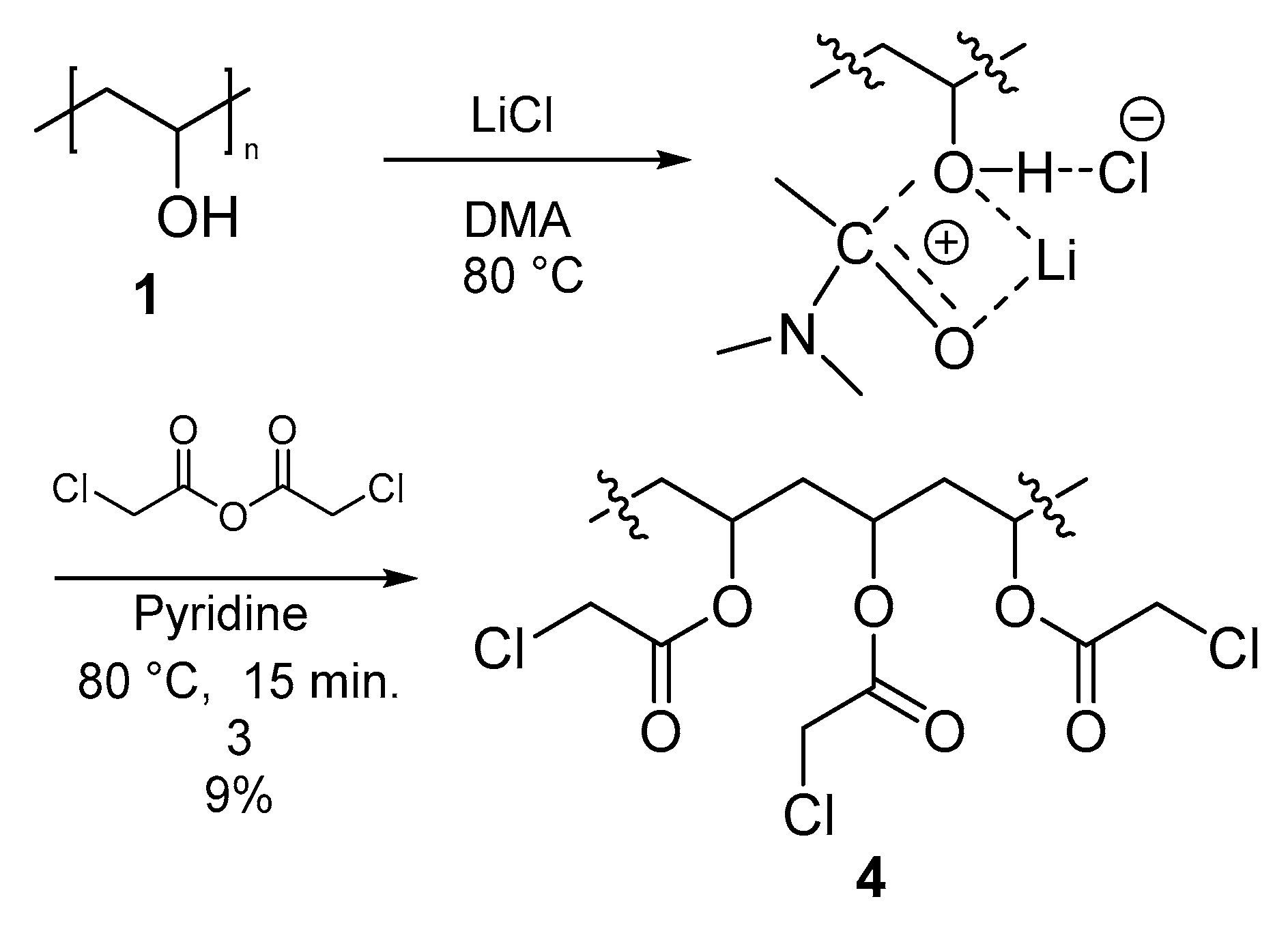
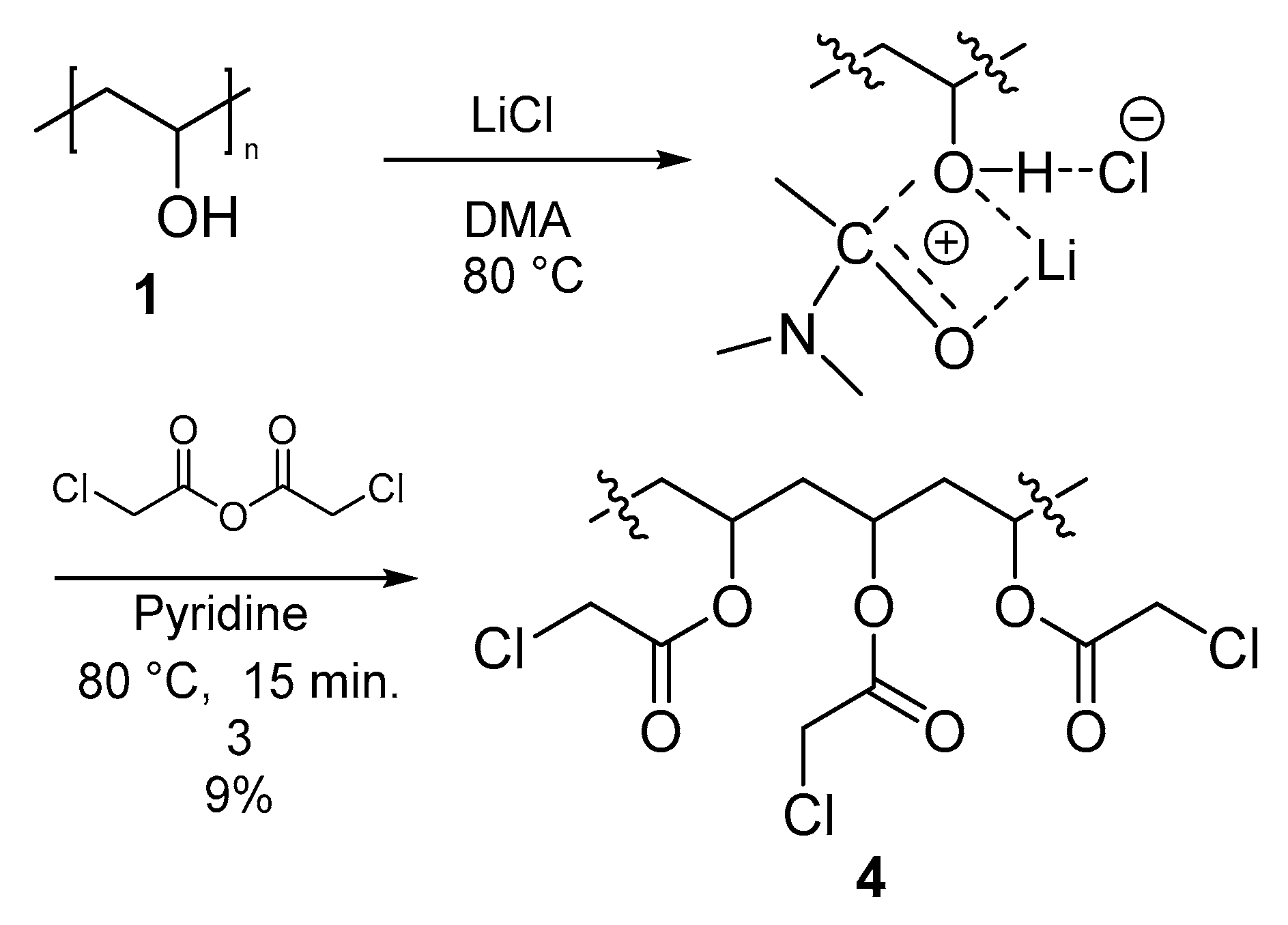
3.3.3. Polymer III (PVA-AcCl) (4)
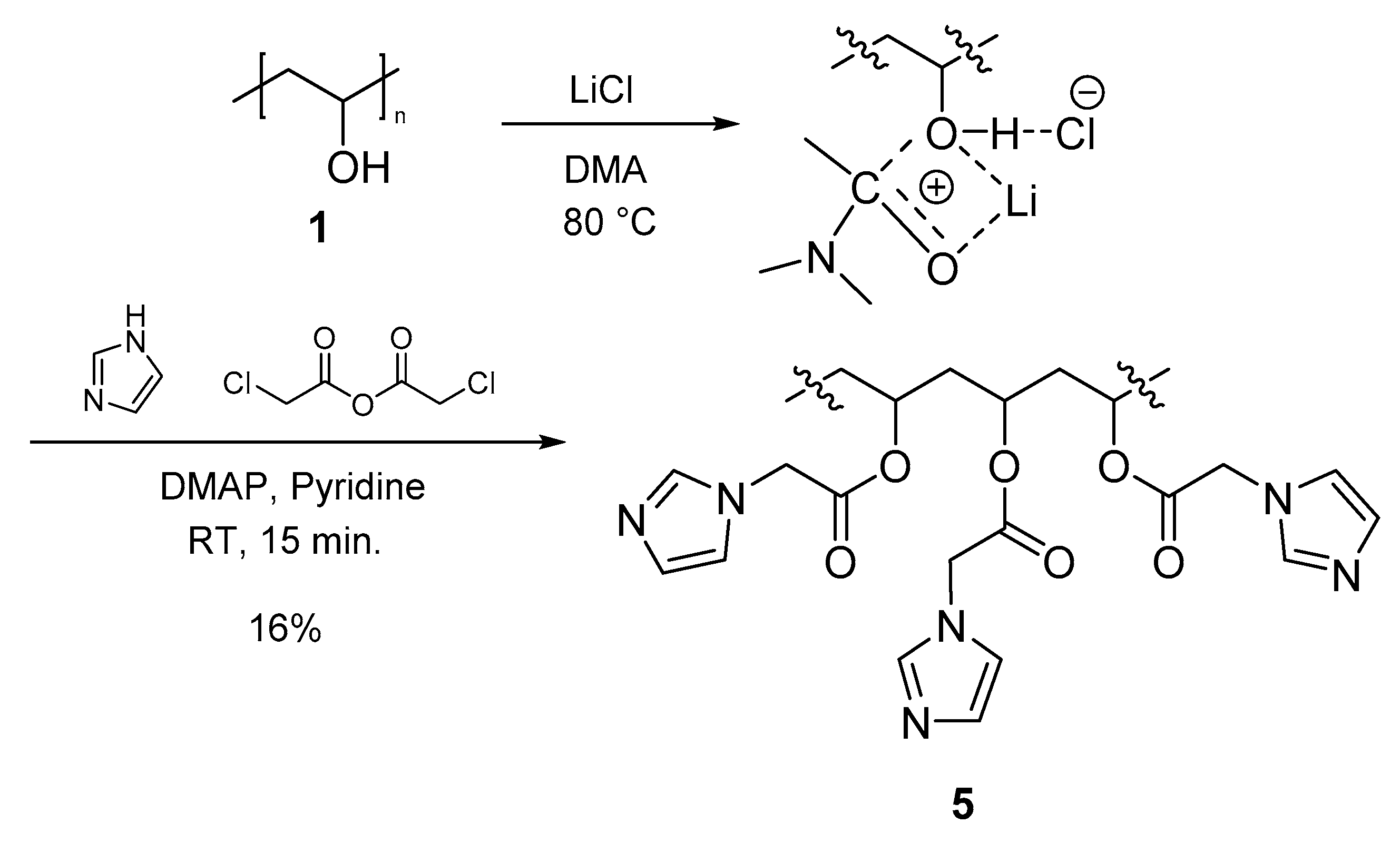
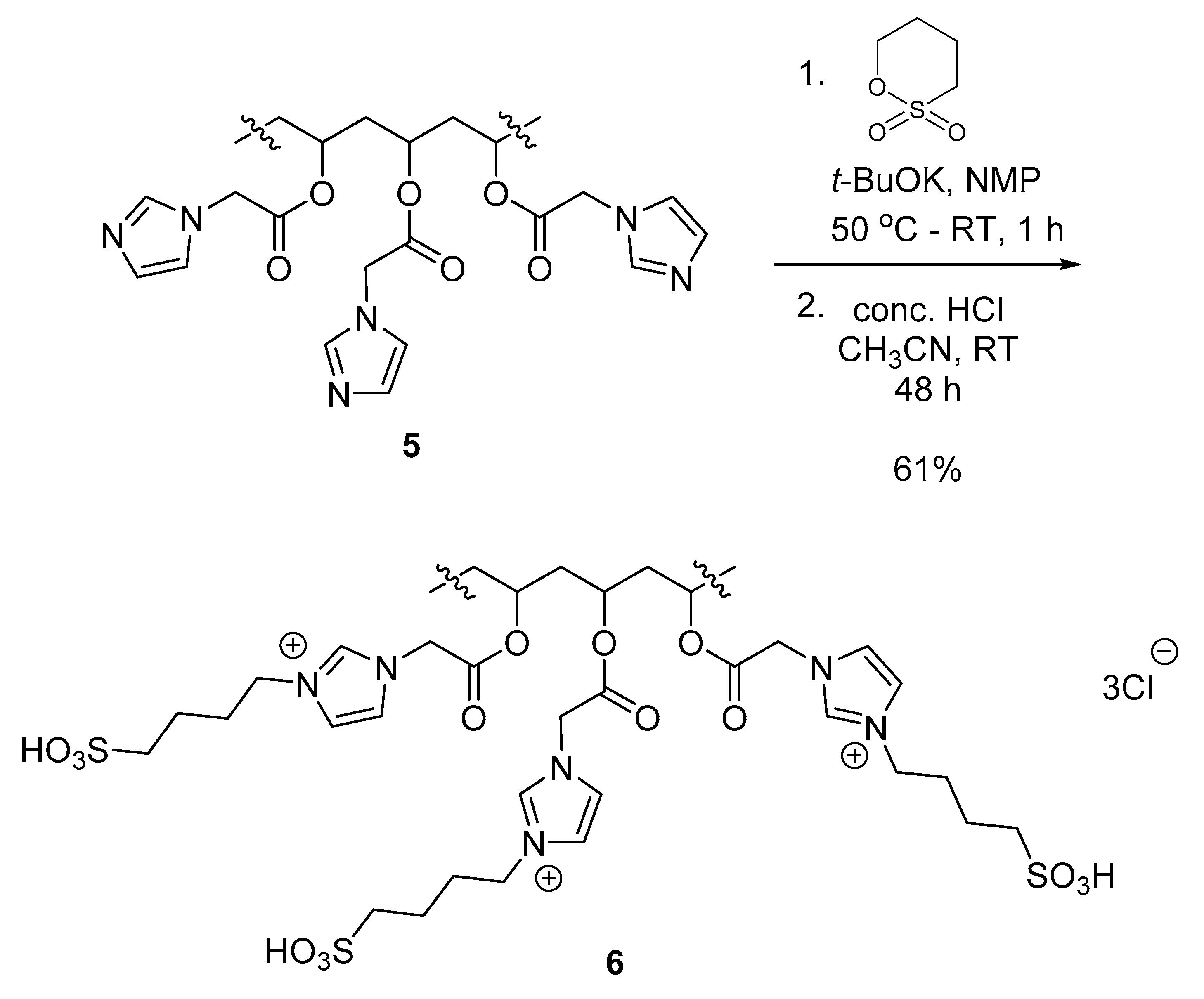
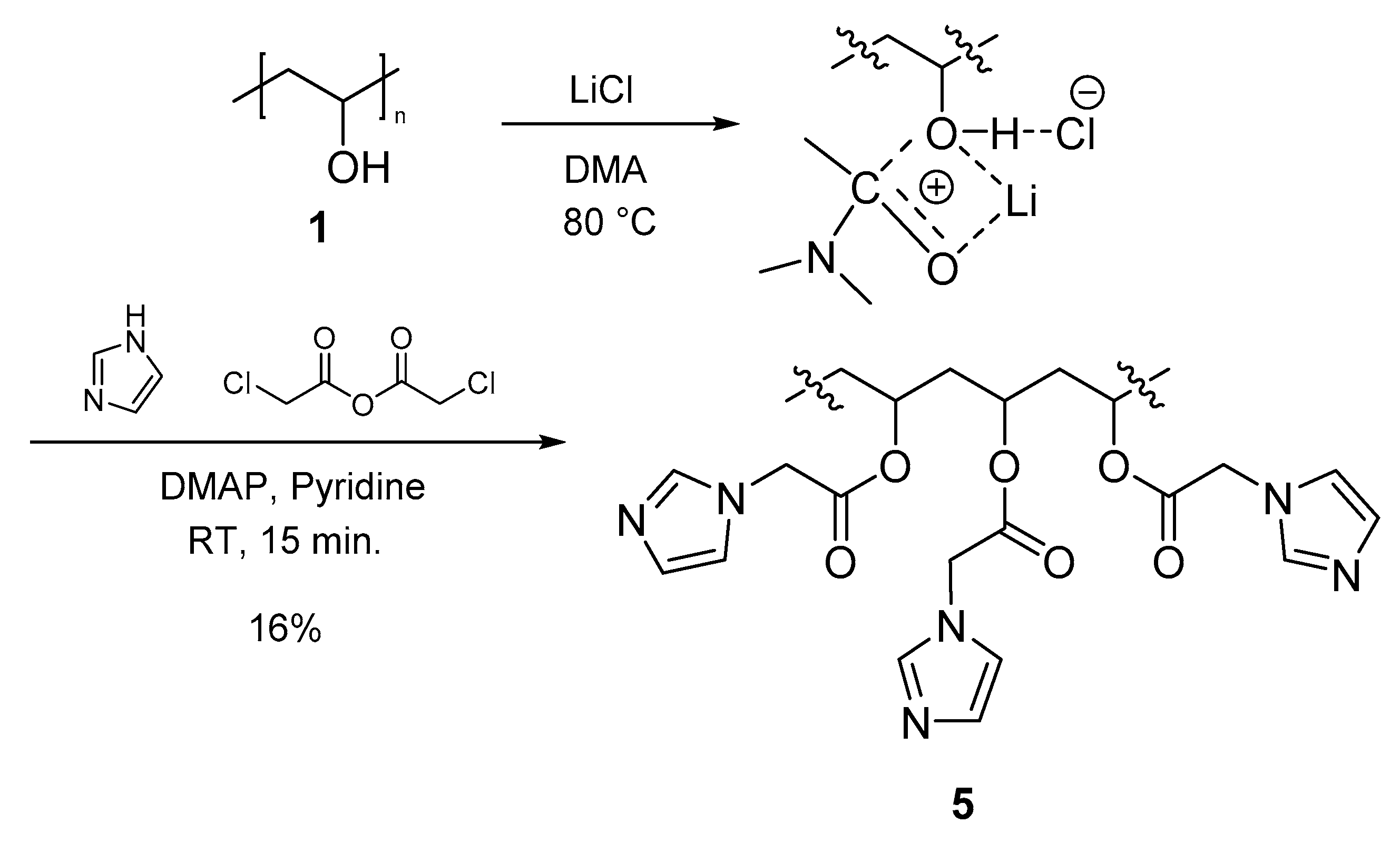
3.3.4. Polymer IV (PVA-AcIm) (5)
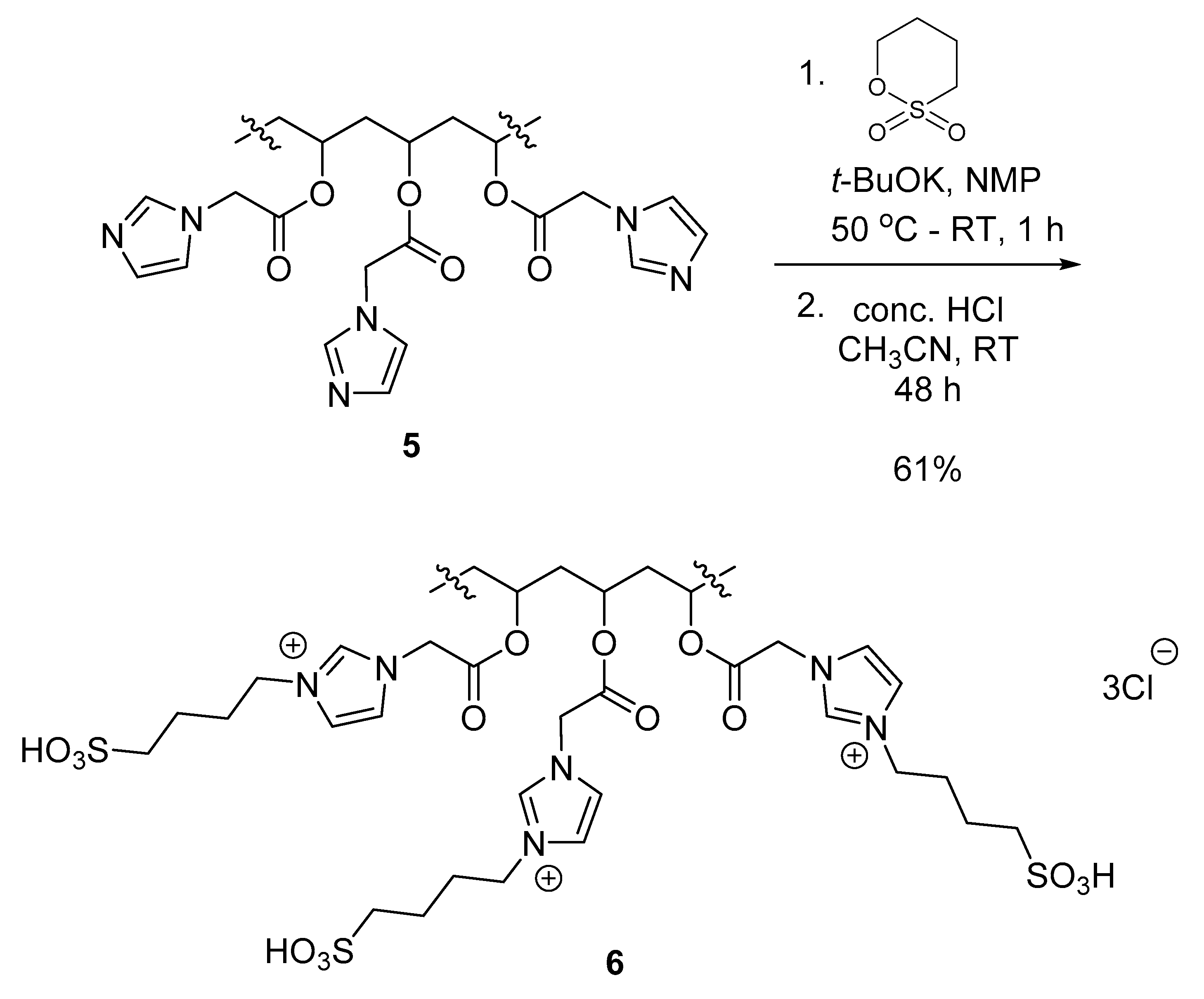
3.4. Modification of Polymer IV (PVA-AcIm) with Sultones
Polymer V (PVA-AcIm-1,4) (6)
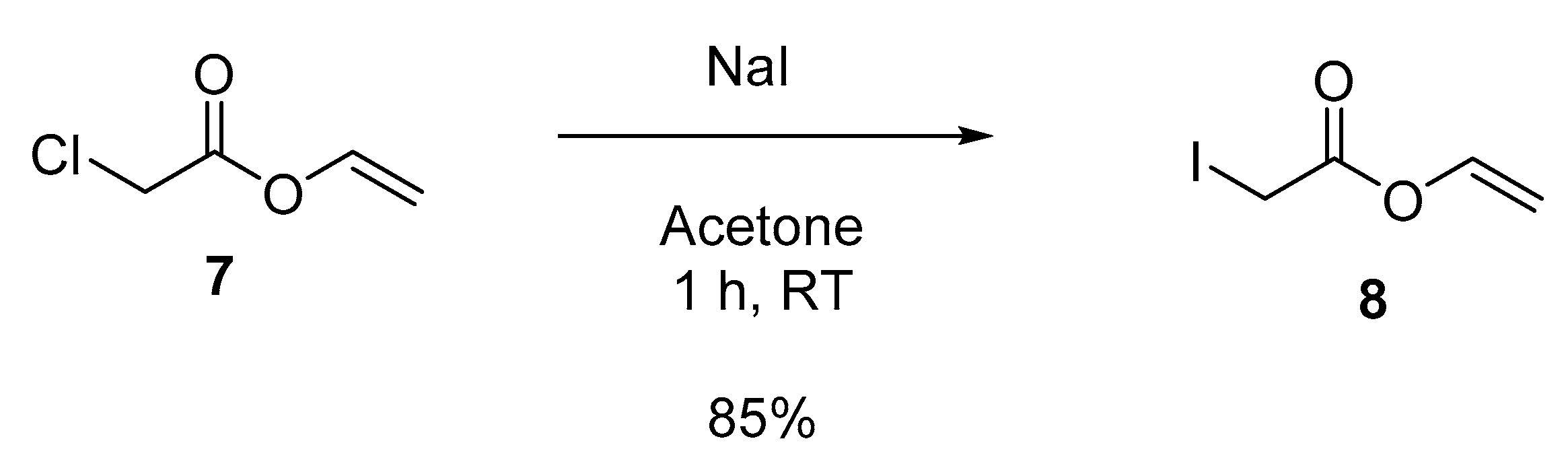
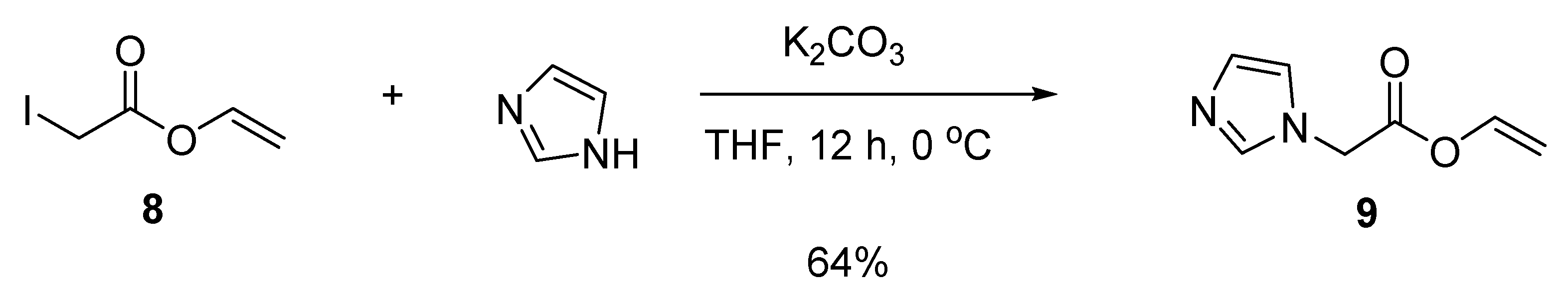
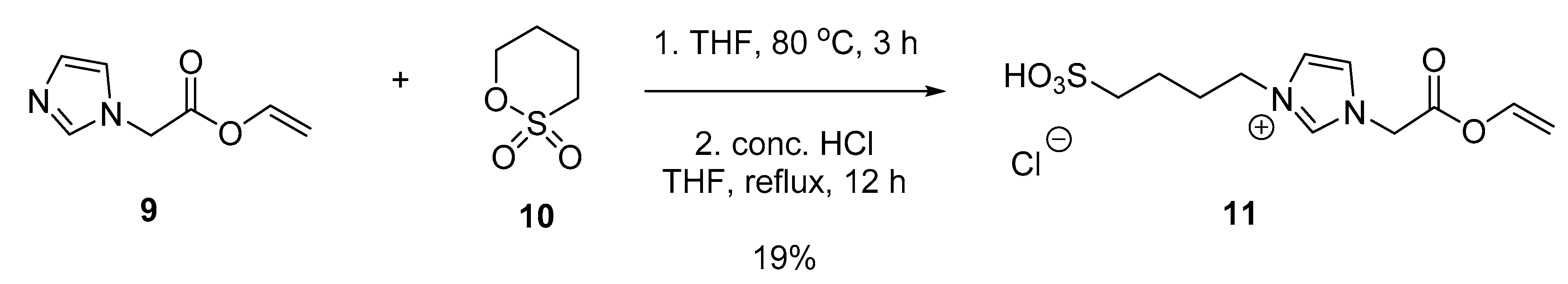
3.5. Synthesis of Vinylimidazole and Vinyl Acetate Copolymers
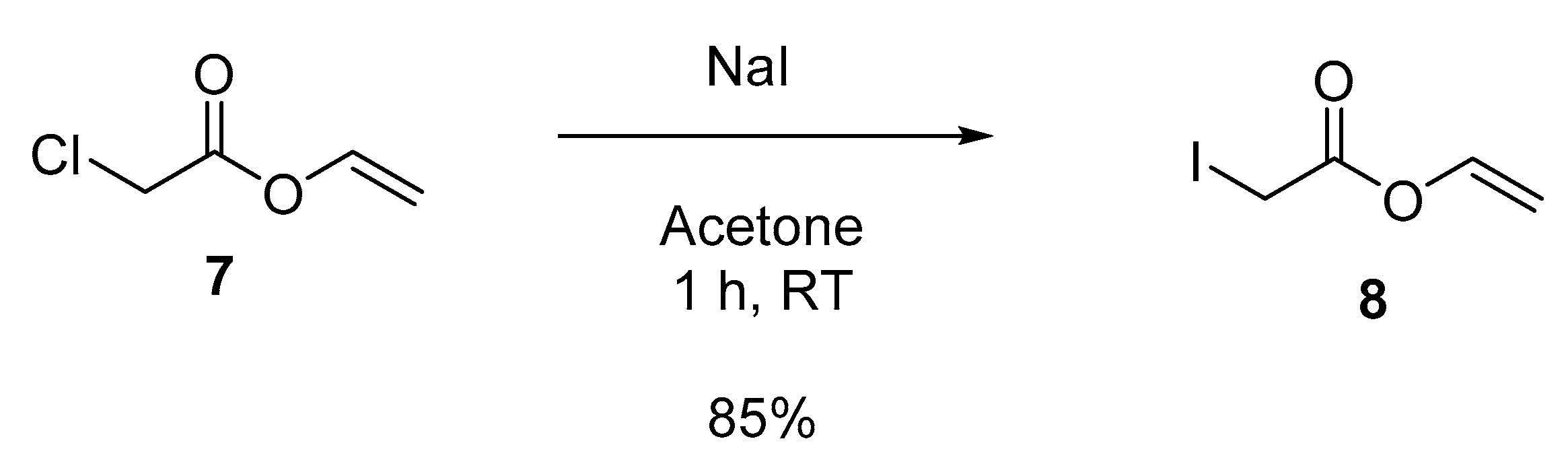
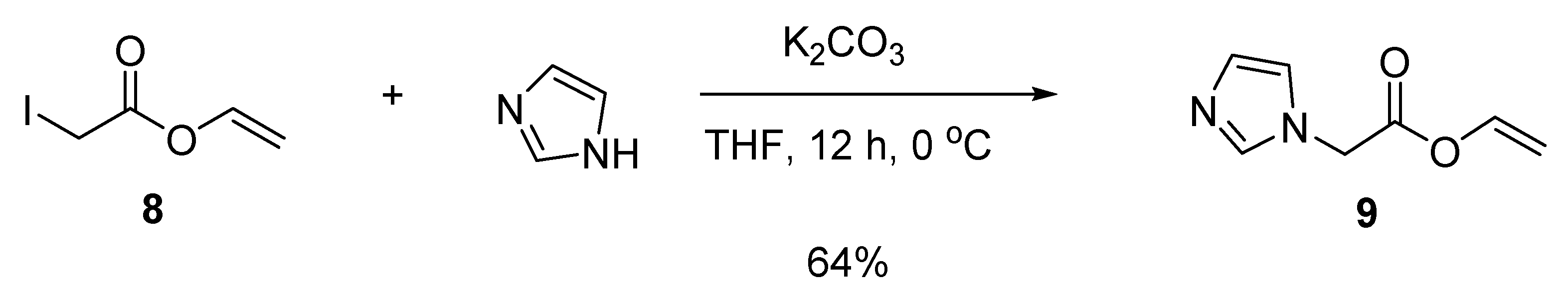
3.5.1. Vinyl 2-iodoacetate (8)
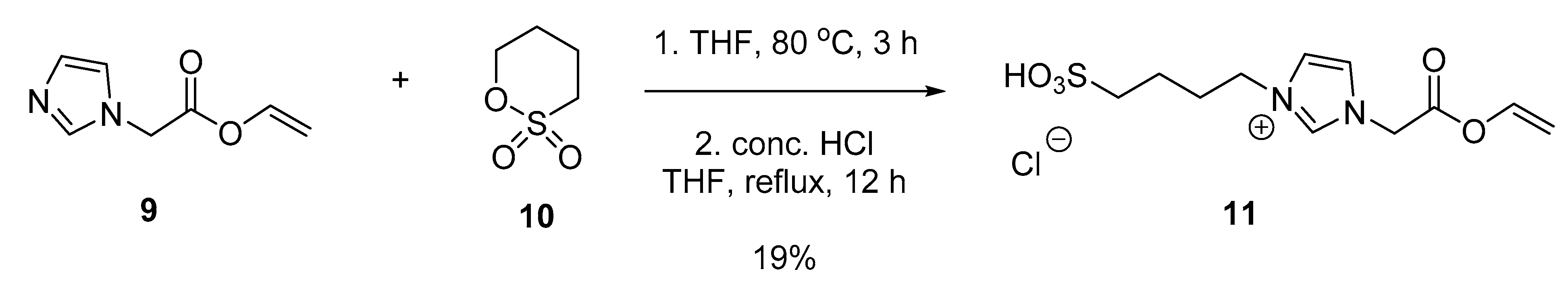
3.5.2. Vinyl 2-(1H-imidazol-1-yl)acetate (9)
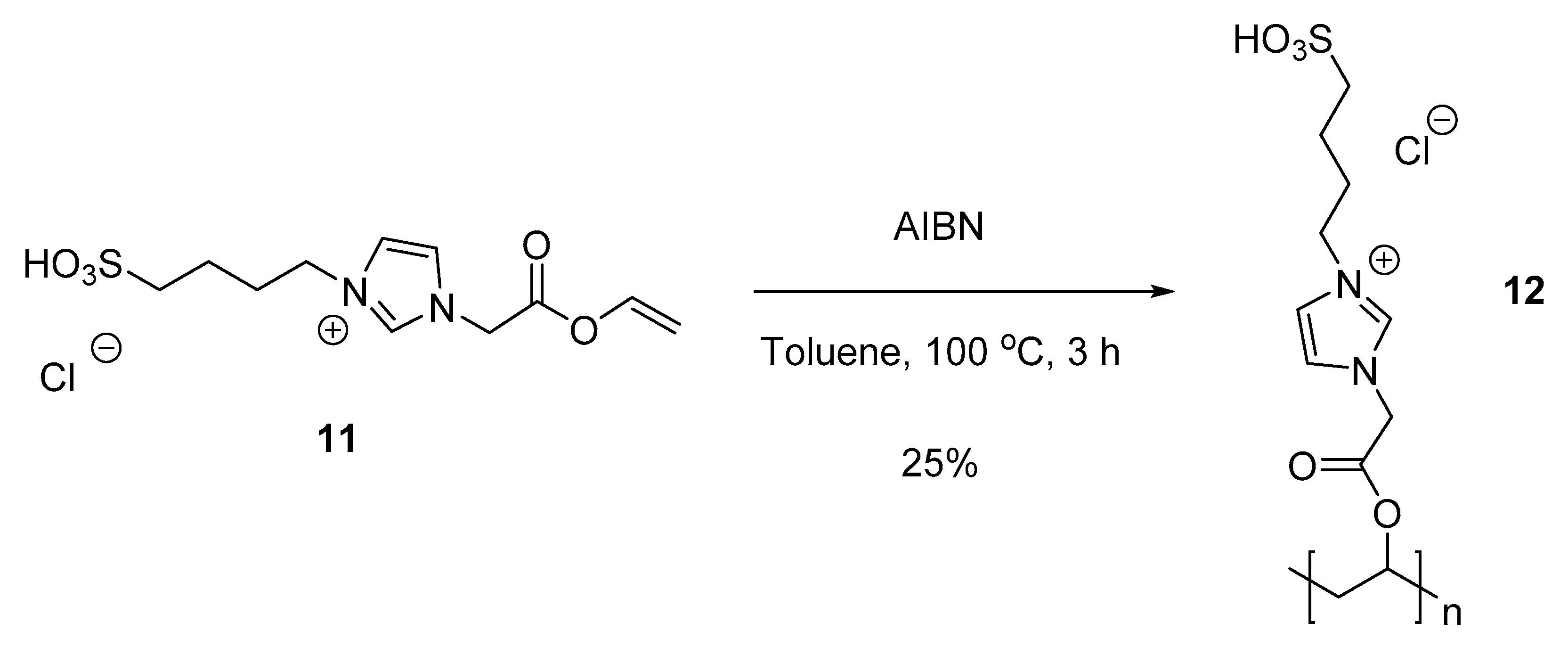
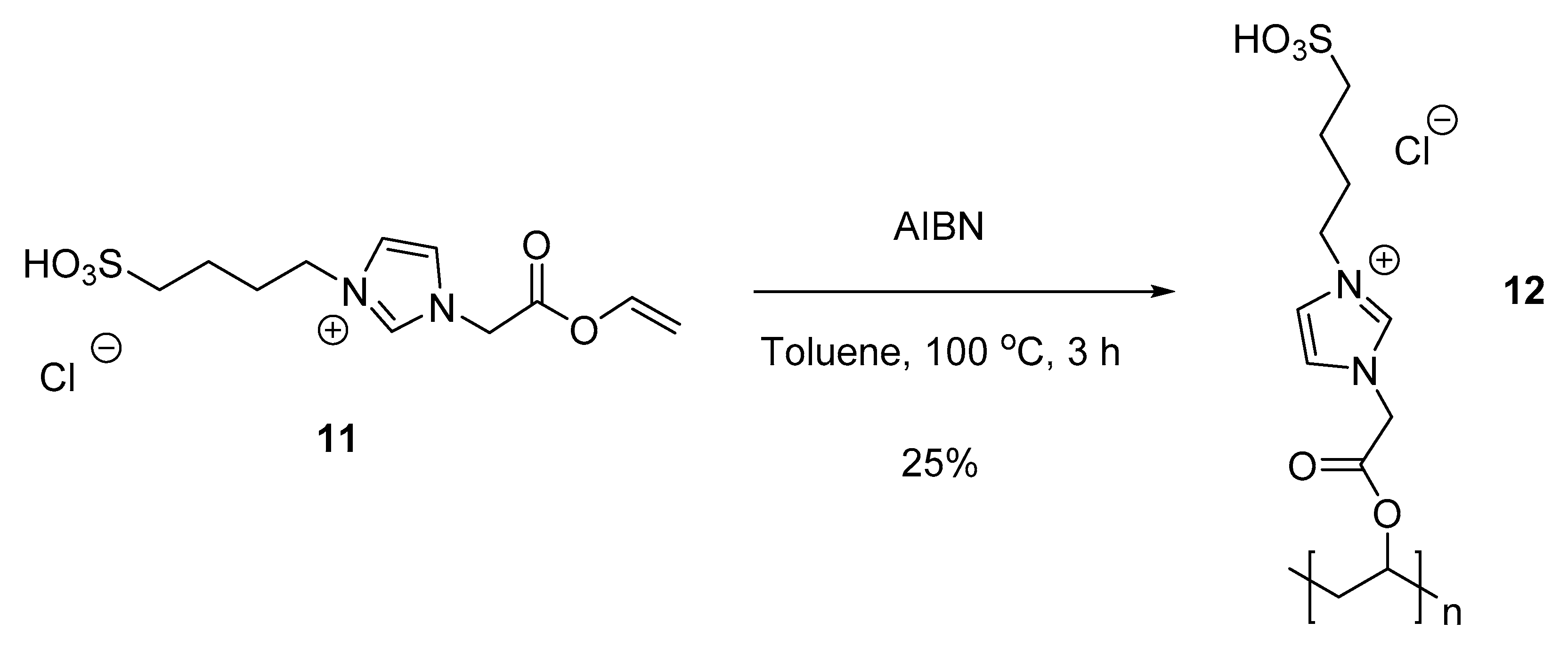
3.5.3. 1-(2-Oxo-2-(vinyloxy)ethyl)-3-(4-sulfobutyl)-1H-imidazol-3-ium chloride (11)
3.5.4. Polymer VI (PVA-ImSO3H) (12)
3.5.5. Polymer VII (PVA-Im) (13)
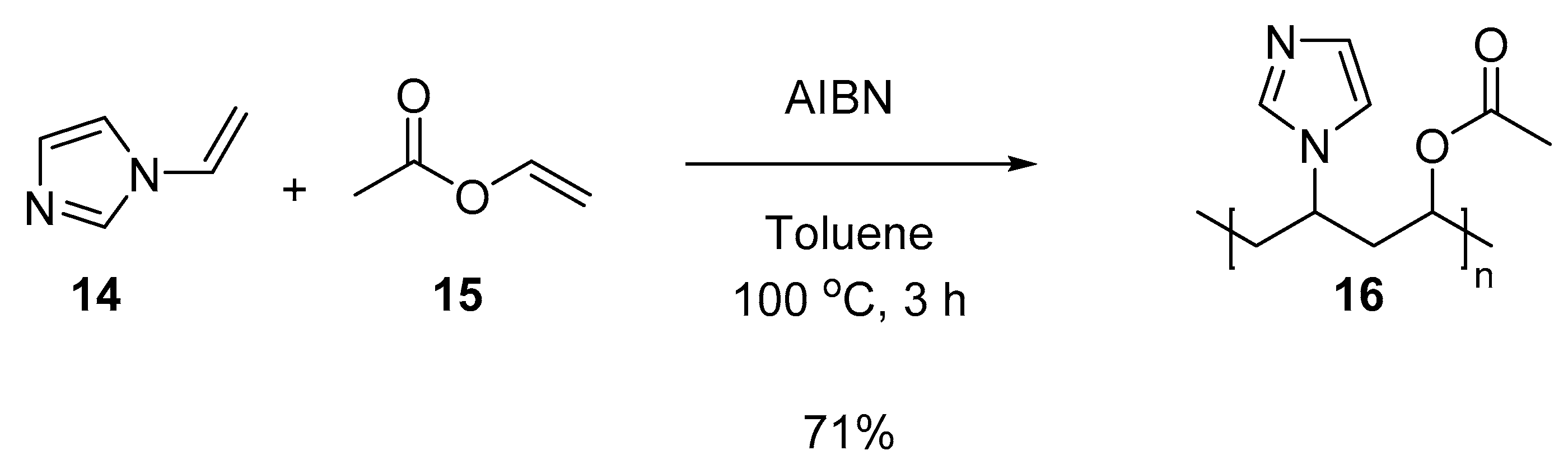
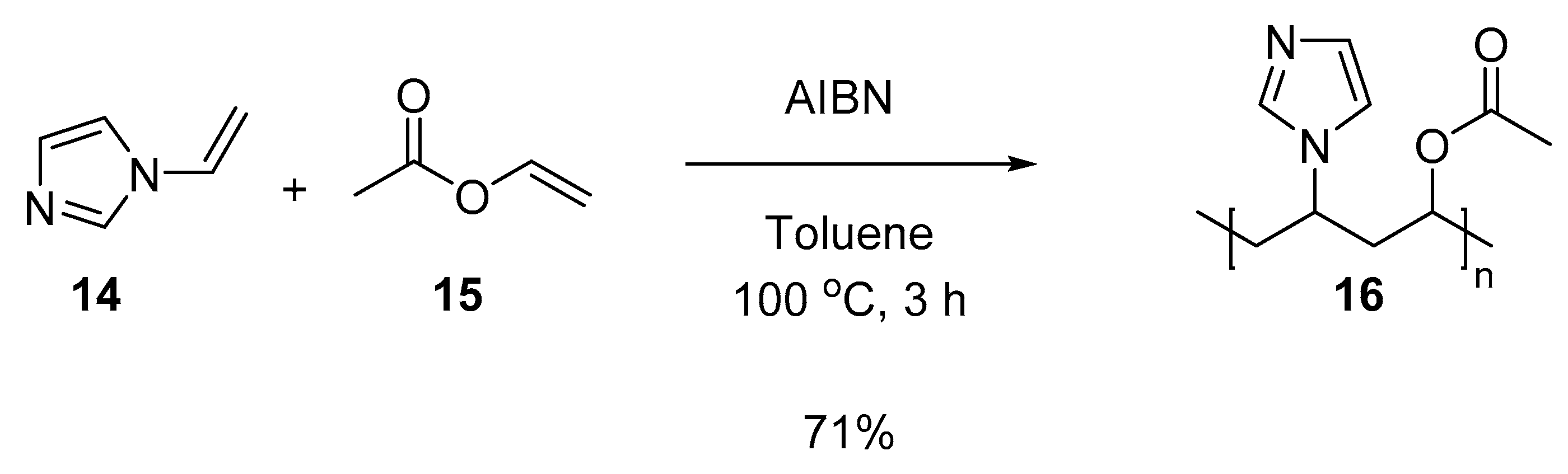
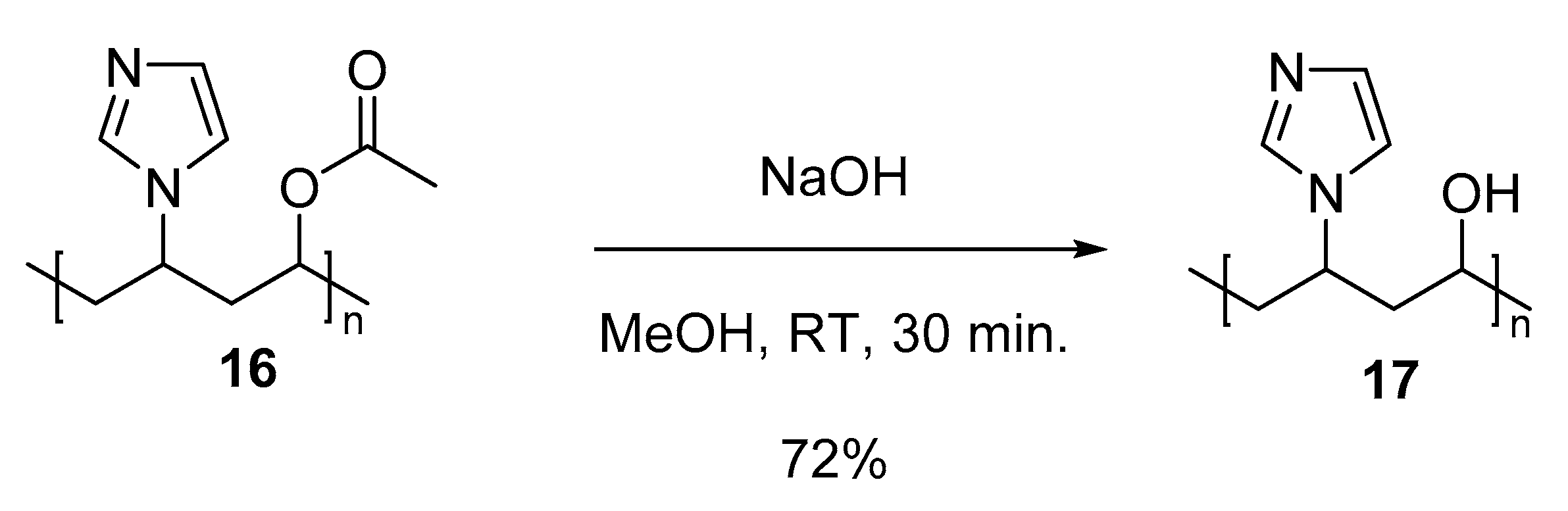
3.5.6. Copolymer I (Co-NVIm-VAc) (16)
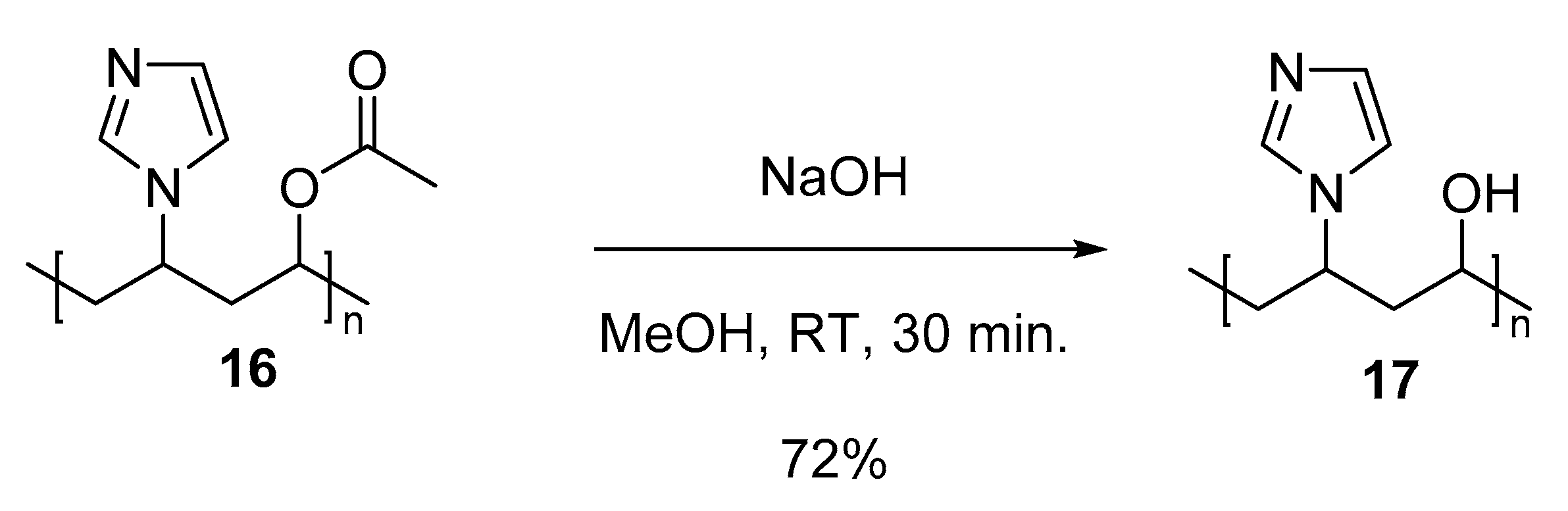
3.5.7. Copolymer II (Co-NVIm-PVA) (17)
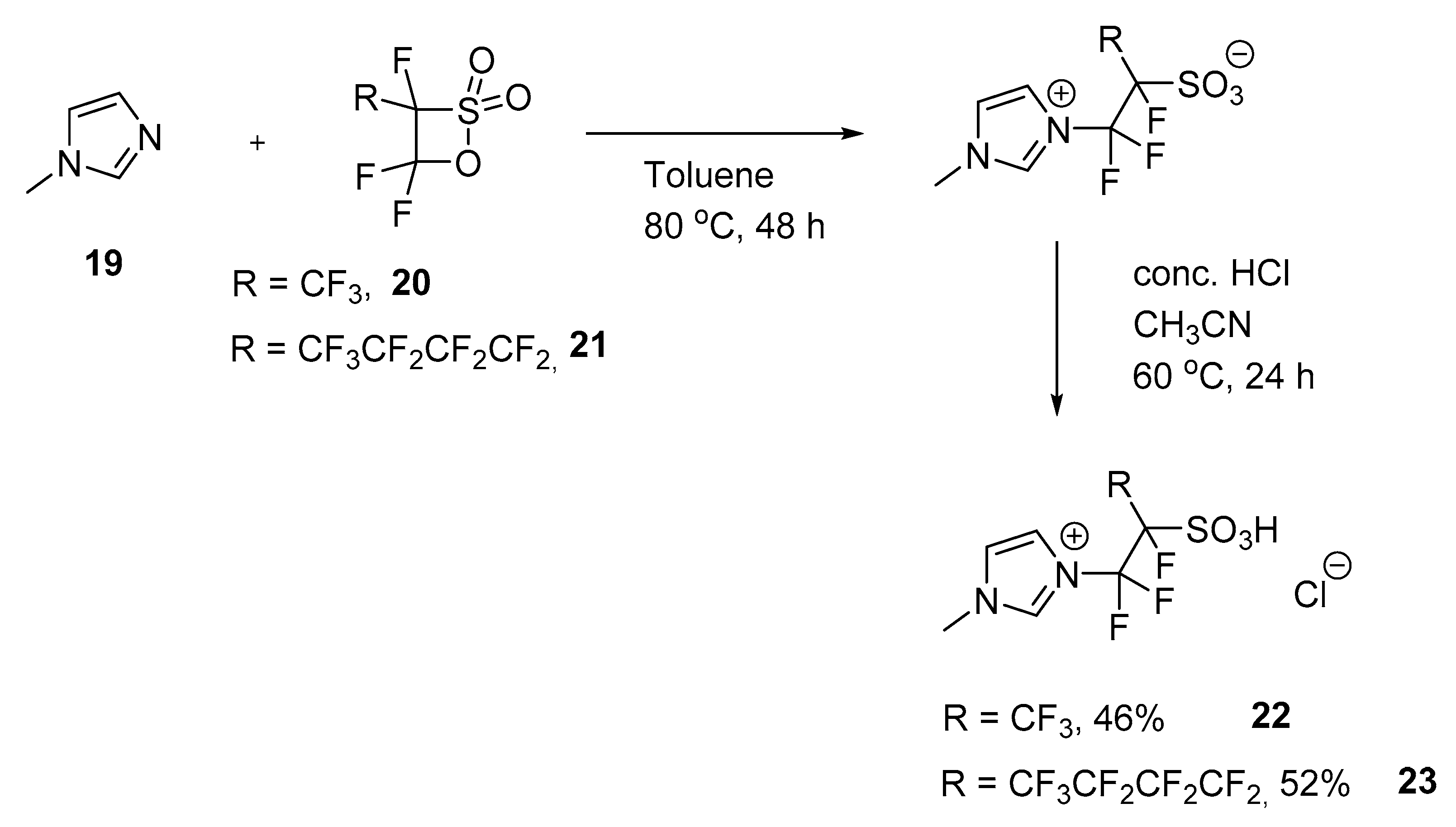
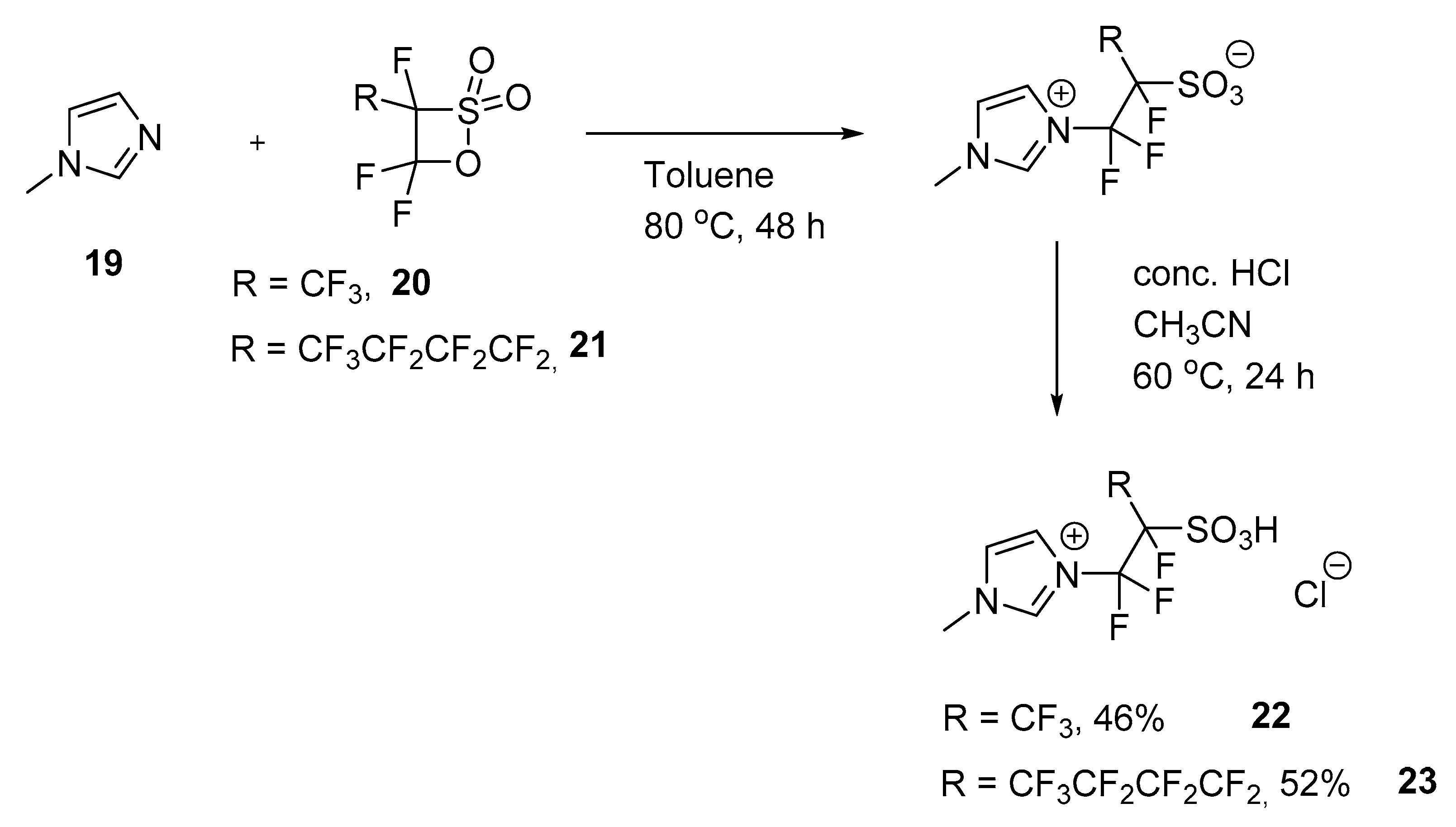
3.6. Reaction of N-Methylimidazole with Perfluorinated Sultones
