1. Introduction
Since the advent of the theoretical description of classical and quantum phase transitions (QPTs), long-range interactions between degrees of freedom challenged the established concepts and propelled the development of new ideas in the field [
1,
2,
3,
4,
5]. It is remarkable that, only a few years after the introduction of the renormalisation group (RG) theory by K.G. Wilson in 1971 as a tool to study phase transitions and as an explanation for universality classes [
6,
7,
8,
9,
10,
11], it was used to investigate ordering phase transitions with long-range interactions. These studies found that the criticality depends on the decay strength of the interaction [
1,
2,
3]. It then took two decades to develop numerical Monte Carlo (MC) tools capable of simulating basic magnetic long-range models with thermal phase transitions following the behaviour predicted by the RG theory [
12,
13]. The results of these simulations sparked a renewed interest in finite-size scaling above the upper critical dimension [
12,
14,
15,
16,
17,
18,
19] since “hyperscaling is violated” [
13] for long-range interactions that decay slowly enough. In this regime, the treatment of dangerous irrelevant variables (DIVs) in the scaling forms is required to extract critical exponents from finite systems.
Meanwhile, a similar historic development took place regarding the study of QPTs under the influence of long-range interactions. By virtue of pioneering RG studies [
20,
21], the numerical investigation of long-range interacting magnetic systems has been triggered [
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38]. In particular, Monte Carlo-based techniques became a popular tool to gain quantitative insight into these long-range interacting quantum magnets [
22,
25,
26,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40]. On the one hand, this includes high-order series expansion techniques, where classical Monte Carlo integration is applied for the graph embedding scheme, allowing extracting energies and observables in the thermodynamic limit [
25,
29]. On the other hand, there is stochastic series expansion quantum Monte Carlo [
39], which enables calculations on large finite systems. To determine the physical properties of the infinite system, finite-size scaling is performed with the results of these computations. Inspired by the recent developments for classical phase transitions [
15,
16,
17,
18,
19,
41], a theory for finite-size scaling above the upper critical dimension for QPTs was introduced [
32,
34].
When investigating algebraically decaying long-range interactions
with the distance
r and the dimension
d of the system, there are two distinct regimes: one for
(strong long-range interaction) and another one for
(weak long-range interaction) [
5,
42,
43,
44,
45]. In the case of strong long-range interactions, common definitions of internal energy and entropy in the thermodynamic limit are not applicable and standard thermodynamics breaks down [
5,
42,
43,
44,
45]. We will not focus on this regime in this review. For details specific to strong long-range interactions, we refer to other review articles such as Refs. [
5,
42,
43,
44,
45]. For the sake of this work, we restrict the discussion to weak long-range interaction or competing antiferromagnetic strong long-range interactions, for which an extensive ground-state energy can be defined without rescaling of the coupling constant [
5].
The interest in quantum magnets with long-range interactions is further fuelled by the relevance of these models in state-of-the-art quantum–optical platforms [
5,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86,
87]. To realise long-range interacting quantum lattice models with a tunable algebraic decay exponent, one can use trapped ions, which are coupled off-resonantly to motional degrees of freedom [
5,
81,
82,
83,
84,
85,
88]. Another possibility is to couple trapped neutral atoms to photonic modes of a cavity [
5,
86,
87]. Alternatively, one can realise long-range interactions decaying with a fixed algebraic decay exponent of six or three using Rydberg atom quantum simulators [
46,
47,
48,
49,
50,
51,
52,
53,
54,
55] or ultracold dipolar quantum atomic or molecular gases in optical lattices [
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73]. Note that, in many of the above-listed cases, it is possible to map the long-range interacting atomic degrees of freedom onto quantum spin models [
5,
52,
89]. Therefore, they can be exploited as analogue quantum simulators for long-range interacting quantum magnets, and the relevance of the theoretical concepts transcends the boundary between the fields.
From the perspective of condensed matter physics, there are multiple materials with relevant long-range interactions [
90,
91,
92,
93,
94,
95,
96,
97,
98,
99,
100,
101,
102,
103,
104,
105,
106]. The compound LiHoF
4 in an external field realises an Ising magnet in a transverse magnetic field [
102,
103,
104,
105]. A recent experiment with the two-dimensional Heisenberg ferromagnet Fe
3GeTe
2 demonstrates that phase transitions and continuous symmetry breaking can be implemented by circumventing the Hohenberg–Mermin–Wagner theorem with long-range interactions [
106]. This material is in the recently discovered material class of 2D magnetic van der Waals systems [
107,
108]. Further, dipolar interactions play a crucial role in the spin ice state in the frustrated magnetic pyrochlore materials Ho
2Ti
2O
7 and Dy
2Ti
2O
7 [
90,
91,
92,
93,
94,
95,
96,
97,
98,
99,
100,
101].
In this review, we are interested in physical systems described by quantum spin models, where the magnetic degrees of freedom are located on the sites of a lattice. We concentrate on the following three paradigmatic types of magnetic interactions between lattice sites: first, Ising interactions, where the magnetic interaction is oriented only in the direction of one quantisation axis; second, XY interactions with a -symmetric magnetic interaction invariant under planar rotations; and third, Heisenberg interactions with a -symmetric magnetic interaction invariant under rotations in 3D spin space. In the microscopic models of interest, a competition between magnetic ordering and trivial product states, external fields, or quasi-long-range order leads to QPTs.
In this context, the primary research pursuit revolves around how the properties of the QPT depend on the long-range interaction. The upper critical dimension of a QPT in magnetic models with non-competing algebraically decaying long-range interactions is known to depend on the decay exponent of the interaction for a small enough exponent, and decreases as the decay exponent decreases [
20,
21]. If the dimension of a system is equal to or exceeds the upper critical dimension, the QPT displays mean-field critical behaviour. At the same time, standard finite-size scaling, as well as standard hyperscaling relations are no longer applicable. Therefore, these systems are primary workhorse models to study finite-size scaling above the upper critical dimension. In this case, the numerical simulation of these systems is crucial in order to gauge novel theoretical developments. Further, QPTs in systems with competing long-range interactions do not tend to depend on the long-range nature of the interaction [
23,
24,
25,
26,
29,
30,
32]. In several cases, long-range interactions then lead to the emergence of ground states and QPTs, which are not present in the corresponding short-range interacting models [
27,
30,
54,
55,
109,
110,
111,
112].
In this review, we are mainly interested in the description and discussion of two Monte Carlo-based numerical techniques, which were successfully used to study the low-energy physics of long-range interacting quantum magnets, in particular with respect to the quantitative investigation of QPTs [
22,
25,
29,
30,
31,
32,
34,
35,
36,
37,
38,
40]. The success of Monte Carlo techniques in this field is due to the occurrence of high-dimensional sums and integrals that commonly arise in the formulation of many-particle statistics. In contrast to many deterministic integration techniques, for which the standard error scales exponentially with the dimension of the underlying integral, the standard error of an integral calculated with Monte Carlo integration does not scale with the dimension of the underlying integral. We further chose to review this topic due to our personal involvement with the application and development of these methods [
25,
29,
30,
31,
32,
34,
35]. On the one hand, we explain in detail how classical Monte Carlo integration can enhance the capabilities of linked-cluster expansions (LCEs) with the pCUT+MC approach (a combination of the perturbative unitary transform approach (pCUT) and MC embedding). On the other hand, we describe how stochastic series expansion (SSE) quantum Monte Carlo (QMC) integration is used to directly sample the thermodynamic properties of suitable long-range quantum magnets on finite systems.
This review is structured as follows. In
Section 2, we review the basic concept of a QPT in a condensed way, focusing on the details relevant for this review. We define the quantum-critical exponents and the relations between them in
Section 2.1. Here, we also have the first encounter with the generalised hyperscaling relation, which is also valid above the upper critical dimension where conventional hyperscaling breaks down. As the SSE QMC method discussed in this review is a finite-system simulation, we discuss the conventional finite-size scaling below the upper critical dimension in
Section 2.2 and the peculiarities of finite-size scaling above the upper critical dimension in
Section 2.3. In
Section 3, we summarise the basic concepts of Markov chain Monte Carlo integration: Monte Carlo sampling, Markovian random walks, stationary distributions, the detailed balance condition, and the Metropolis–Hastings algorithm. We continue by introducing the series-expansion Monte Carlo embedding method pCUT+MC in
Section 4. We start with the basic concepts of a graph expansion in
Section 4.1 and introduce the perturbative method of our choice, the perturbative continuous unitary transformation method, in
Section 4.2. We introduce the theoretical concepts for setting up a linked-cluster expansion as a full graph decomposition in
Section 4.3 and, subsequently, discuss how to practically calculate perturbative contributions in
Section 4.4 and
Section 4.5. We prepare the discussion of the white graph decomposition in
Section 4.6 with an interlude on the relevant graph theory in
Section 4.6.1 and
Section 4.6.2 and the important concept of white graphs in
Section 4.6.3. Further, in
Section 4.7, we discuss the embedding problem for the white graph contributions. Starting from the nearest-neighbour embedding problem in
Section 4.7.1, we generalise it to the long-range case in
Section 4.7.2 and then introduce a classical Monte Carlo algorithm to calculate the resulting high-dimensional sums in
Section 4.7.3. This is followed by some technical aspects on series extrapolations in
Section 4.8 and a summary of the entire workflow in
Section 4.9. In the next section, the topic changes towards the review of the SSE QMC method, which is an approach to simulate thermodynamic properties of suitable quantum many-body systems on finite systems at a finite temperature. First, we discuss the general concepts of the method in
Section 5. We review the algorithm to simulate arbitrary transverse-field Ising models introduced by A. Sandvik [
39] in
Section 5.1. We then review an algorithm used to simulate non-frustrated Heisenberg models in
Section 5.2. After the introduction to the algorithms, we summarise techniques on how to measure common observables in the SSE QMC scheme in
Section 5.3. Since the SSE QMC method is a finite-temperature method, we discuss how to rigorously use this scheme to perform simulations at effective zero temperature in
Section 5.4. We conclude this section with a brief summary of path integral Monte Carlo techniques used for systems with long-range interactions (see
Section 5.5). To maintain the balance between algorithmic aspects and their physical relevance, we summarise several theoretical and numerical results for quantum phase transitions in basics long-range interacting quantum spin models, for which the discussed Monte Carlo-based techniques provided significant results. First, we discuss long-range interacting transverse-field Ising models in
Section 6. For ferromagnetic interactions, this model displays three regimes of universality: a long-range mean-field regime for slowly decaying long-range interactions, an intermediate long-range non-trivial regime, and a regime of short-range universality for strong decaying long-range interactions. We discuss the theoretical origins of this behaviour in
Section 6.1.1 and numerical results for quantum critical exponents in
Section 6.1.2. Since this model is a prime example to study scaling above the upper critical dimension in the long-range mean-field regime, we emphasise these aspects in
Section 6.1.3. Further, we discuss the antiferromagnetic long-range transverse-field Ising model on bipartite lattices in
Section 6.2 and on non-bipartite lattices in
Section 6.3. The next obvious step is to change the symmetry of the magnetic interactions. Therefore, we turn to long-range interacting XY models in
Section 7 and Heisenberg models in
Section 8. We discuss the long-range interacting transverse-field XY chain in
Section 7 starting with the
-symmetric isotropic case in
Section 7.1, followed by the anisotropic case for ferromagnetic (see
Section 7.2) and antiferromagnetic (see
Section 7.3) interactions, which display similar behaviour to the long-range transverse-field Ising model on the chain discussed in
Section 6. We conclude the discussion of the results with unfrustrated long-range Heisenberg models in
Section 8. We focus on the staggered antiferromagnetic long-range Heisenberg square lattice bilayer model in
Section 8.1 followed by long-range Heisenberg ladders in
Section 8.2 and the long-range Heisenberg chain in
Section 8.3. We conclude in
Section 9 with a brief summary and with some comments on the next possible steps in the field.
2. Quantum Phase Transitions
This review is part of the Special Issue with the topic “Violations of Hyperscaling in Phase Transitions and Critical Phenomena”. In this work, we summarise investigations of low-dimensional quantum magnets with long-range interactions targeting, in particular, quantum phase transitions (QPTs) above the upper critical dimension, where the naive hyperscaling relation is no longer applicable. In this section, we recapitulate the relevant aspects of QPTs needed to discuss the results of the Monte Carlo-based numerical approaches. First, we give a general introduction to QPTs. After that, we discuss in detail the definition of critical exponents and the relations among them in
Section 2.1, as well as the scaling below (see
Section 2.2) and above (see
Section 2.3) the upper critical dimension.
Any non-analytic point of the ground-state energy of an infinite quantum system as a function of a tuning parameter
is identified with a QPT [
113]. This tuning parameter can, for instance, be a magnetic field or pressure, but not the temperature. Quantum phase transitions are a concept of zero temperature as there are no thermal fluctuations and all excited states are suppressed infinitely strong such that the system remains in its ground state. There are two scenarios for how a non-analytic point in the ground-state energy can emerge [
113]: First is an actual (sharp) level crossing between the ground-state energy and another energy level. Second, the non-analytic point can be considered as a limiting case of an avoided level crossing. Historically, phase transitions are classified by the lowest order derivative of the free energy that is discontinuous [
113,
114]. Therefore, a first-order phase transition is discontinuous in the order parameter (first derivative) and a second-order phase transition is discontinuous in the response functions (second derivative). Since, in second-order phase transitions, the order parameter is still continuous across the phase transition, we use the term “continuous phase transition” as an equivalent for “second-order phase transition”.
In this review, we are interested in second-order QPTs, which fall into the second scenario. At a second-order QPT, the relevant elementary excitations condense into a novel ground state, while the characteristic length and time scales diverge. Apart from topological QPTs involving a long-range entangled topological phase, such continuous transitions are described by the concept of spontaneous symmetry breaking. On one side of the QPT, the ground state obeys a symmetry of the Hamiltonian, while on the other side, this symmetry is broken in the ground state and a ground-state degeneracy arises.
Following the idea of the quantum-to-classical mapping [
113,
115],
d-dimensional quantum systems can be mapped in the vicinity of a second-order QPT to models of statistical mechanics with a classical (thermal) second-order phase transition in
dimensions. In many cases, the models obtained from a quantum-to-classical mapping are rather artificial [
113]. However, such mappings often allow categorising QPTs in terms of universality classes and associated critical exponents by the non-analytic behaviour of the classical counterparts [
10,
11,
113,
116,
117]. The mapping further illustrates that the renormalisation group (RG) theory is also applicable to describe QPTs [
10,
11,
113,
116,
117].
In the RG theory, each QPT belongs to a non-trivial fixed point of the RG transformation [
10,
11], whereas a trivial fixed point would, for instance, be a fully ordered state with maximal correlation or a fully disordered state with no correlation at all. Critical exponents are connected to the RG flow in the immediate vicinity of these non-trivial fixed points [
10,
11,
113]. The concept of universality classes arises from the fact that different microscopic Hamiltonians can have a quantum critical point that is attracted by the same non-trivial fixed point under successive application of the RG transformation [
10,
11]. Due to this, the QPTs in these models have the same critical exponents.
Another remarkable result of the RG theory is the scaling of observables in the vicinity of phase transitions. Historically, the theory of scaling at phase transitions was heuristically introduced before the RG approach [
118,
119,
120,
121,
122,
123,
124]. The latter provided the theoretical foundation for the scaling hypothesis [
6,
7]. The main statement of the scaling theory is that the non-analytic contributions to the free energy and correlation functions are mathematically described by generalised homogeneous functions (GHFs) [
124]. A function with
n variables
is called a GHF, if there exist
with at least one being non-zero and
such that, for
,
The exponents
are the scaling powers of the variables, and
is the scaling power of the function
f itself. An in-depth summary of the mathematical properties of GHFs can be found in
Appendix B. The most important properties of GHFs are that their derivatives, Legendre transforms, and Fourier transforms are also GHFs. As we will outline in
Section 2.2, the theory of finite-size scaling is formulated in terms of GHFs and relates the non-analytic behaviour at QPTs in the thermodynamic limit with the scaling of observables for different finite system sizes. In this, the variables
are related to physical parameters like the temperature
T, control parameter
, symmetry-breaking field
H, and also irrelevant, more abstract parameters that parameterise the microscopic details of the model like the lattice spacing. Later in this section, we will define irrelevant variables in the context of the RG and GHFs.