3.6.1. IL I (22)
3.6.2. IL II (23)
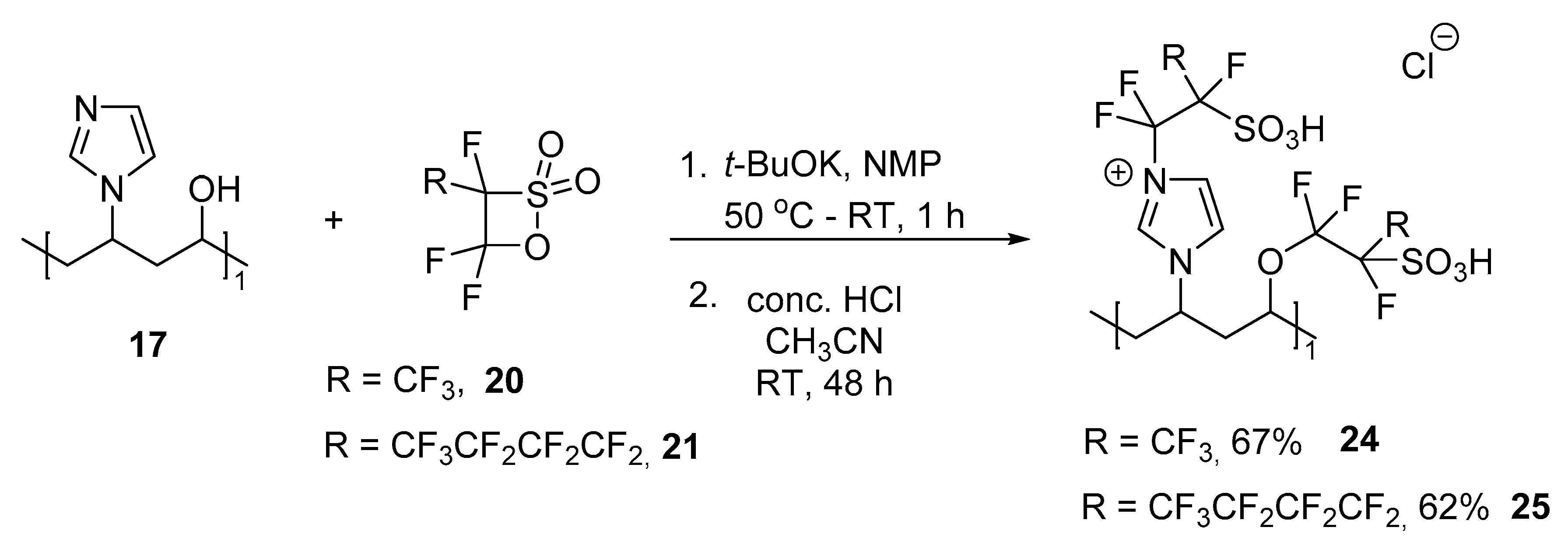
3.7. Reaction of Copolymer II (Co-NVIm-PVA) with Sultones
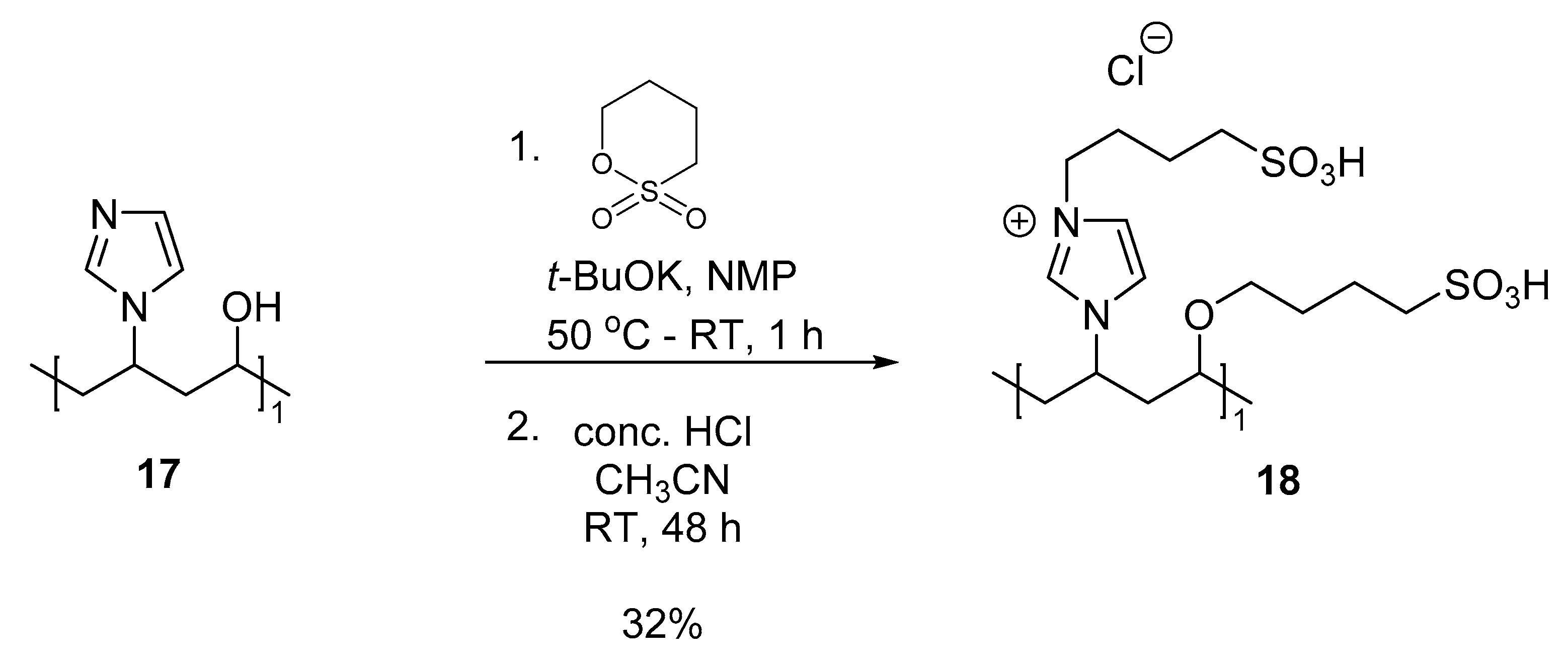
3.7.1. Copolymer III (Co-NVIm-PVA-1,4) (18)
3.7.2. Copolymer IV (Co-NVIm-PVA-6F) (24)
3.7.3. Copolymer V (Co-NVIm-PVA-12F) (25)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zaidi, S.M.J. Research trends in polymer electrolyte membranes for PEMFC. In Polymer Membranes for Fuel Cells; Matsuura, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Ebrahimi, M.; Kujawski, W.; Fatyeyeva, K.; Kujawa, J. A Review on Ionic Liquids-Based Membranes for Middle and High Temperature Polymer Electrolyte Membrane Fuel Cells (PEM FCs). Int. J. Mol. Sci. 2021, 22, 5430. [Google Scholar] [CrossRef]
- Kim, H.K.; Zhang, M.; Yuan, X.; Lvov, S.N.; Chung, T.C.M. Synthesis of polyethylene-based proton exchange membranes containing a polyethylene (PE) backbone and sulfonated poly(arylene ether sulfone) side chains for fuel cell applications. Macromolecules 2012, 45, 2460–2470. [Google Scholar] [CrossRef]
- Özdemir, Y.; Ozkan, N.; Devrim, Y. Fabrication and Characterization of Cross-linked Polybenzimidazole Based Membranes for High Temperature PEM Fuel Cells. Electrochim. Acta 2017, 245, 1–13. [Google Scholar] [CrossRef]
- Lee, H.; Han, J.; Kim, K.; Kim, J.; Kim, E.; Shin, H.; Lee, J.-C. Highly sulfonated polymer-grafted graphene oxide composite membranes for proton exchange membrane fuel cells. J. Ind. Eng. Chem. 2019, 74, 223–232. [Google Scholar] [CrossRef]
- Liew, C.-W.; Ramesh, S.; Arof, A. A novel approach on ionic liquid-based poly(vinyl alcohol) proton conductive polymer electrolytes for fuel cell applications. Int. J. Hydrogen Energy 2014, 39, 2917–2928. [Google Scholar] [CrossRef]
- Bai, H.; Wang, H.; Zhang, J.; Zhang, J.; Lu, S.; Xiang, Y. High temperature polymer electrolyte membrane achieved by grafting poly(1-vinylimidazole) on polysulfone for fuel cells application. J. Membr. Sci. 2019, 592, 117395. [Google Scholar] [CrossRef]
- Moradi, M.; Moheb, A.; Javanbakht, M.; Hooshyari, K. Experimental study and modeling of proton conductivity of phosphoric acid doped PBI-Fe2TiO5 nanocomposite membranes for using in high temperature proton exchange membrane fuel cell (HT-PEMFC). Int. J. Hydrogen Energy 2016, 41, 2896–2910. [Google Scholar] [CrossRef]
- Morancho, J.M.; Salla, J.; Cadenato, A.; Fernández-Francos, X.; Colomer, P.; Calventus, Y.; Ramis, X.; Ruiz, R. Thermal analysis of enhanced poly(vinyl alcohol)-based proton-conducting membranes crosslinked with sulfonation agents for direct methanol fuel cells. J. Appl. Polym. Sci. 2011, 124, E57–E65. [Google Scholar] [CrossRef]