Another aspect relevant for this work is that quantum fluctuations are the driving factor with QPTs [
113]. In general, fluctuations are more important in low dimensions [
117]. The universality class of QPTs for a certain symmetry breaking depends on the dimensionality of the system.
An important aspect regarding this review is the so-called upper critical dimension
. The upper critical dimension is defined as a dimensional boundary such that, for systems with dimension
, the critical exponents are those obtained from mean-field considerations. The upper critical dimension is of particular importance for QPTs in systems with non-competing long-range interactions. For sufficiently small decay exponents of an algebraically decaying long-range interaction
, the upper critical dimension starts to decrease as a function of the decay exponent
[
20,
32,
34]. In the limiting case of a completely flat decay (
) of the long-range interaction resulting in an all-to-all coupling, the model is intrinsically of the mean-field type and mean-field considerations become exact. For a certain value of the decay exponent, the upper critical dimension becomes equal to the fixed spatial dimension, and for decay exponents below this value, the dimension of the system is above the upper critical dimension of the transition [
20,
32,
34]. This makes long-range interacting systems an ideal test bed for studying phase transitions above the upper critical dimension in low-dimensional systems. In particular, long-range interactions can make the upper critical dimension accessible in real-world experiments as the upper critical dimension of short-range models is usually not below three.
Although phase transitions above the upper critical dimension display mean-field criticality, they are still a matter worth studying, since naive scaling theory describing the behaviour of finite systems close to a phase transition (see
Section 2.2) is no longer applicable [
16,
19,
125]. Moreover, the naive versions of some relations between critical exponents, as discussed in
Section 2.1, do not hold any longer [
15,
16]. The reason for this issue are the dangerous irrelevant variables (DIVs) in the RG framework [
126,
127,
128]. During the application of the RG transformation, the original Hamiltonian is successively mapped to other Hamiltonians, which can have infinitely many couplings. All these couplings, in principle, enter the GHFs. In practice, all but a finite number of these couplings are set to zero since their scaling powers are negative, which means they flow to zero under renormalisation. These couplings are, therefore, called irrelevant. This approach of setting irrelevant couplings to zero can be used to derive the finite-size scaling behaviour as described in
Section 2.2. However, above the upper critical dimension, this approach breaks down because it is only possible to set irrelevant variables to zero if the GHF does not have a singularity in this limit [
126]. Above the upper critical dimension, such singularities in irrelevant parameters exist, which makes them DIVs [
127]. We explain the effect of DIVs on scaling in
Section 2.3.
2.1. Critical Exponents in the Thermodynamic Limit
As outlined above, a second-order QPT comes with a singularity in the free energy density. In fact, also, other observables experience singular behaviour at the critical point in the form of power-law singularities. For instance, the order parameter
m as a function of the control parameter
behaves as
in the ordered phase. Without loss of generality, the system is taken to be in the ordered phase for
and the notation
means that
is approaching
from below, i.e., it is approaching in the ordered phase. In the disordered phase
, the order parameter by definition vanishes such that
. The observables with their respective power-law singular behaviour, which is characterised by the critical exponents
, and
z, are summarised in
Table 1 together with how they are commonly defined in terms of the free energy density
f, the symmetry-breaking field
H, which couples to the order parameter, and the reduced control parameter
.
One usually defines reduced parameters like
r that vanish at the critical point not only to shorten the notation, but also to express the power-law singularities independent of the microscopic details of the specific model one is looking at. While the value of
depends on these details, the power-law singularities are empirically known to not depend on the microscopic details, but only on more general properties like the dimensionality, the symmetry that is being broken, and, with particular emphasis due to the focus of this review, on the range of the interaction. It is, therefore, common to classify continuous phase transitions in terms of universality classes. These universality classes share the same set of critical exponents. In terms of the RG, this behaviour is understood as distinct critical points of microscopically different models flowing to the same renormalisation group fixed point, which determines the criticality of the system [
6,
7,
10]. Prominent examples for universality classes of magnets are the 2D and 3D Ising (
symmetry), 3D XY (
symmetry), and 3D Heisenberg (
symmetry) universality classes [
113]. It is important to mention that the dimension in the classifications is referring to classical and not quantum systems, and they should not be confused with each other. In fact, the universality class of a short-range interacting non-frustrated quantum Ising model of dimension
d lies in the universality of the classical
-dimensional Ising model.
There are only a few dimensions for which a separate universality class is defined for the different models. For lower dimensions, the fluctuations are too strong in order for a spontaneous symmetry breaking to occur. In the case of the classical Ising model, there is no phase transition for 1D, while for the classical XY and Heisenberg models with continuous symmetries, there is not even a phase transitions for 2D due to the Hohenberg–Mermin–Wagner (HWM) theorem [
130,
131]. This dimensional boundary is referred to as lower critical dimension
. The lower critical dimension is the highest dimension for which no transition occurs, i.e.,
for the Ising model and
for the XY and Heisenberg model. For higher dimensions
, the critical exponents of the mentioned models do not depend on the dimensionality any longer, and they take on the mean-field critical exponents in all dimensions. The underlying reason is that, with increasing dimensions, the local fluctuations become smaller due to the higher connectivity of the system [
132]. This has been also exploited in diagrammatic and series expansions in
[
133,
134,
135]. This dimensional boundary, at which the criticality becomes the mean-field one, is called upper critical dimension
. Usually, the upper critical dimension is too large to realise a system above its upper critical dimension in the real world. However, long-rang interactions can increase the connectivity of a system in a similar sense as the dimensionality. A sufficiently long-range interaction can, therefore, lower the upper critical dimension to a value that is accessible in experiments.
Finally, it is worth mentioning that the critical exponents are not independent of each other, but obey certain relations [
129], namely
The first relation in Equation (
3) is the so-called hyperscaling relation, whose classical analogue (without
z) was introduced by Widom [
10,
136]. The Essam–Fisher relation in Equation (
4) [
137,
138] is reminiscent of a similar inequality, which was proven rigorously by Rushbrooke using thermodynamic stability arguments. Equation (
5) is called the Widom relation. The last relation in Equation (
6) is the Fisher scaling relation, which can be derived using the fluctuation–dissipation theorem [
10,
129,
138]. Those relations were originally obtained from scaling assumptions of observables close to the critical point, which were only later derived rigorously when the RG formalism introduced to critical phenomena [
10,
129]. Due to these relations, it is sufficient to calculate three, albeit not arbitrary, exponents to obtain the full set of critical exponents.
The hyperscaling relation Equation (
3) is the only relation containing the dimension of the system and is, therefore, often said to break down above the upper critical dimension, where one expects the same mean-field critical exponents independent of the dimension
d [
10]. It, therefore, deserves special focus in this review since the long-range models discussed will be above the upper critical dimension in certain parameter regimes. Personally, we would not agree that the hyperscaling relation breaks down above the upper critical dimension, but we would rather call Equation (
3) a special case of a more general hyperscaling relation:
with the pseudo-critical exponent
(“koppa”) [
34]:
Below the upper critical dimension, the general hyperscaling relation, therefore, relaxes to Equation (
3). Above the upper critical dimension, the relation becomes
which is independent of the dimension of the system. For the derivation of this generalised version of the hyperscaling relation for QPTs, see
Section 2.3 or Ref. [
34]. The derivation of the classical counterpart can be found in Ref. [
15] and is reviewed in Ref. [
41].
2.2. Finite-Size Scaling below the Upper Critical Dimension
Even though the singular behaviour of observables at the critical point is only present in the thermodynamic limit, it is possible to study the criticality of an infinite system by investigating their finite counterparts. In finite systems, the power-law singularities of the infinite system are rounded and shifted with respect to the critical point, e.g., the susceptibility with its characteristic divergence at the critical point
is deformed to a broadened peak of finite height. The peak’s position
is shifted with respect to the critical point
. A possible definition of a pseudo-critical point of a finite system is the peak position
. As the characteristic length scale of fluctuations
diverges at the critical point, the finite system approaching the critical point will at some point begin to “feel” its finite extent and the observables start to deviate from the ones in the thermodynamic limit. As
diverges with the exponent
like
at the critical point, the extent of rounding in a finite system is related to the value of
. Similarly, the peak magnitude of finite-size observables at the pseudo-critical point will depend on how strong the singularity in the thermodynamic limit is, which means it depends on the respective critical exponents
,
,
, and
. The shifting, rounding, and varying peak magnitude are shown for the susceptibility of the long-range transverse-field Ising model in
Figure 1. This dependence of observables in finite systems on the criticality of the infinite system is the basis of finite-size scaling.
In a more mathematical sense, the relation between critical exponents and finite-size observables has its origin in the renormalisation group (RG) flow close to the corresponding RG fixed point that determines the criticality [
139]. Close to this fixed point, one can linearise the RG flow so that the free energy density and the characteristic length
become generalised homogeneous functions (GHFs) in their parameters [
124,
128,
129,
140,
141]. For a thorough discussion of the mathematical properties of GHFs, we refer to Ref. [
124] and
Appendix B. This means that the free energy density
f and characteristic length scale
as functions of the couplings
and the inverse system length
obey the relations:
with the respective scaling dimensions
,
, and
governing the linearised RG flow with spatial rescaling factor
around the RG fixed point, at which all couplings vanish by definition. All of those couplings are relevant except for
u, which denotes the leading irrelevant coupling [
10,
113]. Relevant couplings are related to real-world parameters that can be used to tune our system away from the critical point like the temperature
T, a symmetry-breaking field
H, or simply the control parameter
r. The irrelevant couplings
u do not per se vanish at the critical point like the relevant ones do. However, they flow to zero under the RG transformation and are commonly set to zero in the scaling laws:
by assuming analyticity in these parameters. The generalised homogeneity of thermodynamic observables can be derived from the one of the free energy density
f. For example, the generalised homogeneity of the magnetisation:
can be derived by taking the derivative of
f with respect to the symmetry-breaking field
H.
By investigating the singularity of
in
r via Equation (
13), one can show that the scaling power
of the control parameter
r is related to the critical exponent
by
[
113]. For this, one fixes the value
of the first argument to
in the right-hand side of Equation (
13) by setting
such that
Analogously, further relations between the scaling powers and other critical exponents can be derived by looking at the singular behaviour of the respective observables in the corresponding parameters. Overall, one further obtains
From these equations, one can already tell that the critical exponents are not independent of each other. In fact, the scaling relations
,
and
(see Equations (
3)–(
5)) can be derived from Equation (
16) and
. By expressing the RG scaling powers
in terms of critical exponents, the homogeneity law for an observable
with a bulk divergence
is given by
In order to investigate the dependence on the linear system size
L, the last argument in the homogeneity law is fixed to
by inserting
. This readily gives the finite-size scaling form
with
being the universal scaling function of the observables
. The scaling function
itself does not depend on
L any longer, but in order to compare different linear system sizes, one has to rescale its arguments. To extract the critical exponents from finite systems, the observable
is measured for different system sizes
L and parameters
close to the critical point
. The
L-dependence according to Equation (
19) is then fit with the critical exponents
,
,
, and
z, as well as the critical point
, which is hidden in the definition of
r, as free parameters. It is advisable to fix two of the three parameters
to their critical values in order to minimise the amount of free parameters in the fit. For example, with
and only
, one can extract the two critical exponents
and
alongside
. For further details on a fitting procedure, we refer to Ref. [
32]. If one knows the critical point, one can also set
and look at the
L-dependent scaling
directly at the critical point to extract the exponent ratio
. There are many more possible approaches to extract critical exponents from the FSS law in Equation (
19) [
142,
143,
144]. For relatively small system sizes, it might be required to take corrections to scaling into account [
143,
144].
2.3. Finite-Size Scaling above the Upper Critical Dimension
In the derivation of finite-size scaling below the upper critical dimension, it was assumed that the free energy density is an analytic function in the leading irrelevant coupling u and, therefore, one can set it to zero. However, this is not the case above the upper critical dimension any longer, and the free energy density f is singular at . Due to this singular behaviour u is referred to as a dangerous irrelevant variable (DIV).
As a consequence, one has to take the scaling of
u close to the RG fixed point into account. This is achieved by absorbing the scaling of
f in
u for small
u into the scaling of the other variables [
128]:
up to a global power
of
u. This leads to a modification of the scaling powers in the homogeneity law for the free energy density [
128]:
with the modified scaling powers [
34,
128]:
In the classical case [
128],
was commonly set to 1 by choice. This is justified because the scaling powers of a GHF are only determined up to a common non-zero factor [
124]. However, for the quantum case [
34], this was kept general as it has no impact on the FSS.
As the predictions from the Gaussian field theory and mean field differed for the critical exponents
,
, and
, but not for the “correlation” critical exponents
,
z,
, and
[
145], the correlation sector was thought not to be affected by DIVs at first [
128,
142,
145]. Later, the Q-FSS, another approach to FSS above the upper critical dimension, pioneered by Ralph Kenna and his colleagues, was developed for classical [
15,
19,
125], as well as for quantum systems [
34], which explicitly allowed the correlation sector to also be affected by the DIV. In analogy to the free energy density, the homogeneity law of the characteristic length scale is then also modified to
with
in order to reproduce the correct bulk singularity
. A new pseudo-critical exponent
(“koppa”):
is introduced. This exponent describes the scaling of the characteristic length scale with the linear system size. This non-linear scaling of
with
L is one of the key differences to the previous treatments above the upper critical dimension in Ref. [
128].
Analogous to the case below the upper critical dimension, the modified scaling powers
can be related to the critical exponents:
By using the mean-field critical exponents for the
quantum rotor model, one obtains restrictions for the ratios of modified scaling powers:
Furthermore, one can link the bulk scaling powers
, and
to the scaling power
of the inverse linear system size [
34]:
by looking at the scaling of the susceptibility in a finite system [
34,
128]. This relation is crucial for deriving an FSS form above the upper critical dimension as the modified scaling power
, or rather, its relation to the other scaling powers determines the scaling with the linear system size
L. For details on the derivation, we refer to Ref. [
34]. We want to stress again that the scaling powers of GHFs are only determined up to a common non-zero factor [
124]. Therefore, it is evident that one can only determine the ratios of the modified scaling powers, but not their absolute value. The absolute values are subject to choice. Different choices were discussed in Ref. [
34], but these choices rather correspond to taking on different perspectives and have no impact on the FSS nor the physics.
From Equation (
32) together with Equations (
26) and (
27), a generalised hyperscaling relation:
can be derived. This also determines the pseudo-critical exponent:
Finally, we can express the modified scaling powers in the FSS law for an observable
with power-law singularity
:
For
below the upper critical dimension, Equation (
36) relaxes to the standard FSS law Equation (
19). The scaling in temperature has not yet been studied for finite quantum systems above the upper critical dimension. However, in Ref. [
34], it was conjectured that
based on Equation (
32), which is also in agreement with
z being the dynamical critical exponent that determines the space–time anisotropy
, as we will shortly see. This means that the finite-size gap scales as
with the system size [
34]. Of particular interest is the scaling of the characteristic length scale above the upper critical dimension, for which the modified scaling law Equation (
36) also holds with
. Hence, the characteristic length scale in dependence of the control parameter
r scales like
with the scaling function
. Directly at the critical point
, this leads to
. Comparing this with the scaling of the inverse finite-size gap
verifies that
z still determines the space–time anisotropy. Prior to the Q-FSS [
15,
17], the characteristic length scale was thought to be bound by the linear system size
L [
128]. However, this was shown not to be the case by measurements of the characteristic length scale for the classical five-dimensional Ising model [
14] and for long-range transverse-field Ising models [
34].
For the latter, the data collapse of the correlation length according to Equation (
37) is shown in
Figure 2 as an example.