- Zeng, Y.; Gu, L.; Zhang, L.; Cheng, Z.; Zhu, X. Synthesis of highly proton-conductive poly(arylene ether sulfone) bearing perfluoroalkyl sulfonic acids via polymer post-modification. Polymer 2017, 123, 345–354. [Google Scholar] [CrossRef]
- Elumalai, V.; Annapooranan, R.; Ganapathikrishnan, M.; Sangeetha, D. A synthesis study of phosphonated PSEBS for high temperature proton exchange membrane fuel cells. J. Appl. Polym. Sci. 2018, 135, 45954. [Google Scholar] [CrossRef]
- Beydaghi, H.; Javanbakht, M.; Kowsari, E. Synthesis and Characterization of Poly(vinyl alcohol)/Sulfonated Graphene Oxide Nanocomposite Membranes for Use in Proton Exchange Membrane Fuel Cells (PEMFCs). Ind. Eng. Chem. Res. 2014, 53, 16621–16632. [Google Scholar] [CrossRef]
- Arı, G.A.; Şimşek, Ö. Imidazolium functionalized poly(vinyl alcohol) membranes for direct methanol alkaline fuel cell applications. Polym. Int. 2020, 69, 644–652. [Google Scholar]
- Moulay, S. Review: Poly(vinyl alcohol) Functionalizations and Applications. Polym. Technol. Eng. 2015, 54, 1289–1319. [Google Scholar] [CrossRef]
- Malik, R.S.; Soni, U.; Chauhan, S.S.; Verma, P.; Choudhary, V. Development of functionalized quantum dot modified poly(vinyl alcohol) membranes for fuel cell applications. RSC Adv. 2016, 6, 47536–47544. [Google Scholar] [CrossRef]