3. Monte Carlo Integration
In this section, we provide a brief introduction to Monte Carlo integration (MCI). We focus on the aspects of Markov chain MCI as the basis to formulate the white graph Monte Carlo embedding scheme of the pCUT+MC method in
Section 4 and the stochastic series expansion (SSE) quantum Monte Carlo (QMC) algorithm in
Section 5 in a self-contained fashion. MCI is the foundation for countless numerical applications, which require the integration over high-dimensional integration spaces. As this review has a focus on “Monte Carlo-based techniques for quantum magnets with long-range interactions”, we forward readers with a deeper interest in the fundamental aspects of MCI and Markov chains to Refs. [
146,
147,
148].
MCI summarises numerical techniques to find estimators for integrals of functions
over an integration space
using random numbers. The underlying idea behind MCI is to estimate the integral, or the sum in the case of discrete variables, of the function
f over the configuration space by an expectation value:
with
sampled according to a probability density function
(PDF) and the function
reweighted by the PDF. A famous direct application of this idea is the calculation of the number “pi”, which is discussed in great detail in Ref. [
148].
In this review, MCI is used for the embedding of white graphs on a lattice to evaluate high orders of a perturbative series expansion or to calculate thermodynamic observables using the SSE framework. In both cases, non-normalised relative weights
within a configuration space
arise, which are used for the sampling of the PDF
P:
being oftentimes not directly accessible. In the context of statistical physics,
is often chosen to be the relative Boltzmann weight
of each configuration
. While this relative Boltzmann weight is accessible as long as
is known, the full partition function to normalise the weights is in general not.
In order to efficiently sample the configuration space
according to the relative weights, the methods in this review use a Markovian random walk to generate
. Let
be the random state of a random walk at a discrete step
n. The state
at the next step is randomly determined according to the conditional probabilities
(transition probabilities). These transition probabilities are normalised by
Markovian random walks obey the Markov property, which means the random walk is memory-free and the transition probability for multiple steps factorises into a product over all time steps:
with
the start configuration. We require the Markovian random walk to fulfil the following conditions: First, the random walk should have a certain PDF
defined by the weights
in Equation (
39) as a stationary distribution. By definition,
is a stationary distribution of the Markov chain if it satisfies the global balance condition:
Second, we require the random walk to be irreducible, which means that the transition graph must be connected and every configuration
can be reached from any configuration
in a finite number of steps. This property is necessary for the uniqueness of the stationary distribution [
147]. Lastly, we require the random walk to be aperiodic (see Ref. [
147] for a rigorous definition). Together with the irreducibility condition, this ensures convergence to the stationary distribution [
147].
There are several possibilities to design a Markov chain with a desired stationary distribution [
146,
147,
148,
149,
150,
151]. Commonly, the Markov chain is constructed to be reversible. This means that it satisfies the detailed balance condition:
which is a stronger condition for the stationarity of
P than the global balance condition in Equation (
42). One popular choice for the transition probabilities
that satisfies the detailed balance condition is given by the Metropolis–Hastings algorithm. Most applications of MCI reviewed in this work are based on the Metropolis–Hastings algorithm [
149,
150]. In this approach, the transition probabilities
are decomposed into propositions
and acceptance probabilities
as follows:
The probabilities to propose a move
can be any random walk satisfying the irreducibility and aperiodicity condition. By inserting the decomposition of the transition probabilities Equation (
44) into the detailed balance condition Equation (
43), one obtains for the acceptance probabilities:
where, in the last step, the idea that the unknown normalisation factors (see Equation (
39)) of the PDF cancel was used. The condition in Equation (
45) is fulfilled by the Metropolis–Hastings acceptance probabilities [
150]:
For the special case, for which the proposition probabilities are symmetric
, Equation (
46) reduces to the Metropolis acceptance probabilities:
As an example, we regard a classical thermodynamic system with Boltzmann weights
given by the energies of configurations and the inverse temperature to give an intuitive interpretation of the Metropolis acceptance probabilities in Equation (
47). The proposition to move from a configuration
to a configuration
with a smaller energy
is always accepted independent of the temperature. On the other hand, the proposition to move to a configuration
with a larger energy than
is only accepted with a probability depending on the ratio of the Boltzmann weights. If the temperature is higher, it is more likely to move to states with a larger energy. This reflects the physics of the system in the algorithm, focusing on the low-energy states at low temperatures and going to the maximum entropy state at large temperatures.
5. Stochastic Series Expansion Quantum Monte Carlo
In this section, we discuss the method of stochastic series expansion (SSE) QMC. This class of QMC algorithms is closely related to path integral (PI) QMC and samples configurations according to the Boltzmann distribution of a quantum mechanical Hamiltonian. This sampling is achieved by extending the configuration space in the imaginary-time direction by operator sequences. The objective is to evaluate thermal expectation values for operators at a finite temperature on a finite system.
The canonical partition function of a system with a quantum mechanical Hamiltonian
can be expressed as
with
being the inverse temperature and the sum over an arbitrary orthonormal basis
. The task is to bring Equation (
211) into the form of
with all weights
required to be non-negative.
Of course, for a Hamiltonian that is traced over in its eigenbasis (or a classical system), Equation (
211) is already in the form of Equation (
212), and the system can be directly sampled by a Metropolis–Hastings algorithm (see Equation (
46)). For a general quantum mechanical problem, we do not have access to the eigenstates of a system and require a reformulation of Equation (
211).
The SSE QMC idea resolves this issue in the following way: Given a Hamiltonian
, a computational orthonormal basis
is chosen in which the trace is evaluated. Further, there should exist a decomposition of the Hamiltonian:
into operators
.
and
are chosen such that the following two conditions are met:
No-branching rule:
ensuring that no superpositions of basis states are created by acting with
.
Non-negative real matrix elements in the computational basis:
The second condition is not strictly necessary, but makes sure that no sign problem arises, which would lead to exponentially hard computational complexity. In general, it is not necessarily possible to find a computational basis in which this condition can be fulfilled for all Hamiltonians. However, if the negative matrix elements contribute in such a way that they always occur in pairs and the minus signs cancel, the condition can be relaxed without inducing a sign problem. We will encounter this case for the antiferromagnetic Heisenberg models in
Section 5.2.
For operators
that are diagonal in the computational basis, the conditions (
214) and (
215) never pose a problem as diagonal operators intrinsically obey the no-branching rule and can always be made non-negative by adding a suitable constant to the Hamiltonian. The main difficulty is to find a computational basis in which the off-diagonal matrix elements are non-negative, which is not necessarily possible, as mentioned above. In particular, for fermionic or frustrated systems, negative signs typically occur.
In order to reformulate Equation (
211) in the form of Equation (
212), a high-temperature expansion for the partition function is performed:
In general, the evaluation of
is not feasible. The way the SSE tackles this expression is by expanding the product of sums as
as a sum over all occurring operator sequences
resulting from the exponentiation. The additional dimension created by the operator sequence is usually referred to as imaginary time in analogy to path-integral formulations. Inserting Equation (
219) into Equation (
218), one obtains
Note that each of the summands in Equation (
220) is non-negative by design due to the condition (
215) and can be interpreted as the relative weight of a configuration. By comparing Equation (
220) with Equation (
212), we see that it is of a suitable form for a Markov chain Monte Carlo sampling. We identify the direct product of the set of all basis states
with the set of all sequences as the configuration space:
The weight of a configuration is given by
In the next step, we discuss the structure of each configuration consisting of a computational basis state
and an operator sequence
. Regarding Equation (
220), we stress that the action of the product of operators in the sequence onto the basis state is crucial. Due to the no-branching rule (see Equation (
214)), the action of
onto the basis state can be interpreted as a discrete propagation of the state
in imaginary time according to the operators in the sequence. We define the state at propagation index
to be
Due to the periodic boundary condition of the trace and the orthogonality of the basis states
, only sequences for which
have a non-zero weight (see Equation (
222)).
At this point, we can demonstrate why it does not matter if some matrix elements are negative in some instances, e.g., for some antiferromagnetic spin models on bipartite lattices. If a matrix element of an operator is negative, but it is ensured by the Hamiltonian and the periodicity of the trace that there is always an even number of negative matrix elements in a sequence with non-vanishing weight, then the definition of weights as in Equation (
222) is nevertheless possible.
In the discussion so far, we considered sequences of all possible lengths. In order to formulate algorithms sampling the configuration space efficiently, a scheme with a fixed sequence length
can be introduced, in which all sequences with
are padded with identity operators
to length
and all sequences
are discarded. The physical justification to discard all sequences above a certain fixed length
is that they are exponentially suppressed for sufficiently large
. In short, this is the case because the mean operator number
is proportional to the mean energy
and the variance is related to the specific heat in the following way:
A derivation and discussion of these statements can be found in Refs. [
39,
210,
211,
212,
213]. From Equation (
224), we can infer that the infinite sum over all sequence lengths can be truncated at a finite
. From rearranging Equation (
224) and using the extensivity of the mean energy, the mean sequence length scales as
proportional to the inverse temperature and system size
N. As the mean sequence length has a finite value, the idea is to choose an
great enough to be able to sample all but a negligible amount of operator sequences. Further, we will argue that the introduction of a large enough cutoff results in an exponentially small and negligible error. In the limit of
, the specific heat has to vanish. Therefore, the variance of the mean sequence length is proportional to
. From this, it is concluded that the weights of sequences vanish exponentially for large enough sequence lengths
n [
212].
We, therefore, introduce a large enough cutoff for the sequence length
and consider operator sequences with a fixed length
. The expression for the partition function can then be written as
The new configuration space includes all sequences of length
where the shorter sequences are padded by inserting unity operators. The random insertion of unity operators into a sequence of
non-trivial operators results in
sequences of length
. The modified prefactor in Equation (
225) accounts for this overcounting. Although Equation (
225) is, strictly speaking, an approximation to the partition function, we want to note that, in practice, this does not cause a systematic error as
can be chosen large enough in a dynamical fashion such that, during the finite simulation time, no sequence of length
would occur. Therefore, we will not consider the fixed-length scheme as an approximation below.
To summarise: Following the argumentation discussed in this section, it is possible to bring any partition function into the form of Equation (
225), whereby all the summands are non-negative if a suitable decomposition and computational basis can be found. The next step is to implement a Markov chain MC sampling on the configuration space. As the configuration space largely depends on the model, the sampling is performed in a model-dependent way. In
Section 5.1, we will introduce an algorithm to sample the (LR)TFIM. In
Section 5.2, we will further describe an algorithm to sample unfrustrated (long-range) Heisenberg models. In
Section 5.3, the measurement of a variety of operators is discussed. Further, we will discuss how to sample systems at effectively zero temperature in
Section 5.4 as this review considers the zero-temperature physics of quantum phase transitions. In
Section 5.5, we give a short overview of other MC algorithms for long-range models based on path integrals.
5.1. Algorithm for Arbitrary Transverse-Field Ising Models
In this section, we describe the SSE algorithm to sample arbitrary transverse-field Ising models (TFIMs) of the form
as introduced by A. Sandvik in Ref. [
39]. The Pauli matrices
with
describe
N spins 1/2 located on the lattice sites
. The transverse-field strength at a lattice site
i is
, and the Ising couplings between sites
i and
j have the strength
. For
, the interaction is antiferromagnetic and it is energetically favourable for the spins to anti-align, while for
, the interaction is ferromagnetic and it is energetically favourable for the spins to align.
Choosing the
-eigenbasis
for the SSE formulation avoids the sign problem for arbitrary Ising interactions. The Hamiltonian is decomposed using the following operators:
We call
a trivial operator,
a field operator,
a constant operator, and
an Ising operator. The operator
is associated with the site
i, even though it is proportional to
. With these operators, we can rewrite Equation (
226) up to an irrelevant constant as
Note that Equation (
231) is a sum over field, constant, and Ising operators. The constant operators are not part of the original Hamiltonian, but will be relevant for algorithmic purposes. The trivial operators (Equation (
227)) are not relevant to express the Hamiltonian, but are necessary for the fixed-length sampling scheme. The proposed decomposition fulfils the no-branching property, and there are no negative matrix elements of the operators in the computational basis due to the positive constant
that is added to the Ising operators. The possible matrix elements for Ising operators
acting on a pair of spins
are given by
This implies that only sequences where (anti)ferromagnetic Ising bonds are placed on (anti)aligned spins have a non-vanishing weight.
The partition function (Equation (
225)) in the fixed-length scheme reads
The propagated states
only change in the imaginary-time propagation if a field operator, the only off-diagonal operator in the chosen basis, acts on it. A configuration only has a non-zero weight if all the Ising operators in the sequence are placed on spins that have the correct alignment. Constant operators are included in the Hamiltonian for algorithmic purposes, which will become important in the off-diagonal update described below.
Before going into the description of the Markov chain to sample the configuration space, it is illustrative to visualise a configuration with non-vanishing weight defined by a state
and an operator sequence
. In
Figure 14, an exemplary configuration with non-vanishing weight is illustrated for the one-dimensional TFIM and visualisations of the different operators Equations (
228)–(
230) in such a configuration are shown.
Before going into the details of the algorithm, we introduce the main concept of the update scheme and the crucial obstacles that are encountered in setting up an efficient algorithm [
39]. Each step of the Markov chain sampling of configurations is performed by performing a so-called diagonal update followed by an off-diagonal quantum cluster update [
39]. In the diagonal update, one iterates over the sequence and exchanges trivial operators with constant or Ising operators and vice versa while propagating the states along the sequence. In the off-diagonal update, constant operators are exchanged with field operators while preserving the weight of the configuration. The main obstacle that is circumvented with this update procedure is the following: It is a non-trivial and non-local task to insert off-diagonal operators into the sequence without creating a configuration with vanishing weight. The first problem is that one cannot simply insert a single field operator into the sequence as it breaks the periodicity of the propagated states
, leading to a vanishing weight due to the orthonormality of the computational basis. Therefore, field operators can only occur in an even number for each site to preserve the periodicity in imaginary time. The second issue involves Ising operators placed on a pair of sites
i and
j. If one places a field operator at one of the sites before and behind the Ising operator in the sequence, this preserves the periodicity in imaginary time, but the matrix element of the Ising operator becomes zero as the spins will be misaligned with respect to the sign of the Ising coupling (see Equation (
232)). These issues can be tackled by a non-local off-diagonal update [
39], which we will thoroughly discuss after the diagonal update.
In the diagonal update, the number of non-trivial operators
n is altered by exchanging trivial operators with constant and Ising operators in the operator sequence and vice versa. This does not change the states
, including the state
. Starting from propagation step
and state
, one iterates over the sequence step-by-step and conducts the exchange of trivial operators with non-trivial diagonal operators. If a field operator is encountered at the current propagation step, the state is propagated by flipping the respective spin, and the iteration proceeds. If a diagonal operator is encountered, a local update following the Metropolis–Hastings algorithm as described in
Section 3 is performed and the total transition probability is made of a proposition probability
and an acceptance probability
.
If a trivial operator is encountered at propagation step
k, a non-trivial diagonal operator gets proposed with the probability
taking into account the weight
of the proposed operator with the normalising constant
C:
The weight is essentially given by the matrix elements of the respective operators
with the special case that the Ising operators are a priori handled as if they were allowed. At a later stage, it is checked if the spins are correctly aligned at the considered propagation step
k and state
, and the operator gets rejected if this is not the case. This has the benefit that one does not need to check every single bond for correct alignment for the insertion of a single operator, which scales like
for long-range models. This sampling can be performed in a constant time complexity in the number of elements in the distribution by using the so-called walker method of aliases [
214]. Instructions on how to set up a walker sampler from a distribution of discrete weights and how to draw from this distribution can be found in
Appendix C or in Ref. [
214]. Drawing from the discrete distribution of weights, an operator
gets proposed to be inserted. On the other side, if a non-trivial diagonal operator is encountered, it is always proposed to be replaced by a trivial operator:
The acceptance probabilities are then chosen according to the Metropolis–Hastings algorithm in Equation (
46). For a constant field operator
, this gives the acceptance probability:
Similarly, for an Ising operator
, one has
up to a factor
, which is either 1 or 0 depending on if the spins at the current propagation step
k are correctly aligned or misaligned. An Ising bond with misaligned spins would lead to a vanishing weight of the configuration and is, therefore, not allowed.
Up to this factor, the acceptance probability is the same independent of the type of non-trivial diagonal operator that is proposed to be inserted as the operator weight arising in the weight for the newly proposed configuration cancels with the same factor in the proposition probability. This is because we already chose to propose an operator by considering its respective weight . As the acceptance probabilities do not differ, one can, therefore, also first check if one accepts to insert any non-trivial diagonal operator and, only if this is accepted, draw the precise operator to be inserted. Of course, if the chosen operator is an Ising operator, it still has to be checked if the spins are correctly aligned to prevent a non-vanishing weight.
The cancellation of the operator weights
also makes it easier to perform the reverse process and replace a non-trivial diagonal operator with a trivial one in the sense that the acceptance probability for inserting
:
does not depend on the current non-trivial diagonal operator
.
If a proposition gets rejected, the iteration along the sequence continues and the procedure starts again for the next operator. After each diagonal update sweep (iteration over the whole sequence) during the equilibration, trivial operators are appended to the end of the sequence such that
[
39]. This allows dynamically adjusting the sequence length
for the fixed-length scheme to ensure a sufficiently long sequence.
For an efficient implementation of the off-diagonal quantum cluster update, it is crucial to introduce the concept of operator legs. An operator at position
p in the sequence has legs with the numbers
. For an Ising operator, these legs are associated with two legs per site, one upper and one lower leg (see
Figure 15a).
For a constant or field operator, only two of the four legs are real vertex legs since these operators act only on a single site (see
Figure 15b). The remaining two legs are called ghost legs and are considered solely due to algorithmic reasons as it is numerically beneficial to let every operator have the same number of legs. This allows calculating the propagation index
p of an operator from the leg numbers using integer division with
. Trivial operators can be described with four ghost legs or can be ignored entirely in the sequence for the off-diagonal update.
For the chosen representation of the Ising model within the SSE, one can subdivide the configuration into disjoint clusters that extend in space, as well as in imaginary time with Ising operators acting as bridges in real space and with constant and field operators acting as delimiters in imaginary time. These clusters of spins can be flipped by replacing the delimiting constant operators with field operators and vice versa without changing the weight . If the cluster winds around the boundary in imaginary time, the respective spins in state have to be updated as well. In order to flip half of the configuration on average and obtain a good mixing, all clusters are constructed and the probability to flip a specific cluster is chosen to be for each cluster separately.
For the construction of the clusters, the propagated spin states along imaginary time are not needed. The entire problem can be dealt with in the language of vertices with legs. The relevant information for the cluster formation is which legs are connected. This information about the connection between legs of different operators is stored in a doubly linked list. The list is set up in the following way: At the index of a vertex leg
i, we store the index
j of the leg it is connected to, i.e.,
and vice versa
. This segmentation is illustrated for an exemplary configuration in
Figure 16 together with the doubly linked leg list for the depicted configuration. The doubly linked list can already be set up during the diagonal update when the sequence is traversed either way. An efficient algorithm to set up the data structure is described in Refs. [
213,
215].
In comparison to general off-diagonal loop updates [
215,
216,
217], the formation of clusters in the presented off-diagonal cluster update for the TFIM is fully deterministic. The whole configuration is divided into disjoint clusters, all of which will be built and flipped with probability
. It is beneficial to already decide whether a cluster is flipped or not before constructing the cluster so the constant and field operators and spin states can already be processed during the construction. A leg that is processed during the construction of a cluster is marked as visited in order to not process the same leg twice. This is also the reason why all the clusters have to be constructed even if they will not be flipped, as otherwise, the same cluster would get constructed starting from another leg later on. Further, we introduce a stack for the legs that were visited during the construction, but whose connections are yet to be processed.
The formation of each cluster starts by choosing a leg that has not yet been visited in the current off-diagonal update. At the beginning of the cluster formation, it is randomly determined if the cluster is flipped or not. If the leg corresponds to an Ising vertex, the cluster branches out to all four legs of the vertex. This means that all four legs of the operator are put on the stack. If the leg corresponds to a constant or field operator, the type of operator is exchanged if the cluster is flipped and only this primal leg is put on the stack. Next, the following logic is repeated until the stack is empty. We pop a leg l from the stack. If the leg has been visited already, we continue with a new leg from the stack. Else, we determine the new leg using the doubly linked list. We mark both legs l and as visited. If one passes by the periodic boundary in imaginary time while going from leg l to and the cluster will be flipped, the corresponding site belonging to the legs in the state has to be flipped. If belongs to an Ising operator, we add all legs of the Ising operator that have not been visited yet to the stack. If belongs to a constant or field operator, we exchange the operator if the cluster is said to be flipped. This procedure is repeated until the stack is empty. After that, one proceeds with the next cluster starting from a leg that has not yet been visited in the current cluster update. If no such leg is left, the cluster update is finished.
In addition to this cluster update, spins that have no operators acting on them in the sequence can be flipped with a probability of (thermal spin flip).
In summary, the off-diagonal update exchanges field and constant operators with each other and changes the state . Combining the diagonal update with the off-diagonal cluster update samples the entire configuration space.
5.2. Algorithm for Unfrustrated Heisenberg Models
In this section, we describe the algorithm to sample arbitrary unfrustrated spin-1/2 Heisenberg models with the SSE framework [
213]. By unfrustrated Heisenberg models, we mean Hamiltonians:
written as a sum over three-component interactions between two sites
and
connected by bond
b, where each bond is either ferromagnetic (F) with
or antiferromagnetic (AF) with
, with the property that there is no loop of lattice sites connected by bonds that contains an odd number of antiferromagnetic bonds, as this would lead to frustration. The spin operators are a compact notation for a three-component vector of spin operators
. The coupling strengths
can have a priori arbitrary amplitudes. It is crucial to look at unfrustrated Heisenberg models in order to define non-negative weights for configurations as the off-diagonal components of the antiferromagnetic operators have a negative matrix element. In an unfrustrated model, these matrix elements always occur an even number of times in the operator sequence of any valid configuration, which makes it possible to construct a non-negative SSE weight [
213].
For the SSE algorithm, the
-eigenbasis
is chosen, but the
- or
-eigenbasis would work the same way. The Hamiltonian is decomposed into
using the following operators:
The diagonal operators
and
in Equations (
246) and (
248) are defined in the same fashion as for the TFIM up to the factor of
due to the usage of spin operators
instead of Pauli matrices
with
. The contribution to the weight of a sequence of these operators is either
if the bond fulfils the (anti)ferromagnetic condition and zero otherwise. Although the expressions for the ferromagnetic and antiferromagnetic bonds look the same, we distinguish between these two bonds to highlight that these objects behave differently within the off-diagonal update.
As
for antiferromagnetic bonds, the off-diagonal operators
do not fulfil the non-negativity of matrix elements (see Equation (
215)) in the computational basis. Therefore, they must always appear in an even number of times in the operator sequence to avoid the sign problem. For the ferromagnetic bonds, this restriction is not necessary.
Analogous to the TFIM, the operator sequence additionally contains trivial operators
, which are not part of the Hamiltonian, but are used to pad the sequence to a fixed length
. In contrast to the TFIM, there is no need for further artificial operators like the constant field operators in the TFIM used to limit the cluster in the cluster update. In the case of the Heisenberg model, the non-local off-diagonal update is constructed in the form of loops instead of a cluster with several branches that need to be limited in imaginary time. An exemplary SSE configuration for a Heisenberg chain using the decomposition from above can be seen in
Figure 17.
Similar to the sampling of the TFIM in
Section 5.1, each step of the Markov chain sampling of configurations is achieved by performing a diagonal update followed by a non-local off-diagonal update. The diagonal update exchanges trivial operators with diagonal operators, while the off-diagonal update exchanges diagonal bond operators with the respective off-diagonal bond operators.
Due to the structure of the diagonal operators (see Equations (
246) and (
248)), similar to the TFIM, the diagonal update is performed similarly to the diagonal update of the TFIM described in
Section 5.1. Nonetheless, to make the description of the algorithm for unfrustrated Heisenberg models self-contained, we recapitulate the diagonal update and adapt it to the Heisenberg case.
The diagonal update exchanges trivial operators with diagonal operators in the sequence and vice versa. It will, therefore, not change the states
, including the state
. Starting from
at
, the sequence is again iterated over by
k. If the operator at the current position
k in the sequence is an off-diagonal operator operator, the state
is propagated by applying the off-diagonal operator and the iteration proceeds. If one encounters diagonal or trivial operators in the sequence, the Metropolis–Hastings algorithm (see
Section 3) is performed and the transition probability to an altered configuration is again split into a proposition probability
and an acceptance probability
.
Analogous to the algorithm for the TFIM, if a trivial operator is encountered, a non-trivial diagonal operator gets proposed with the probability:
taking into account the weight
of the proposed operator and the normalising constant
C:
The diagonal update for the Heisenberg model only differs from the one of the TFIM by the detailed probabilities
and the normalising constant
C. The factor of
in the bond weights in comparison to the Ising model comes from using spin operators in contrast to Pauli matrices in the Hamiltonian. This sampling according to the weight
can be performed in a constant-time complexity in the number of elements in the distribution by using the walker method of aliases [
214]. An introduction on how to set up a walker sampler from a distribution of discrete weights and how to draw from this distribution can be found in
Appendix C or in Ref. [
214]. Drawing from the discrete distribution of weights, an operator
gets proposed to be inserted. On the other side, if a non-trivial diagonal operator is encountered, it is always proposed to be replaced by a trivial operator:
exactly as for the TFIM. The acceptance probabilities are then chosen according to the Metropolis–Hastings algorithm Equation (
46). The respective Metropolis–Hastings probabilities with which the proposed replacements at position
k in the operator sequence are accepted are given by the ratios of the configuration weights before and after the potential replacement and the ratio of the proposition probabilities:
As in the case of the TFIM, the specific matrix element for the propagated states and has to be considered, which is either , and cancels with this factor in the acceptance probability, or is 0, and the acceptance probability vanishes. Intuitively, this factor simply checks if the spins are correctly aligned or misaligned with respect to the sign of the coupling and only allows the operator to be inserted if the spins and of the propagated state are correctly aligned. If the proposition is accepted, the replacement of the operator at index k is performed, and one continues with the next propagation index in imaginary time. If the proposition is denied, the current operator remains, and one continues with the next propagation index in imaginary time.
As for the TFIM, a fixed-length scheme is used and the sequence length
limiting the amount of non-trivial operators
n has to be dynamically adjusted. During the thermalisation of the sampling procedure, trivial operators are appended to the end of the sequence after each diagonal update (iteration over the whole sequence) such that
[
39].
In order to efficiently implement an off-diagonal loop update, it is crucial to introduce the concept of operator legs. An operator at position p in the sequence has four legs with the numbers , two legs (an upper and a lower leg) for each of the spin sites. Trivial operators can be described with four ghost legs or can be ignored entirely in the sequence for the off-diagonal update.
For the unfrustrated Heisenberg models, the off-diagonal update is performed using a loop update. During the update, loops are constructed in the space–time configuration (see
Figure 17 for an example) and the spin states on the loop are flipped along the way, which changes the operator vertex types. Thereby, diagonal operators become off-diagonal operators and vice versa. It is noteworthy that the weight of a configuration is not modified by this since the weights of diagonal and off-diagonal operators are the same in the isotropic Heisenberg model. Therefore, flipping loops with a fixed probability satisfies detailed balance as long as the probability to construct the reverse loop is the same.
For the Heisenberg model, the creation of the loops is deterministic. Whenever a loop enters an operator during the loop construction, there is only one exit leg that creates a valid operator for the bond type (AF or F). Flipping the spins along the loop exchanges an diagonal operator
with its off-diagonal counterpart
and vice versa. The entrance leg and exit leg never belong to the same site. For ferromagnetic operators, the propagation direction of the loop in imaginary time remains the same (see
Figure 18). For antiferromagnetic operators, the propagation direction of the loop in imaginary time changes (see
Figure 18). When the loop crosses the periodic boundary in imaginary time at a site
i, the computational basis state
has to be updated by flipping the spin at site
i. As each leg only belongs to one loop, the configuration can be split into disjoint loops. Therefore, it is possible to construct all loops during the off-diagonal update and flip each loop independently with a probability of
.
Combining the diagonal and off-diagonal update allows sampling arbitrary unfrustrated Heisenberg models. This includes antiferromagnetic Heisenberg ladders and bilayer systems with an unfrustrated long-range interaction.
5.3. Observables
In this section, we introduce some observables that can be easily measured within the SSE formalism introduced. Throughout this section, we do not use the fixed-length scheme, but keep the general form with operator sequences of fluctuating length to keep the notation short and simple. The sampling of observables can be performed analogously in the fixed-length scheme. When implementing the formulas for the observables in the fixed-length scheme, one just has to keep in mind that the sequences used in the formulas are the same ones, but without the padded trivial operators. This means, for instance, that a sum over all propagation steps p becomes a sum over all non-trivial propagation steps in the fixed-length scheme.
Up to this point in
Section 5, we have focused on the partition function of a quantum-mechanical Hamiltonian:
expressed as a sum over non-negative weights
of a configuration
. However, we are actually not interested in calculating the partition function itself, but rather, the expectation values of observables.
Analogous to the partition function, quantum mechanical expectation values can be expanded into a high-temperature series:
It is important to realise that configurations with a non-vanishing weight
constituting the partition function do not necessarily contribute to the expectation value
with the same weight or a non-vanishing weight at all [
213]. In general, only for operators
A that are diagonal in the computational basis
, the expectation value can be written as
with
. For instance, the magnetisation in the
z-direction or any
n-th moment thereof is such a diagonal operator when using the
-eigenbasis
as a computational basis. The statistics of the MC estimates for such observables can be improved by realising that [
218]
with
being the sequence obtained from cyclically permuting
for
p times [
218]. This means that the expectation value Equation (
257) can be expressed as
where
is additionally averaged over imaginary time with
.
For off-diagonal operators, one needs to find customised formulas for the expectation value. However, some of these expectation values are accessible in a quite general way. For instance, one can show that the mean energy has a rather simple formula [
210,
211,
218]:
Moreover, for the operators
(see Equation (
213)), one finds [
210,
211,
218]
where
is the amount of operators
occurring in the operator sequence. Similarly, one can derive a formula for the heat capacity [
210,
211,
218]:
However, for small temperatures, the heat capacity is calculated as the small difference of large numbers.
Linear Response and Correlation Functions
An important class of observables is linear response functions like susceptibilities. The linear response of an observable
A to a perturbation
tuned by perturbation parameter
is given by a Kubo integral [
212,
219]:
Once again, if
A or
B are off-diagonal operators, one has to consider case by case and find customised formulas that can be used to sample the specific observables within the SSE formulation. We will focus on the case where
A and
B are diagonal in the chosen computational basis. Important examples are the magnetic susceptibility or its local version of spin–spin correlation functions averaged over imaginary time, i.e., the correlation function at zero frequency when regarding Equation (
264) as a Laplace transformation from imaginary time to frequency space. To make this type of observable accessible to a sampling in the SSE formulation, the imaginary-time correlation function
is expanded in temperature analogous to the partition function:
where, in the last step, the operators
A and
B were replaced by their respective eigenvalues
and
of the state
at the propagation steps
.
By replacing the sum over
k by a sum over
and inserting the decomposition of the Hamiltonian, this takes the form
where, in the last step, the average over imaginary time was taken in order to improve the statistics. From Equation (
269), the connection between the discrete propagation steps of the SSE and the continuous imaginary time
becomes apparent [
211,
212]. An imaginary-time separation
corresponds to a binomial distribution of separations
l of SSE propagation steps:
which is peaked around
[
211]. If one is interested in the spectral properties of the system, one can use this formula for sampling imaginary-time correlation functions
. However, there are more efficient ways to calculate imaginary-time correlation functions by embedding the SSE configuration into continuous imaginary time. We refer to Refs. [
212,
220] for details, as this is out of the scope of this review.