- Wong, C.Y.; Wong, W.Y.; Loh, K.S.; Daud, W.R.W.; Lim, K.L.; Khalid, M.; Walvekar, R. Development of Poly(Vinyl Alcohol)-Based Polymers as Proton Exchange Membranes and Challenges in Fuel Cell Application: A Review. Polym. Rev. 2020, 60, 171–202. [Google Scholar] [CrossRef]
- Chan, L.W.; Hao, J.S.; Heng, P.W.S. Evaluation of Permeability and Mechanical Properties of Composite Polyvinyl Alcohol Films. Chem. Pharm. Bull. 1999, 47, 1412–1416. [Google Scholar] [CrossRef] [Green Version]
- Guirguis, O.W.; Moselhey, M.T.H. Thermal and structural studies of poly (vinyl alcohol) and hydroxypropyl cellulose blends. Nat. Sci. 2012, 4, 57–67. [Google Scholar] [CrossRef] [Green Version]
- de Dicastillo, C.L.; Jordá, M.; Catalá, R.; Gavara, R.; Hernández-Muñoz, P. Development of Active Polyvinyl Alcohol/β-Cyclodextrin Composites to Scavenge Undesirable Food Components. J. Agric. Food Chem. 2011, 59, 11026–11033. [Google Scholar] [CrossRef]
- Martinez-Felipe, A.; Moliner, C.; Imrie, C.; Ribes-Greus, A. Characterization of crosslinked poly(vinyl alcohol)-based membranes with different hydrolysis degrees for their use as electrolytes in direct methanol fuel cells. J. Appl. Polym. Sci. 2011, 124, 1000–1011. [Google Scholar] [CrossRef]
- Rhim, J.-W.; Park, H.B.; Lee, C.-S.; Jun, J.-H.; Kim, D.S.; Lee, Y.M. Crosslinked poly(vinyl alcohol) membranes containing sulfonic acid group: Proton and methanol transport through membranes. J. Membr. Sci. 2004, 238, 143–151. [Google Scholar] [CrossRef]