To get to the linear response function
in Equation (
263), the imaginary time integral of in Equation (
269) has to be calculated. This integral can be analytically calculated:
by performing
l partial integrations. Inserting Equation (
269) into the formula (
263) and performing the imaginary time integral Equation (
274) yield
One can further separate the
term while using the periodicity
in imaginary time and rewrite the sums over
l and
p as a product of two sums. This eventually yields [
210]
From an algorithmic perspective, the two sums in Equation (
278) need to be calculated by traversing the operator sequence. The effort for measuring
, therefore, scales like the algorithm with complexity
when the observables
,
, and
are not calculated from scratch at every propagation step
p, but gradually updated and averaged while propagating the state through the sequence.
For the special case of the susceptibility, one needs to set
and
. The last term
in Equation (
278) can be dropped for simulations of finite systems due to
. Explicitly, this gives
for the susceptibility. Once again, we want to stress that this formula is not formulated in the fixed-length scheme and the average over the propagation index
p, therefore, refers to the propagation steps of non-trivial operators. Averaging over all propagation steps in the fixed-length scheme is incorrect.
For the spin–spin correlation functions
, the operators are set to
and
, leading to
where it was already used that
for any finite system. Similar to the zero-frequency spin–spin correlation function, one can also calculate an equal-time spin–spin correlation function:
Sampling the correlations among all sites
i and
j at all propagation steps
p leads to a complexity of
, which is worse than the computational complexity
of the SSE QMC updates. Even though one could eliminate the average over imaginary time in the case of Equation (
282) to reduce the complexity to
at the expense of statistical accuracy, this is not possible for Equation (
280) as the sum over imaginary time is intrinsic in the definition of the zero-frequency correlation function. However, by realising that the correlations
between two sites
i and
j are not altered at the order
-times in imaginary time, but only
, one can measure the correlation functions Equations (
280) and (
282) in
time. This is achieved by traversing imaginary time while memorising the propagation step last
of the last preceding spin flip operator for every site separately. When a spin flip occurs at a site
j at propagation step
p, one needs to update the correlation function:
for every site
i before the local magnetisation is propagated with
. One further needs to update last
. In order to avoid that the measurement dominates the simulation for large systems, we only measure the correlations between a fixed site
and
. This finally reduces the complexity of the measurement to
.
Correlation functions are observables that contain much information about a system’s state and, in particular, its order. Moreover, its decay at the critical point with the distance between two spins is connected to the critical exponent
(anomalous dimension). Away from the critical point, the correlation function usually decays exponentially with long-range models being one exception to this. The length scale of this exponential decay is given by the correlation length
, which itself is an interesting quantity at a continuous phase transition as its divergence is the reason for systems becoming scale-free at the critical point. As the correlation function does not decay exponentially for long-range system, which we focus on in this review, we will use the more general term “characteristic length scale” to refer to the length scale at which the correlations switch to their long-distance behaviour. In particular, with respect to the Q-FSS for systems above the upper critical dimension, the characteristic length scale is a crucial quantity as its scaling was predicted to be different from the standard FSS with
instead of
(see
Section 2.3 or Ref. [
34] for details on scaling above the upper critical dimension in quantum systems). This quantity, therefore, played a crucial role in confirming the Q-FSS hypothesis. However, the characteristic length scale is a subtle quantity, which is hard to extract or even define on a finite lattice. For a finite system, the definition of a characteristic length scale is not unique, and there are several definitions for
, which will converge to
for
[
221]. For long-range systems, finding a suitable definition for the characteristic length scale is even more difficult, as the correlation function decays algebraically even away from the critical point [
222].
Common definitions that are tailored for correlation lengths, which specify the exponential decay of a correlation function at long distances, such as the second moment:
therefore, might yield
in an infinite system not only at the critical point, but also away from the critical point [
223].
One possible definition for long-range quantum systems is [
224]
with
being the Fourier transform of
from real space to momentum space and
the smallest wavevector fitting on the finite lattice. By inserting the Gaussian propagator
for long-range interactions [
20,
21] into Equation (
285), one obtains
so that the momentum dependency cancels. Another definition for the same quantity, but using the equal-time Gaussian propagator
[
212], is
By inserting the scaling of the gap
for the
n-component quantum rotor model in the long-range mean-field regime [
20,
21], which is relevant for most applications discussed below, one obtains the scaling of
in the thermodynamic limit to be
which is the singularity one expects from the characteristic length scale.
5.4. Sampling at Effectively Zero Temperature
The SSE QMC approach can be used to stochastically calculate the thermal average of operators for systems of a finite size at a finite temperature. We discussed in
Section 2 that QPTs are a concept defined in the thermodynamic limit at zero temperature. Further, we presented in
Section 2.2 and
Section 2.3 how the critical point, as well as critical exponents can be extracted from certain observables calculated on finite systems at zero temperature. The goal of many works that use SSE QMC sampling is to calculate the expectation value of these observables on finite systems at an effective zero temperature [
32,
34]. We define the notion of effective zero temperature as a temperature at which, in practice, only the ground state contributes to the thermal average, as all excited states are exponentially suppressed [
32,
34]. This definition can be applied only to gapped systems as gapless systems at infinitely small, but finite temperature lead to a thermal average over the infinitesimally low-lying excitations. As the SSE QMC approach is sampling finite systems, there cannot be a non-analytic point in the ground-state energy associated with a second-order QPT [
113]. Closely related with this statement, the ground state of a finite system cannot spontaneously break the symmetry of the Hamiltonian, as it is the case at a second-order QPT [
113]. An intuition to this statement can be built from a perturbative point of view. On a finite system, there is always a finite-order perturbative process coupling the hypothetical ground states of a symmetry-broken phase. Due to this finite-order coupling, there will always be a level repulsion between the states and no true degeneracy occurs. This means that, on a finite system, there is always a finite-size energy gap between the ground state and the excitations. As discussed in
Section 2.2 on a finite system, there is a pseudo-critical point close to the parameter values of the QPT in the thermodynamic limit. For a system with gapped phases on both sides of the QPT, we expect the relevant finite-size gap to be the smallest in the vicinity of the pseudo-critical point. The finite-size gap at the pseudo-critical point is expected scales as
with the linear system size
L to the power of the dynamical correlation length exponent
z [
34,
113,
129,
212] and
for a QPT below the upper critical dimension (see
Section 2.3). This statement can be directly derived from the finite-size scaling form of the energy gap:
Note that the scaling dependence for systems above the upper critical dimension has not been confirmed yet by numerical studies. To perform simulations at effective zero temperature, there are two main approaches in the contemporary literature. Firstly, one just scales the simulation temperature with
L for different linear system sizes. This is a valid approach for many systems as
is a common value for the critical exponent, indicating a space–time symmetry in the correlations. For long-range interacting systems such as the LRTFIM or unfrustrated long-range Heisenberg models, one has
, which makes the scaling sufficient, but overly ambitious [
21]. An improved version of this naive approach is to scale the simulation temperature with
for different linear system sizes. This follows the expected scaling of the finite-size gap. The correct prefactor of the scaling is not known and accounted for in the scaling, and corrections to scaling for small system sizes can lead to errors. A more sophisticated approach to determine a suitable effective zero temperature for a finite system is discussed in Refs. [
32,
34,
225]. This technique was introduced in Ref. [
225] as the beta-doubling method. The idea is to study the convergence of observables
for a fixed system size and parameter value
r at successively doubled inverse temperatures
of the simulation. The fraction of
is considered with
being the largest considered
value. This quantity is used to probe the convergence of the observable
towards zero temperature. The temperature convergence can be gauged using a plot, as demonstrated in
Figure 19. Note that the value of
is always at one. Therefore, as a rule of thumb, one can validate simulation temperatures for which at least the two points prior to the last point also have a value close to one.
Although the beta-doubling scheme has more overhead than a naive scaling with , it provides multiple advantages. First of all, one does not need to know the dynamical critical exponents z beforehand. Secondly, in most cases, it is easy to implement: if one uses either way a simulated annealing scheme by successively cooling the simulation temperature during the thermalisation of the algorithm, the beta-doubling scheme can be easily incorporated in this procedure. Thirdly, the beta doubling provides a direct demonstration that a simulation is sampling zero-temperature properties. It captures intrinsically the relevant scaling , as well as potentially large prefactors and corrections to this scaling. Fourthly, it does not need to be applied to all parameter values r, as it suffices to perform the beta-doubling procedure only for the parameter values where the relevant finite-size gap is expected to be the smallest and use this temperature for the whole parameter range.
In general, the beta-doubling scheme can help to reduce computational cost by performing simulations at a tailored effective zero temperature. If computational time is not a critical factor, in many practical applications, one can use the temperature that is sufficient for the largest system and sample all smaller systems at the same temperature.
5.5. Overview: Path Integral Quantum Monte Carlo Algorithms for Long-Range Models
The QMC methods discussed so far are both based on the SSE formulation [
39,
213]. In parallel with the development of the SSE-based QMC methods for long-range systems, path integral (PI) QMC methods operating in continuous imaginary time have been introduced for extended Bose–Hubbard models with long-range density–density interactions [
215,
226,
227,
228] (see (
290)). The main application of these methods in the context of long-range interactions is the determination and classification of ground states [
64,
229,
230]. Since we focus on reviewing QPTs in long-range interacting systems, which are usually simulated using the SSE, we only skim over the basic concepts relevant for the PI QMC methods. One motivation for the development of these methods was the study of the Mott insulator to superfluid transition in the presence of disorder [
231,
232,
233]. Further, long-range density–density interactions were added to the algorithms [
226]. A great application case for these algorithms is the study of extended Bose–Hubbard models [
64,
229,
230], which are an effective description of ultracold atomic or molecular gases trapped in optical lattice potentials [
56,
58,
61,
62,
64,
68,
229,
234,
235,
236,
237,
238]. For a comprehensive explanation on how to derive effective Bose–Hubbard models for these systems, see Refs. [
56,
236,
237,
238].
A basic Hamiltonian studied with the world-line QMC methods in this context [
226,
227,
228] reads
with (hard-core) bosonic degrees of freedom on the lattice site
i described by creation
, annihilation
, and particle number
operators. The coupling
describes a chemical potential and
the density–density interaction between particles at sites
i and
j, and the hopping amplitude
.
PI QMC methods formulated in imaginary time are based on the PI formulation of the partition function:
using the Suzuki–Trotter decomposition [
115]. The PI formulation is used to extend the configuration space to all imaginary-time trajectories
of computational basis states [
39,
226], which are periodically closed in
and which are connected by an imaginary-time evolution
. These methods can be implemented in discrete [
239] or continuous [
226,
240] imaginary time.
In the SSE approach, the configuration space is extended using the operator sequences arising from a high-temperature expansion of the exponential in the partition function. These configurations can be regarded as a series of discretely propagated states
with an artificial propagation index
p labelling its position in the sequence. This introduction of an additional dimension to obtain a configuration space that can be sampled using MC in the PI and SSE QMC approach hints that both approaches are closely related. Studies of the close relationships between the SSE and PI representations can be found in Refs. [
211,
241]. Concepts developed in one of the two pictures can be equivalently formulated in the other one. For example, the concept of loops [
215,
216,
242] and directed loops [
215,
217,
242] can be applied in both formulations. The sampling of the Heisenberg model as described in
Section 5.2 can be transferred to the PI QMC picture, as it is a direct application of the directed loop idea [
217,
225].
Since we will focus in this review on quantum-critical properties of magnetic systems, we only briefly summarise the major applications of PI QMC methods for extended (hard-core) Bose–Hubbard models with dipolar density–density interactions [
64,
229,
230]. The PI QMC is used in this field to determine ground-state phase diagrams studying the emergence of solid, supersolid, and superfluid phases [
64,
229,
230]. It has been demonstrated in Ref. [
64] that long-range interactions stabilise more solid phases than the chequerboard solid with a filling of
present in systems with nearest-neighbour interactions. It has also been shown that long-range interactions lead to the emergence of supersolid ground states [
64]. The great benefit of the QMC simulations in comparison to other methods (e.g., mean-field calculations [
111]) is the quantitative nature of these methods. Therefore, the numerical study of these models is highly relevant, since experimental progress in cooling and trapping atoms and molecules with dipolar electric or magnetic moments [
67,
69,
72,
73,
243] enables experiments to realise extended Hubbard models with long-range interactions [
68]. We expect the implementation of SSE QMC techniques for this kind of model to be straightforward using the directed loop approach [
217,
225].
6. Long-Range Transverse-Field Ising Models
In this section, we review the ground-state quantum phase diagrams of the long-range transverse-field Ising model (LRTFIM) with algebraically decaying long-range interactions. We emphasise how the Monte Carlo-based techniques introduced in this work are a reliable way to obtain critical exponents of quantum phase transitions (QPTs) in this model. The Hamiltonian of the LRTFIM is given by
with Pauli matrices
and
describing spins
located on the lattice sites
. The Ising coupling is tuned by the parameter
J. For
, the Ising interaction is antiferromagnetic and, for
, ferromagnetic. The amplitude of the transverse field is denoted by
h. The positive parameter
governs the algebraic decay of the Ising interaction. Here,
d denotes the spatial dimension of the system and
is a tunable real-valued parameter. The limiting cases of the algebraically decaying long-range interaction are the nearest-neighbour interaction for
and an all-to-all coupling for
. As a side note, there is literature where
is used as a parameter to tune the decay of the long-range interaction. For this review, we stay with
in order to treat systems with different spatial dimensions
d on the same footing.
The Hamiltonian (
292) can be treated using the pCUT+MC method as described in
Section 4. A generic, perturbative starting point for the LRTFIM is the high-field limit. The transverse field term is regarded as the unperturbed Hamiltonian and the Ising interaction as the perturbation. Using the Matsubara–Matsuda transformation [
25,
29,
89]:
the Hamiltonian (
292) can be brought into a hard-core bosonic quasi-particle picture:
Here,
is a constant,
N is the number of sites, and
is a hard-core bosonic quasiparticle annihilation (creation) operator. The perturbation parameters are given by
In the pCUT language, the perturbation associated with the perturbation parameter
can be written as
, where the hopping processes
are contained in
and the pair creation (annihilation) processes are in
(
).
Further, the Hamiltonian (
292) is of the form to straightforwardly apply the SSE QMC algorithm reviewed in
Section 5.1 (see Equation (
226)). We recall that this QMC algorithm is sign-problem-free for arbitrary, potentially frustrated, Ising interactions.
The studied effects of algebraically decaying long-range Ising interactions on the ground-state phase diagram can be categorised into three scenarios:
First, for ferromagnetic interactions, there is a QPT with
symmetry breaking between a ferromagnetic low-field phase and a symmetric
x-polarised high-field phase. The intriguing effect of long-range interaction is the emergence of three regimes with distinct types of universality of the QPT. For large values of the parameter
, the universality class is the same as in the nearest-neighbour model. For intermediate
values of
, the critical exponents change as a function of
and the criticality can be described by a non-trivial long-range theory. For
, the long-range interaction lowers the upper critical dimension below the physical dimension of the model and the transition becomes a long-range mean-field transition. We review the recent studies investigating this model in
Section 6.1 with a particular focus on the
-theory in
Section 6.1.1 and on numerical studies investigating the critical properties in
Section 6.1.2. The aspects regarding FSS above the upper critical dimension are summarised in
Section 6.1.3.
The second relevant category is antiferromagnetic Ising interactions where the nearest-neighbour couplings form a bipartite lattice. In this case, there is a QPT with
symmetry breaking between an antiferromagnetic low-field phase and a symmetric
x-polarised high-field phase. The current state of literature indicates no change in the universality class of the phase transition in dependence of the long-range interaction. We summarise and review the recent progress in studying this case in
Section 6.2.
The last aspect is scenarios where the nearest-neighbour couplings do not form a bipartite lattice. In this case, the long-range interactions may substantially alter the quantum phase diagram of the nearest-neighbour model. We summarise and review the recent progress regarding this case in
Section 6.3.
6.1. Ferromagnetic Long-Range Transverse-Field Ising Models
The ferromagnetic TFIM with nearest-neighbour interactions in dimensions is a paradigmatic model to display a QPT in a D-Ising universality class. This transition describes the non-analytic change in the ground-state between a symmetry-broken low-field phase and a symmetric x-polarised high-field phase. By adding algebraically decaying long-range interactions to this model, one can study how such interactions can alter non-universal, as well as universal properties of a QPT and which new features can emerge.
In the case of ferromagnetic interactions, the algebraically decaying long-range interaction stabilises the symmetry-broken phase and the QPT shifts to larger
h values [
25,
29,
32,
34]. An intuitive way to understand this behaviour is that, due to the long-range interaction, more connections are introduced that align the spins in the
z-direction, and the energy cost for a single spin flip increases. In the limit
, the critical value of the transverse field diverges as the cost for a single spin flip becomes extensive.
In this review, we focus exclusively on the regime of so-called weak long-range interactions with an algebraic decay exponent
[
21]. The research field of strong long-range interactions considers
[
5,
42,
43,
44,
45]. For weak long-range interactions, the Hamiltonian (
292) is well defined in the thermodynamic limit, and common properties such as the ground-state additivity and thermodynamic quantities are well defined [
5]. For strong long-range interactions, the energy of the ferromagnetically aligned state is superextensive, and in order to study the model in the thermodynamic limit, an appropriate rescaling of the Ising coupling with the system size is required [
5].
The significance of the ferromagnetic LRTFIM comes from its paradigmatic nature of displaying changes in the universal behaviour due to the long-range interaction [
20,
21,
25,
29,
32]. It has been known since the advent of the theory of classical phase transitions that algebraically decaying long-range interactions are a potential knob to alter the fixed-point structure of the renormalisation flow and, therefore, change the universal properties (e.g., critical exponents) [
1,
2,
140]. Coming from the limit
of nearest-neighbour interactions, the QPT of a
d-dimensional model remains in the
D-Ising universality class until
(
is the anomalous dimension critical exponent of the nearest-neighbour universality class). This means that the critical exponents are constant as a function of
and the fixed point associated with the QPT remains the one of the nearest-neighbour model. For
, the RG fixed point associated with the QPT changes to the one of a non-trivial long-range interacting theory, and the critical exponents become
-dependent [
21]. The upper critical dimension of this theory with long-range interaction is lowered with respect to the short-range model as
. If the (fixed) spatial system dimension
d is larger than or equal to the
-dependent upper critical dimension, the universality class describing the QPT enters a long-range mean-field regime. The three different regimes, as well as their boundaries in dependence of the dimensionality of the system are visualised in
Figure 20.
In the following, we recapitulate the basic field-theoretical arguments leading to the distinction between three universality regimes (see
Section 6.1.1). We will further emphasise the most relevant aspects for the two long-range regimes. In the non-trivial, intermediate regime, the precise values of the critical exponents depend on
and need to be determined numerically. Therefore, we review the recent numerical studies computing the critical exponents in
Section 6.1.2. To conclude the discussion, we outline how to utilise ferromagnetic long-range Ising models to study QPTs above the upper critical dimension (see
Section 6.1.3).
6.1.1. -Theory for Quantum Rotor Models with Long-Range Interactions
Starting with the short-range interacting action (see Equation (
A3)) of the
n-component quantum rotor model, Dutta et al. [
20] introduced an action for long-range interacting rotor models, by adding the algebraic decay between interacting fields as an additional term to the action in real space. Note the the action for the Ising model is equivalent to the one-component (scalar) rotor model [
113]. By performing a Fourier transformation of the resulting action, the authors obtained the action:
with
,
from the decay exponent
of the microscopic model, and
coupling constants as in the nearest-neighbour case. The
term, which is new compared to the nearest-neighbour theory, results from the Fourier transformation of the added long-range interacting term:
of the order-parameter field
[
40]. As the relevant modes for the QPT are the ones with long wavelengths, it is clear that, for
, the
term gives the leading contribution in
q; the
term can be neglected and, therefore, the system is, in this case, described by the short-range interacting theory. In contrast to this, a different critical behaviour distinct from the short-range case is possible for
. With the same argument as above, the
term becomes negligible for
. In the following, we will focus on this regime, where the behaviour differs from the nearest-neighbour case. In order to gain insight into the QPT affected by long-range interactions, an investigation of the Gaussian theory is a good starting point to enter the framework of perturbative renormalisation group calculations [
20,
21,
113] like the
-expansion in Ref. [
20]. From Equation (
296), one can directly read off the propagator of the Gaussian theory as
From field-theoretical power-counting arguments, Dutta et al. [
20] derived several properties of the Gaussian theory. Using the divergence of the mass renormalisation term, the lower critical dimension below which no phase transition is occurring can be derived as
[
20]. By regarding the Gaussian propagator, one finds directly that
as
is defined by the deviation from 2 of the leading power by which the momentum enters the propagator (see Equation (
A37)). Note that, in the long-range case, the Gaussian theory has an
. From further power-counting analysis, it is possible to derive more critical exponents from the Gaussian theory [
20]:
In the limit
, the short-range mean-field exponents are recovered [
20]. With the argument that the hyperscaling relation still holds directly at the upper critical dimension, Dutta et al. [
20] derived the upper critical dimension by inserting the long-range Gaussian exponents (see Equation (
299) and
).
In addition to the investigation of the Gaussian part of the action in Equation (
296), one can derive non-trivial exponents below the upper critical dimension by deriving perturbative corrections to the Gaussian exponents in an
-expansion around the upper critical dimension [
113]. Dutta et al. [
20] established with a one-loop renormalisation group expansion of flow equations that the Gaussian fixed point is stable for
. Therefore, the long-range Gaussian exponents (see Equation (
299)) are valid for
. For
, the correction indicates a flow to a non-trivial fixed point. The first-order corrections of the
-expansion for
and
are provided [
20], and the first-order corrections to
and
z are argued to be zero [
20]. It should be noted that the dynamic correlation exponent
for
and, therefore, correlations are not isotropic in the space and imaginary-time direction [
20,
21]. “For any value of
,
sticks to its mean-field value
, because the renormalization does not generate new
terms” [
20]. This also fits well with the claim that
, because, then, there is no discontinuity at the boundary [
1,
2,
3,
21,
244].
A recent study by Defenu et al. [
21] with a functional renormalisation group approach investigated the flow of couplings in an effective action with anomalous dimension effects. This analysis showed that the boundary between the short-range and the non-trivial long-range regime does not occur at
, but at
. So, it is shifted towards lower
by the anomalous dimension of the short-range criticality. This anomalous dimension consideration in [
21] is mainly driven by the following argument: From the functional renormalisation group ansatz, the authors derived two flow equations for the coupling of the long-range coupling
(connected to
a in Equation (
296)) and the short-range coupling
(connected to
b in Equation (
296)):
The first flow Equation (
301) has fixed points for
or
. The first fixed point results in long-range exponents, whereas the second fixed point means that the long-range coupling becomes irrelevant.
In addition to this anomalous dimension effect, the authors in [
21] also calculated critical exponents in the non-trivial intermediate phase with their functional renormalisation group technique. They also compared their results with Monte Carlo simulations of the dissipative non-Ohmic spin chains, which can be mapped to the same behaviour as the long-range interacting quantum Ising model [
245], showing reasonable agreement.
6.1.2. Critical Exponents and Critical Points for One- and Two-Dimensional Systems
The criticality of the ferromagnetic LRTFIM has been studied by a wide variety of methods with different strengths and weaknesses. Overall, all methods qualitatively agree with each other and confirm the three expected regimes from the field-theoretical analysis [
20,
21], namely a long-range mean field, a short-range regime, and an intermediate non-trivial long-range regime, which connects the two limiting regimes with monotonously changing exponents. In the limit
, the value of the critical field
diverges as the energy cost for a single spin flip in the low-field phase becomes extensive and the quantum fluctuations introduced by the transverse field fail to compete with the Ising interaction. In the limit
, the value of
converges towards the exact value for the nearest-neighbour transverse-field Ising model [
25,
29,
32,
246].
In the case of the LRTFIM on the one-dimensional linear chain, the model has been studied by perturbative continuous unitary transformation with Monte Carlo embedding (pCUT + MC) [
29,
31,
34,
247], the functional renormalisation group (FRG) [
21], the density matrix renormalisation group (DMRG) [
28,
248], as well as stochastic series expansion (SSE) [
32,
34,
247,
249], path integral (PI) [
33] QMC, and QMC with stochastic parameter optimisation (QMC+SPO) [
250].
The critical value of the transverse field was calculated with the pCUT method [
29,
31,
34] by estimating the gap closing using DlogPadé extrapolation of the perturbative gap series coming from the high-field phase, by the DMRG using finite-size scaling of the fidelity susceptibility [
28], and by SSE QMC calculations using finite-size scaling of the magnetisation [
32,
249]. The finite-temperature transition points were studied by PI QMC calculations [
33] for
in the long-range mean field regime and
in the strong long-range regime.
Depending on the method, different critical exponents were extracted. Only in the case of pCUT + MC and SSE QMC calculations [
34,
247], all three regimes were investigated. By extracting three independent critical exponents, the authors were able to provide the full set of critical exponents for each method individually. In the case of the pCUT + MC approach [
34,
247], the exponents were computed by (biased) DlogPadé extrapolants of high-order series of the gap (
), the one-quasiparticle static spectral weight (
), and the control-parameter susceptibility (
). In SSE QMC studies [
34,
247], the exponents were calculated using data collapses of the magnetisation (
and
) and the order-parameter susceptibility (
). The DMRG study [
28] applied the method of data collapse to the fidelity susceptibility to extract the single exponent
in the non-trivial long-range regime. Another DMRG study [
248] extracted critical exponents by using finite-size scaling for the energy gap (
z) and the squared order parameter (
and
). Using the FRG [
21], the exponents
and
z were calculated, from which one can additionally calculate the exponent
. In the PI QMC study [
33], the finite-temperature criticality of the LRTFIM was investigated for
in the long-range mean field regime and
in the strong long-range regime. The study includes the extraction of an exponent
, which can be related to
, from the Binder cumulant and classical order-parameter susceptibility via finite-size scaling, as well as the exponent
from finite-size scaling of the classical order-parameter susceptibility. In
Figure 21, the critical field
and the canonical critical exponents are plotted for the discussed studies.
The two-dimensional case of the square lattice is less studied. The critical values of the transverse field were only calculated by the pCUT + MC [
29] and SSE QMC method [
32,
249]. There is so far no study that has extracted the full set of critical exponents altogether. With pCUT [
29], the gap exponents
were also extracted by DlogPadé extrapolants of the gap series. In the FRG study [
21], the critical exponents
z and
were calculated, and in the SSE QMC study [
32], the critical exponents
and
were extracted from the data collapses of the magnetisation. These results are summarised in
Figure 22. Note that we present the results from pCUT + MC calculations from Ref. [
29] with one additional order to the maximal order in the perturbation parameter.
Overall, the different methods all perform well even for relatively small
. For the pCUT and SSE QMC studies, where all three regimes were simulated, the limiting cases of long-range mean-field criticality and nearest-neighbour criticality were correctly reproduced, which underlines the reliability of the presented results in the intermediate regime. The main challenge that the methods are facing is to sharply resolve the regime boundaries due to rounding effects, which are probably due to the limited length scales in the simulation. Additionally, the boundary to the long-range mean field regime is spoiled by logarithmic corrections to scaling at the upper critical dimension. Even in the case of pCUT + MC, which is a method operating in the thermodynamic limit, there is an intrinsic limit of the length scale due to the finite order of the series. In particular, the rounding makes it hard to verify (or falsify) the claim that the boundary between the short-range and non-trivial long-range regime is shifted
. For 1D, the boundary is shifted to
, which could be resolvable in extensive simulation. However, in 2D, the boundary is only marginally shifted to
, which is probably not resolvable by the reviewed methods. The best boundary resolution is probably given by the SSE QMC study [
34]. If one is interested in studying the shift of the boundary, one could, therefore, push the SSE QMC simulations of the one-dimensional chain to larger systems for
. Another difficulty for the methods operating on finite systems is the breakdown of the common hyperscaling relation and standard FSS above the upper critical dimension, which affects the processing of data in the long-range mean-field regime for
. The scaling above the upper critical dimension was already outlined in
Section 2.3. However, we will recapitulate the most important points in the following
Section 6.1.3, not only because this modified scaling was used in several of the above-mentioned methods to extract critical exponents [
32,
33,
34], but also because the LRTFIM constitutes a testing ground for quantum Q-FSS scaling and the modified scaling of the correlation length was confirmed in a study of the LRTFIM [
34].
6.1.3. Scaling above the Upper Critical Dimension
As discussed in
Section 6.1.1, for
, the action describing the QPT and its universality changes from short range to long range. In this long-range regime, the upper critical dimension
was found to depend on the value of
[
20,
21]. This means that the connectivity of the system increases for decreasing
and a smaller spatial dimension
is already sufficient to reach or even exceed the upper critical dimension. The sigma below which the model is above its upper critical dimension is
. In this regime, it is not possible to correctly extract all critical exponents from the standard FSS, and the common hyperscaling relation:
becomes invalid as the critical exponents become independent of the dimension
d. The origin of this discrepancy comes from the effect of dangerous irrelevant variables that one has to take into account in the derivation of scaling above the upper critical dimension. When doing so, one obtains a modified hyperscaling relation:
with the exponent
. This relation can be used for the conversion of different critical exponents [
34,
35].
Additionally, the standard FSS scaling form of an observable
with power-law singularity
is modified to [
34]
following the formulation of the Q-FSS for classical [
15,
16,
17,
18,
19,
41] and quantum [
34] systems. Finite-size methods that rely on the method of data collapse or other FSS-based techniques have to use this adapted formula to extract all the critical exponents successfully [
34]. For a more elaborate description of quantum Q-FSS, please refer to
Section 2.3 or to Ref. [
34].
On the other side, the LRTFIM can also be used as a testing ground for quantum Q-FSS. One key difference that distinguishes the Q-FSS from standard FSS is that the correlation sector is also affected by the DIV in the case of the Q-FSS. This leads to a modified scaling of the characteristic length scale
with the system size instead of
, as it is for standard FSS. The characteristic length scale was measured in the SSE QMC study [
34] in the long-range mean-field regime, and the exponent
was extracted by a data collapse with fixed mean field value
. The extracted values coming from Ref. [
34] are presented in
Figure 23 and are clearly in line with the predictions made by the Q-FSS.
6.2. Antiferromagnetic Long-Range Transverse-Field Ising Models on Bipartite Lattices
In this section, we review results for the antiferromagnetic LRTFIM on lattices where the coupling structure of the nearest-neighbour couplings forms a bipartite lattice. A lattice—or, more generally, a graph—is called bipartite if the sites can be split into two disjoint sets A and B so that there are no edges between sites within each of the two sets A and B. In the case of a lattice, A and B are called bipartite sublattices. Note that, only in the nearest-neighbour limit on bipartite lattices, there is an exact duality between the ferromagnetic and antiferromagnetic TFIM, and the quantum-critical properties coincide. This duality comes from a sublattice rotation of along the x-axis for the spins in one of the two sublattices. In the nearest-neighbour limit, there is an x-polarised symmetric high-field phase and a symmetry-broken low-field phase. The low-field phase is adiabatically connected to the zero-field limit. In the zero-field limit, there are two ground states associated with the states where the spins on the sites in one set of the bipartite sublattices are pointing in one direction while the others point in the other direction. Note that these states can be directly mapped onto the two ferromagnetic ground states by the above-mentioned sublattice rotation. As for the ferromagnetic nearest-neighbour TFIM, the QPT between these two phases is of D-Ising universality.
The antiferromagnetic long-range interaction beyond the nearest-neighbours induces a hierarchy of competing interactions. Due to the long-range interactions, there is no longer a bipartite coupling structure as the spins cannot be aligned such that the energy of all interactions is minimised. Nevertheless, there is evidence that the two ground states in the low-field phase of the nearest-neighbour limit are adiabatically connected to the ground states in the low-field phase for all
[
23,
24,
25,
26,
29,
31,
32,
111].
For the all-to-all connected case
, every state with zero
z-magnetisation is a ground state of the system [
30,
40] at
. The Hamiltonian (
292) can then be written in terms of total spin
with
as
with the total number of spins
[
30,
40]. For all finite transverse fields
, the ground state of Equation (
306) is directly located in the
x-polarised phase [
40,
254].