- Dadfar, S.M.M.; Kavoosi, G.; Dadfar, S.M.A. Investigation of mechanical properties, antibacterial features, and water vapor permeability of polyvinyl alcohol thin films reinforced by glutaraldehyde and multiwalled carbon nanotube. Polym. Compos. 2014, 35, 1736–1743. [Google Scholar] [CrossRef]
- Boroglu, M.S.; Celik, S.U.; Bozkurt, A.; Boz, I. The synthesis and characterization of anhydrous proton conducting membranes based on sulfonated poly(vinyl alcohol) and imidazole. J. Membr. Sci. 2011, 375, 157–164. [Google Scholar] [CrossRef]
- Jansen, J.C.; Clarizia, G.; Bernardo, P.; Bazzarelli, F.; Friess, K.; Randová, A.; Schauer, J.; Kubicka, D.; Kacirková, M.; Izak, P. Gas transport properties and pervaporation performance of fluoropolymer gel membranes based on pure and mixed ionic liquids. Sep. Purif. Technol. 2013, 109, 87–97. [Google Scholar] [CrossRef]
- Izák, P.; Köckerling, M.; Kragl, U. Solute transport from aqueous mixture throught supported ionic liquid membrane by pervaporation. Desalination 2009, 199, 96–98. [Google Scholar] [CrossRef]
- Lin, B.; Yuan, W.; Xu, F.; Chen, Q.; Zhu, H.; Li, X.; Yuan, N.; Chu, F.; Ding, J. Protic ionic liquid/functionalized graphene oxide hybrid membranes for high temperature proton exchange membrane fuel cell applications. Appl. Surf. Sci. 2018, 455, 295–301. [Google Scholar] [CrossRef]