The natural questions arising for the QPT in the antiferromagnetic LRTFIM on the bipartite lattices are similar to the ferromagnetic LRTFIM: How does the algebraically decaying long-range interaction influence the critical field value of the QPT? Is there a change in the universality class of the QPT depending on the precise values of the decay exponent ?
Regarding the critical field values in one-dimensional chains, there are MC-based studies using the SSE QMC (see
Section 5) [
32] and pCUT+MC (see
Section 4) [
25,
31], as well as the DMRG [
24,
26,
248] and matrix product states [
23]. We summarise the results regarding critical field values and selected critical exponents of the studies mentioned above in
Figure 24. In two-dimensional systems, there are MC-based studies for the square lattice using SSE QMC (see
Section 5) [
32] and pCUT+MC (see
Section 4) [
29] (for selected results, see
Figure 25). Variational methods in two dimensions such as projected entangled pair states [
255,
256] or DMRG calculations [
257] have not been applied for antiferromagnetic systems on bipartite lattices. Entanglement-based methods become more challenging from the increased entanglement due to the area law in two dimensions [
258]. Further, there are reports about the violation of the area law for one-dimensional systems with long-range interactions [
23,
24]. On general grounds, one cannot efficiently represent algebraically decaying long-range interactions in these techniques [
259,
260]. However, there exist DMRG calculations for antiferromagnetic LRTFIMs on non-bipartite lattices [
27,
54,
55,
109].
In general, it has been numerically demonstrated that the critical field values decrease monotonically as a function of
from the nearest-neighbour value at
to zero at
[
23,
24,
25,
26,
29,
31,
32,
248]. Regarding the QPT on the antiferromagnetic chain, the established picture is that it is of the
D-Ising type for all
[
23,
24,
25,
26,
31,
32,
248]. In the regime of ultra-long-range couplings
, recent finite-size DMRG findings by G. Sun [
26] and R. Puebla et al. [
248] suggest that the
D-Ising universality even holds for any
. The only study indicating a change in criticality below the boundary of
uses matrix product states generalising the time-dependent variational principle (TDVP) [
23]. Note that there is no further theoretical motivation for this boundary besides this single numerical study, which does not provide convincing evidence for its existence. The more recent results from Refs. [
26,
248] challenge these findings and establish the
D-Ising universality for all
. The studies using MC-based techniques [
25,
31,
32] cannot extract reliable critical exponents in this regime. In the SSE QMC study [
32], the increasing competition of the long-range interactions is the major problem making the numerical simulations in this regime impractical. If the long-range interaction becomes more prominent, this leads to similar algorithmic challenges as in frustrated systems [
32,
40,
261,
262]. The autocorrelation times of the SSE QMC algorithm increase as more and more bond operators are present in the operator sequence, and the field operators diffuse only slowly [
32]. Nevertheless, following the trend of the results in Ref. [
32], the scenario of a single universality class across the entire
range is plausible. Regarding the pCUT+MC studies [
25,
31], the main obstacle to extract information about the quantum criticality in the regime
is the fact that the critical field value is decreasing. Refs. [
25,
31] use a perturbative expansion around the high-field limit; therefore, if the critical field value increases, then the extrapolation of the gap series becomes increasingly challenging.
Figure 24.
Critical field values and exponents from numerical studies of the antiferromagnetic LRTFIM on the chain. The upper panel displays critical field values
; the lower left panel displays values for the critical exponents
and
and, the lower right panel, values for the critical exponents
. The data points “SSE QMC (2021)” and “SSE QMC
(2021)” for
,
, and
are from Refs. [
32,
249]. The data points “pCUT+MC (2019)” for
and
are from Ref. [
29]. The data points “TDVP (2012)” for
,
, and
originate from Ref. [
23]. The exponents
and
are calculated from the scaling dimensions in Ref. [
23] under the assumption of
, which is reasonable according to [
248]. The data points “DMRG (2016)” for
are from Ref. [
24]. The data points “DMRG (2017)” for
and
originate from Ref. [
26]. The data points “DMRG (2019)” for
are from Ref. [
248]. The values for the critical exponents of the transition in the nearest-neighbour model
,
, and
[
246,
251,
252,
263] are given by the black dashed lines.
Figure 24.
Critical field values and exponents from numerical studies of the antiferromagnetic LRTFIM on the chain. The upper panel displays critical field values
; the lower left panel displays values for the critical exponents
and
and, the lower right panel, values for the critical exponents
. The data points “SSE QMC (2021)” and “SSE QMC
(2021)” for
,
, and
are from Refs. [
32,
249]. The data points “pCUT+MC (2019)” for
and
are from Ref. [
29]. The data points “TDVP (2012)” for
,
, and
originate from Ref. [
23]. The exponents
and
are calculated from the scaling dimensions in Ref. [
23] under the assumption of
, which is reasonable according to [
248]. The data points “DMRG (2016)” for
are from Ref. [
24]. The data points “DMRG (2017)” for
and
originate from Ref. [
26]. The data points “DMRG (2019)” for
are from Ref. [
248]. The values for the critical exponents of the transition in the nearest-neighbour model
,
, and
[
246,
251,
252,
263] are given by the black dashed lines.

To summarise, there are studies indicating a change from the
D-Ising criticality on the chain for small
values [
23,
24]. These studies also report a possible breakdown on the area law of the entanglement entropy for the ground state in this regime [
23,
24], which is a vital prerequisite for the methods they implemented. Two more recent studies [
26,
248] demonstrate no change in the universality class, which is backed by [
25,
30,
31]. For two-dimensional systems, the universality class is demonstrated to be
D-Ising for
[
29,
32], and no trend is reported that the universality class should change for smaller values of
.
Figure 25.
Critical field values and exponents from numerical studies of the antiferromagnetic LRTFIM on the square lattice. The upper panel displays critical field values
; the lower left panel displays values for the critical exponents
and
and, the lower right panel, values for the critical exponents
. The data points “SSE QMC (2021)” for
,
, and
are from Refs. [
32,
249]. The data points “pCUT+MC (2019)” for
and
originate from Ref. [
29]. The values for the critical exponents of the transition in the nearest-neighbour model
,
, and
[
253] are given by the dashed lines.
Figure 25.
Critical field values and exponents from numerical studies of the antiferromagnetic LRTFIM on the square lattice. The upper panel displays critical field values
; the lower left panel displays values for the critical exponents
and
and, the lower right panel, values for the critical exponents
. The data points “SSE QMC (2021)” for
,
, and
are from Refs. [
32,
249]. The data points “pCUT+MC (2019)” for
and
originate from Ref. [
29]. The values for the critical exponents of the transition in the nearest-neighbour model
,
, and
[
253] are given by the dashed lines.
In general, a great algorithmic challenge that remains for the MC-based methods with regard to the antiferromagnetic LRTFIM on bipartite lattices is a reliable study of the regime of small values . Here, as discussed above, all of the commonly applied methods have a handicap in some way.
6.3. Antiferromagnetic Long-Range Transverse-Field Ising Models on Non-Bipartite Lattices
We start with an antiferromagnetic nearest-neighbour interacting TFIM on a non-bipartite lattice (e.g., the sawtooth chain, triangular lattice, Kagome lattice, pyrochlore lattice) at zero field. In the case of non-bipartite lattices, one can always find an odd number of Ising bonds (edges) that form a closed loop. The presence of a loop of odd length means it is impossible to satisfy all antiferromagnetic couplings simultaneously. The phenomenon of not being able to minimise all interactions due to geometrical lattice constraints is called geometrical frustration [
264]. A notion for the strength of geometric frustration can be defined using the resulting ground-state degeneracy [
265]. A theoretical tool to access the ground-state degeneracy is through Maxwell counting arguments [
265,
266,
267]. For systems with a large degree of frustration, e.g., the antiferromagnetic nearest-neighbour TFIM on the triangular or Kagome lattice, there is a finite residual entropy per site in the thermodynamic limit [
168,
268,
269,
270,
271].
In general, extensively degenerate ground-state spaces due to frustration pose a great resource for emergent exotic quantum phenomena. Quantum fluctuations introduced, e.g., by a transverse field result in a breakdown of the extensive ground-state degeneracy and the potential emergence of non-trivial ground states [
270,
271]. In general, it is useful to think about the low-field physics as a degenerate perturbation theory problem on the zero-field ground-state space [
30,
32,
270,
271]. We will later review that this is a reasonable framework in order to treat the breakdown of the degenerate subspace due to fluctuations and algebraically decaying long-range interactions on the same footing [
30,
32,
112].
Perturbing extensively degenerate ground-state spaces with fluctuations may result in several distinct scenarios. First, a distinct symmetry-broken order can emerge for infinitesimal perturbations (order-by-disorder) [
168,
270,
271,
272,
273]. Further, a direct realisation of a symmetry-unbroken phase may occur (disorder-by-disorder). This phase can either be trivial [
168,
274] or exotic, e.g., quantum spin liquids [
99,
100,
101,
275,
276].
For the first part of this section, we review the antiferromagnetic LRTFIM on the triangular lattice [
27,
29,
30,
32,
40,
110,
277]. In the nearest-neighbour limit of that model, the zero-field case has an extensively degenerate ground state [
268,
270,
271,
273]. When adding an infinitesimal transverse field, this ground-state degeneracy breaks down and a
-symmetry-broken clock order emerges from an order-by-disorder mechanism [
270,
271,
273]. The effective Hamiltonian describing the breakdown of the degeneracy in leading order can be expressed as a quantum dimer model [
270,
271,
273]. By investigating the degenerate ground-state space, it was observed that there are local spin configurations in which spins can be flipped without leaving the ground-state space. These configurations, called flippable plaquettes, consist of a spin surrounded by six spins with alternating orientation. It can be easily seen that flipping the spin in the middle of the flippable plaquette results in turning the plaquette by
(see
Figure 26).
The effective low-field quantum dimer model [
270,
271,
273], consisting of the plaquette rotation term, is, therefore, the hexagonal-lattice version of the Rokhsar–Kivelson quantum dimer model with
and
[
270,
271,
278]:
From this mapping, the nature of the state at
was predicted, as well as the breakdown of the order with a 3D-XY QPT [
270,
271]. Further numerical studies extended the insights into this system [
168,
262,
273]. The critical field value at which the phase transition between the symmetric
x-polarised paramagnetic high-field phase and the symmetry-broken gapped clock-ordered low-field phase occurs is at
[
168,
273].
To infer the nature of the full phase diagram including the transverse field and the long-range interaction, the next step is to discuss the zero-field limit of the Ising model with long-range interactions. As the antiferromagnetic long-range interactions introduce a hierarchy of constraints due to the interaction between sites further apart, the long-range interaction breaks the ground-state degeneracy of the nearest-neighbour zero-field as well [
30,
110,
277,
279]. It is important to emphasise that the long-range interaction lifts the degeneracy in a different way and stabilises a gapped stripe phase breaking the translational symmetry in a different way than the clock order promoted by the transverse field [
30,
110,
277,
279,
280]. This plain stripe state is sixfold degenerate by rotations around
, as well as spin flips [
30,
110,
277,
279]. The spins are aligned in straight lines with alternating
z-orientation [
30,
110,
277,
279]. An important aspect is that these stripe states are gapped and, therefore, stable against finite transverse fields [
30,
110,
277].
Regarding the limit of large transverse fields, pCUT+MC was used to investigate the QPT between the
x-polarised high-field phase and the clock-ordered phase [
29,
30]. Similar to the antiferromagnetic models on the bipartite lattices, the critical field value is reduced by the long-range interaction towards smaller field strengths [
29,
30] (see
Figure 27). On the triangular lattice, the universality class of the transition from the high-field to a clock-ordered phase is of the (2+1)D-XY type [
270,
271,
273]. It is reported to remain in this category for all
values investigated (
) [
29]. For small
values, the extrapolation of high-field series expansion is inconclusive [
29,
30]. Infinite DMRG (iDMRG) investigations on triangular lattice cylinders suggest that the clock order vanishes for small values of
and a direct transition to a low-field stripe phase occurs [
27]. A first-order phase transition between the
x-polarised high-field phase and a low-field stripe phase would be consistent with the incapability of the high-field gap to track the transition [
27,
29,
30,
277]. Note that the stripe phase determined from the iDMRG study [
27] does not agree with subsequent studies focusing on the low-field ground states of the model [
30,
32,
277,
279,
280].
Complementary to the high-field analysis and numerical iDMRG studies, approaches in the low-field limit have also been conducted [
30,
110,
277] (see
Figure 27). As already mentioned, it was discussed in the literature for some time [
30,
279,
280] and recently demonstrated using a unit cell-based optimisation technique [
110] that gapped plain stripes are the zero-field ground state as soon as long-range interactions are present. Therefore, in a phase diagram with a
and a transverse-field axis, the clock order phase is wedged between the high-field phase from above and the plain stripe phase from below [
27,
29,
30,
277] (see
Figure 27). The extent of both the stripe phase and the high-field phase increases with stronger long-range interactions [
27,
29,
30,
277]. This behaviour was also demonstrated perturbatively from the nearest-neighbour zero-field limit by treating the long-range interaction, as well as the transverse field as perturbations on the degenerate subspace [
30,
277].
We sketch a qualitative picture of the quantum phase diagram in
Figure 28. The precise ground state at small values of
is still an open research question. We believe that there is some evidence that, after a certain value of
, only a transition between the high-field phase and the low-field stripes remains, which is expected to be of first order. However, there is, by now, no numerical technique to make reliable statements in this regime. In this context, the high-field series expansions cannot detect a first-order phase transition from the elementary excitation gap. There is an SSE QMC study of the system by S. Humeniuk [
40] applying the algorithm discussed in
Section 5.1. The direct QMC simulation was used to determine the critical point between the high-field phase and the clock-ordered phase for
[
40]. For smaller transverse fields, the author identified a “region dominated by classical ground states [
40]”, which are adiabatically connected to the zero-field states. Therefore, there is a qualitatively sketch of a phase diagram in Ref. [
40] similar to
Figure 28. Nevertheless, the naive application of the SSE QMC approach as discussed in
Section 5.1 is not as efficient as for models with interactions that are not competing [
40,
261,
262]. However, there are ideas on how to implement efficient quantum cluster algorithms for frustrated Ising models in a transverse field [
261,
262], which need to be adjusted for long-range interacting Ising models.
The antiferromagnetic LRTFIM on the triangular lattice is a well-studied example of the interplay between long-range interactions and order-by-disorder. Recently, several more examples arose from the field of Rydberg atom quantum simulators [
54,
55,
109,
110,
112]. In Rydberg atom simulators, atoms are positioned in a desired configuration using optical tweezers [
52] and are laser-driven to a Rydberg state [
52]. The Rydberg blockade mechanism leads to an algebraically decaying interaction between Rydberg excitations, which decays with
[
52]. Using the Matsubara–Matsuda transformation [
89], the excitations/non-excitations of a Rydberg atoms are associated with spin degrees of freedom [
52]. The density–density interaction transforms into an Ising interaction [
110,
112]. Rydberg atom quantum simulators are capable of simulating the antiferromagnetic LRTFIM with
and a longitudinal field [
52,
54,
55,
109]. Order-by-disorder scenarios are described for the Kagome lattice at a filling of
Rydberg excitations [
54], the Ruby lattice at a filling of
Rydberg excitations [
55,
109], and at vanishing longitudinal field [
112]. Regarding these examples, the order-by-disorder was studied for a long-range interaction truncated after the third-nearest neighbours [
54,
55,
109,
112]. Considering the remaining long-range interaction as a perturbation to the degenerate ground-state space for most of these mechanisms, a similar scenario as for the triangular lattice is expected [
54,
55,
109,
110]. Using the same unit cell-based optimisation method as for the triangular lattice, Koziol et al. [
110] have calculated ground states of the zero-field model using the full untruncated long-range interaction. For the Kagome lattice at a filling of
Rydberg excitations [
54] and the Ruby lattice at a filling of
Rydberg excitations [
54,
55,
109], gapped ground states were determined [
110], which do not coincide with the order arising from the order-by-disorder mechanism. Interestingly, in all these examples, the zero-field ground state determined by the long-range interaction possess none of the motifs relevant for the leading-order order-by-disorder mechanism [
110]. For example, on the triangular lattice, the plain stripe states contain zero flippable plaquettes, which are the relevant motif to lower the energy at a finite transverse field in leading order. Recently, an order-by-disorder mechanism for the antiferromagnetic
-
-
TFIM on the Ruby lattice was introduced by A. Duft et al. [
112]. The remarkable aspect of the mechanism is that adding long-range interactions does stabilise the same order, which is also selected by the quantum fluctuations [
112]. A general theory, under which conditions the long-range interactions stabilise the same order as quantum fluctuations or stabilise a completely disjoint order, is yet to be found.