- Lin, B.; Cheng, S.; Qiu, L.; Yan, F.; Shang, S.; Lu, J. Protic Ionic Liquid-Based Hybrid Proton-Conducting Membranes for Anhydrous Proton Exchange Membrane Application. Chem. Mater. 2010, 22, 1807–1813. [Google Scholar] [CrossRef]
- Thomson, J.; Dunn, P.; Holmes, L.; Belieres, J.-P.; Angell, C.A.; Gervasio, D. A Flourinated Ionic Liquid as a High-Performance Fuel Cell Electrolyte. ECS Trans. 2008, 13, 21–29. [Google Scholar] [CrossRef]
- Eguizábal, A.; Lemus, J.; Pina, M. On the incorporation of protic ionic liquids imbibed in large pore zeolites to polybenzimidazole membranes for high temperature proton exchange membrane fuel cells. J. Power Sources 2013, 222, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Tosh, B.; Saikia, C.N.; Dass, N.N. Development of a nonaqueous solvent system for poly(vinyl alcohol) and its characterization. J. Appl. Polym. Sci. 1999, 74, 663–669. [Google Scholar] [CrossRef]
- Jantas, R.; Draczyński, Z.; Herczyńska, L.; Stawski, D. Poly(vinyl alcohol)-Salicylic Acid Conjugate: Synthesis and Characterization. Am. J. Polym. Sci. 2012, 2, 79–84. [Google Scholar] [CrossRef]
- Hicks, J.C.; Mullis, B.A.; Jones, C.W. Sulfonic Acid Functionalized SBA-15 Silica as a Methylaluminoxane-Free Cocatalyst/Support for Ethylene Polymerization. J. Am. Chem. Soc. 2007, 129, 8426–8427. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; He, Q.; Gao, N.; Yuan, T.; Zhang, S.; Yang, H. Novel proton exchange membranes based on cardo poly(arylene ether sulfone/nitrile)s with perfluoroalkyl sulfonic acid moieties for passive direct methanol fuel cells. J. Power Sources 2014, 261, 38–45. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glińska, P.; Wolan, A.; Kujawski, W.; Rynkowska, E.; Kujawa, J. The Synthesis of Poly(Vinyl Alcohol) Grafted with Fluorinated Protic Ionic Liquids Containing Sulfo Functional Groups. Molecules 2021, 26, 4158. https://doi.org/10.3390/molecules26144158
Glińska P, Wolan A, Kujawski W, Rynkowska E, Kujawa J. The Synthesis of Poly(Vinyl Alcohol) Grafted with Fluorinated Protic Ionic Liquids Containing Sulfo Functional Groups. Molecules. 2021; 26(14):4158. https://doi.org/10.3390/molecules26144158
Chicago/Turabian StyleGlińska, Patrycja, Andrzej Wolan, Wojciech Kujawski, Edyta Rynkowska, and Joanna Kujawa. 2021. "The Synthesis of Poly(Vinyl Alcohol) Grafted with Fluorinated Protic Ionic Liquids Containing Sulfo Functional Groups" Molecules 26, no. 14: 4158. https://doi.org/10.3390/molecules26144158
APA StyleGlińska, P., Wolan, A., Kujawski, W., Rynkowska, E., & Kujawa, J. (2021). The Synthesis of Poly(Vinyl Alcohol) Grafted with Fluorinated Protic Ionic Liquids Containing Sulfo Functional Groups. Molecules, 26(14), 4158. https://doi.org/10.3390/molecules26144158