Conclusively, the breakdown of extensively degenerate ground-state spaces due to an interplay of long-range interactions and quantum fluctuations is a highly vibrant and relevant field. Long-range interactions are relevant for a wide range of quantum–optical quantum–simulation platforms, including cold atoms [
52,
70,
72] and ions [
74,
75,
76,
77,
79,
80,
81,
82,
84]. These platforms can be used to realise exotic phases of matter, e.g., quantum spin liquids [
54,
55,
109], glassy behaviour [
283], or clock-ordered states [
29,
30,
112,
277]. To understand the emergence of these exotic states of matter, simplified models are oftentimes considered [
54,
55,
109,
112,
283], which truncate the long-range interaction. Therefore, it is imminent to understand the effects of the full long-range interactions on the mechanisms driving the emergence of these exotic phases. At the moment, the commonly used tools to investigate the phase diagrams of these frustrated systems consist of the following:
The derivation of effective models [
30,
54,
55,
109,
112]. These can be used in order to predict exotic emergent phases and the nature of QPTs [
30,
54,
55,
109,
112].
DMRG calculations [
27,
54,
55,
109], which are used to obtain a numerical insight into the ground-state phase diagrams.
QMC calculations, in particular the SSE, as discussed in
Section 5.1, can be used for an unbiased sampling of ground-state properties [
40]. In order to omit the slowdown of the algorithm due to the geometric frustration, algorithmic improvements are required [
261,
262]. It is still an open research question how to set up an algorithm that samples long-range interaction and frustration efficiently.
High-order high-field series expansions using a graph decomposition. This is also a very capable tool to track the first QPT coming from the high-field limit. With this method, the critical field value, as well as critical exponents can be determined [
29,
30,
112]. It is also possible to infer information about the phase on the other side of the phase transition by studying the momentum at which the elementary excitation gap is located [
29,
30,
112]. An MC embedding of white graphs can be performed to study the entire algebraically decaying long-range interaction [
29,
30], while an ordinary graph decomposition can be used for systems with truncated interactions [
112].
7. Long-Range Transverse-Field XY Chain
In this section, we review results on the XY chain with long-range interactions in a transverse field from Ref. [
31]. The Hamiltonian of the long-range transverse-field anisotropic XY model (LRTFAXYM) on a one-dimensional chain is given by
with Pauli matrices
and
describing spin-
degrees of freedom on the
i-th lattice site of the chain. The transverse-field strength is given by
, and the strength of the coupling between sites
i and
j is given by
. The coupling is (anti)ferromagnetic for
(
). We include a continuous interpolation parameter
to tune the system from the XY chain with isotropic interactions (
) to Ising interactions (
). The exponent
determines the decay of the long-range interaction. The limit of short-range interactions is recovered for
and the limit of all-to-all couplings for
.
Similar to the discussion of the transverse-field Ising model, it is useful to express Equation (
308) in terms of hard-core bosonic operators
,
using the Matsubara-Matsuda transformation [
89]
We obtain
in units of
with
being the unperturbed ground-state energy per site,
N the number of sites, and
the perturbation parameter associated with the perturbation
(second sum) taking the form
in the pCUT language, so that a high-order series expansion about the high-field limit can be performed.
In the limit , the ground state is a nondegenerate z-polarised state that serves as an unperturbed reference state. Elementary excitations are called local spin flips at an arbitrary site i, which can be annihilated (created) by the hard-core bosonic operator . Upon increasing , these quasiparticles (qps) become spin flips dressed by quantum fluctuations induced by the perturbation . In the limit of , the system exhibits ferromagnetic or antiferromagnetic magnetic order depending on the sign of J in the x-direction (y-direction) for (). When tuning the interpolation parameter from pure Ising to isotropic XY interactions, ordering in the x-direction starts to compete with ordering in the y-direction.
In order to better appreciate the results for
, we briefly discuss the quantum phase diagram in
Figure 29 of the model (
308) with nearest-neighbour interactions [
284,
285,
286,
287,
288,
289,
290,
291,
292,
293]. For any
, the system with nearest-neighbour interactions undergoes a (1+1)D-Ising QPT at
from the paramagnetic symmetric high-field phase to the (anti)ferromagneti- cally ordered low-field phase, where the ground state spontaneously breaks the
symmetry of the Hamiltonian [
284]. The transition along the
line between the two distinct
symmetry-broken phases is of the (Ising)
2 type, which is conformally and
invariant [
284]. These critical lines meet at
and
in multicritical points
with critical exponents
and
[
284].
For
, the Hamiltonian (
310) becomes particle-conserving as the terms annihilating (creating) two quasiparticles
cancel each other, making the isotropic XY Hamiltonian
symmetric. Due to this particle-conserving nature of Equation (
310), there are no quantum fluctuations dressing the symmetric
ground state for
and the perturbative treatment of the high-field dispersion becomes exact in first order of the perturbation theory [
31]. This distinguished symmetry property of the
case motivates that we review the properties of the isotropic transverse-field XY chain in the ferromagnetic and antiferromagnetic case first in
Section 7.1. Subsequently, we discuss the anisotropic XY chain for ferromagnetic interactions in
Section 7.2 and for antiferromagnetic interactions in
Section 7.3. Here, we show improved results of the ones shown in Ref. [
31] since we noticed right after their publication that the Monte Carlo runs were performed with a suboptimal choice of the simulation parameters.
7.1. Isotropic Long-Range XY Chain in a Transverse Field
To consider the isotropic XY chain,
is set in the Hamiltonians in Equations (
308) and (
310). The quantum criticality of this model was studied in Ref. [
31] in an analytical fashion, by evaluating the ground-state energy and the dispersion of the elementary excitations analytically. Setting
, Equation (
310) reads
since the pair creation and annihilation terms
cancel, as stated above. The perturbation
acting on the unperturbed
z-polarised ground state does not introduce any quantum fluctuations. Therefore, the
z-polarised state becomes an exact eigenstate for arbitrary
and stays in the ground state until a QPT occurs [
31]. Analogously, the one-quasiparticle dispersion is exact in first-order perturbation theory. The one-particle dispersion in the symmetric high-field phase reads [
31]
The critical value
can be determined from the dispersion (
312) via the closing of the quasiparticle gap
. The critical exponent
associated with the gap closing can be extracted from the dominant power-law behaviour of the gap near
:
Further, it is possible to separate the dynamic
z and static
exponent by evaluating the 1qp dispersion
at the quantum-critical point such that
which allows the extraction of the critical dynamic exponent
z. For ferromagnetic XY interactions, the minimum of the dispersion (
312) is located at the momentum
. Therefore, the gap series is given by
and
. The Riemann-zeta function
is convergent for all
and diverges for
. This mathematical observation coincides with the qualitative behaviour found in ferromagnetic Ising systems discussed in
Section 6.1 in the sense that
. Since the expression for the gap
is linear in
, the critical exponent is
, independent of the
value. Using the expression for the dispersion (
312), the knowledge about
, and the definition of the dynamic critical exponent (
314), Adelhardt et al. [
31] evaluated
z and, therefore, also
as a function of
. The results of their calculations are presented in
Figure 30. The exponents resulting from the consideration of the dispersion can be categorised into two distinct regimes: For
, the critical exponents of the nearest-neighbour transition are found (
and
). For
, the exponent
z decreases linearly from 2 to 0 and
increases like
from
to
∞. Note that this linear decrease of
z and the divergence of
appear similar to the behaviour of the ferromagnetic LRTFIM at small
values (see
Section 6.1) [
20,
21]. To explain this behaviour, a quantum field theory was proposed in Ref. [
31]. The idea was to add a
term (analogous to [
20,
21]) to the well-studied bosonic action used for the study of the isotropic short-range transition [
113,
231,
284,
294]. The suggested action reads
with
being a complex
c-number order-parameter field of the transition,
, and the real constants
and
r [
31]. The notation of Equation (
315) is taken from Refs. [
31,
231]. From power counting, one obtains the critical exponents:
from the Gaussian part of the action in Equation (
315) [
31]. Following the arguments of Ref. [
231], these exponents hold “presumably” [
231] for
below the upper critical dimension
. Refs. [
231,
294] provide arguments that the self-energy vanishes for every order in
u and the renormalisation of
u can be performed in all orders via ladder diagrams [
31]. The vanishing self-energy is explained by the fact that, in each diagram, every pole in
lies in the complex upper-half plane. The frequency integral can be deformed into the lower half-plane to give zero [
294]. The fact that the free propagator of the field theory is not changed by a self-energy is a manifestation of the absence of fluctuations that do not preserve the particle number [
31]. The predictions of the field theory (
315) for
z in Equation (
316) and
in Equation (
317) are in perfect agreement with the results of the high-field excitation gap (see
Figure 30) [
31].
Note that, in the nearest-neighbour case, the low-field ground state does not break the
symmetry of the Hamiltonian due to the Hohenberg–Mermin–Wagner (HMW) theorem, ruling out continuous symmetry breaking [
113,
130,
131,
284,
295,
296]. Long-range interactions are a known mechanism to circumvent the HMW theorem [
22,
35,
297,
298,
299,
300,
301,
302,
303,
304,
305,
306]. With the high-field approach discussed above, it is not possible to determine the nature of the low-field ground state and if a continuous symmetry breaking occurs for sufficiently small decay exponents. We are not aware of studies addressing this topic.
For the remainder of this section, we discuss the antiferromagnetic isotropic XY chain in a transverse field along the same lines as for the ferromagnetic case. The minimum of the dispersion is at momentum
, and the gap can be expressed as
with
being the Dirichlet eta function (also known as the alternating
function) [
31]. The Dirichlet eta function
is convergent for all
; therefore, the definition of an excitation gap is also possible in the regime of strong long-range interactions (
). Analogous to the discussion of the Ising interaction in
Section 6.2, the extent of the low-field phase is diminishing for decreasing
values. For
, the energy gap closes at diverging
[
31]. With a similar analysis to the ferromagnetic case, the same critical exponents
and
are found along the entire
line [
31]. This is also analogous to the antiferromagnetic LRTFIM discussed in
Section 6.2, where the nearest-neighbour criticality is believed to be the correct universality class for all values of
.
7.2. Ferromagnetic Anisotropic Long-Range XY Chain in a Transverse Field
The fundamental difference between the isotropic
and anisotropic
XY chain in a transverse field is that, for
, the Hamiltonian is no longer
-invariant (particle-conserving); see Equation (
310). Therefore, a perturbative treatment of the anisotropic model with long-range interaction from the high-field limit requires the entire procedure described in
Section 4 to perform pCUT+MC calculations. In Ref. [
31], this procedure was applied to obtain the critical values
and critical exponents
from the gap. We review these results in this section, followed by the results for the antiferromagnetic case in
Section 7.3.
Critical values
and critical exponents
for several
values as a function of
are depicted in
Figure 31. The overall
dependence of
for
is similar to the
case (see the inset of
Figure 31). However, the low-field ferromagnetic phase becomes more stable for increasing
values [
31]. Note that the perturbative parameter is
; therefore, a QPT at smaller
values means a larger extent of the phase in
. In Ref. [
307], the QPT of the anisotropic LRTFAXYM was studied with exact diagonalisation, and the critical point was determined for
, which coincides perfectly with the value from pCUT+MC (see
Figure 31). For all
, Adelhardt et al. [
31] identified the same three regimes for the universality of the QPT as for the ferromagnetic LRTFIM. The gap exponent behaves analogously to the LRTFIM (
), as depicted in
Figure 31. Solely the
exponent behaves in a special manner, being
for all
values, in full agreement with the existence of a multicritical point in the nearest-neighbour model. For
, the first domain identified in Ref. [
31] is the long-range mean-field regime for
with
. For large values of
, the gap exponent is
and the transition belongs to the 2D-Ising universality class [
31]. Third, in the intermediate regime, the critical exponents vary continuously between the two values in a non-trivial fashion [
31].
The criticality regimes for
can be understood using the same field-theoretical argument (see
Section 6.1.1) as for the ferromagnetic LRTFIM, since the underlying symmetry that is spontaneously broken is, in both cases, the same
symmetry of the Hamiltonian. Therefore, at
, the upper critical dimension as a function of
becomes smaller than one and the transition enters the regime above the upper critical dimension. The boundary between nearest-neighbour criticality and the intermediate long-range non-trivial regime is expected to be at
[
21]. The data from Ref. [
31], presented in
Figure 31, are in good agreement with these three regimes. Nevertheless, the changes between the regimes cannot be determined accurately by the pCUT+MC approach. In
Figure 31, we can observe that the interfaces between the nearest-neighbour and intermediate long-range regime become more pronounced for increasing
values, while at the interface between the long-range mean-field and intermediate regime, the deviation is the same for all values of
. The reason for the distinct deviations at the nearest-neighbour interface may be due to the finite nature of the perturbative series or may also be indicative of different corrections to scaling. At
, where the system dimension equals the upper critical dimension of the transition, there are multiplicative logarithmic corrections to the critical exponents [
125,
135,
308,
309,
310]. From the biased DlogPadé extrapolations at
, Adelhardt et al. [
31] determined these corrections to scaling at the upper critical dimension with a qualitatively good agreement for different
values.
To summarise the findings, we provide the following: Similar to the ferromagnetic LRTFIM, the ferromagnetic LRTFAXYM is a paradigmatic toy model extending the LRTFIM. With the pCUT+MC method described in
Section 4, the critical exponents as a function of the decay exponent
can be determined as demonstrated in Ref. [
31]. The LRTFAXYM displays three distinct regimes of quantum criticality including a mean-field regime above the upper critical dimension for
for all
. The sensitivity of the series expansion method suffices to even study corrections to scaling at the upper critical dimension. The limiting case
is especially interesting, as in the high-field limit, the ground state contains no quantum fluctuations, the dispersion is exactly solvable in first-order perturbation theory, the underlying QFT is different from the case
, and two instead of three critical regimes were found. We would expect that the setup of an SSE QMC algorithm following the directed loop idea [
215,
217,
311] is conceptually possible for the model.
7.3. Antiferromagnetic Anisotropic Long-Range XY Chain in a Transverse Field
For frustrated antiferromagnetic interactions, we begin our discussion with the Ising case at
. Here, we have already discussed in
Section 6.2 that, for the antiferromagnetic LRTFIM, there is strong numerical evidence that it remains in the 2D-Ising universality class for all
. It was also demonstrated that it is possible to determine critical gap exponents up to
with the pCUT+MC method [
29,
31,
35].
Following the same procedure as for the ferromagnetic model, the critical values
and the critical gap exponents
were studied in Ref. [
31], and we summarise these results in
Figure 32. The critical values
shift towards larger values when decreasing
, eventually diverging for
. It is possible to reliably determine the critical exponents for the anisotropic XY chain until
, but it becomes increasingly challenging to extract the exponents for smaller
due to the shifting of
[
31]. For
, the critical values
behave qualitatively in a similar way to
. However, the larger
, the more the high-field phase is stabilised (see the inset of
Figure 32). As a consequence, we observe more reliable exponent estimates in the regime
for
values closer to zero. In general, the observations in Ref. [
31] are in line with a constant gap exponent
within the limitations of the method.
This promotes the scenario that the nearest-neighbour 2D-Ising universality class remains the correct universality class for all considered
values and
. For
, the critical exponents
and critical values
were determined analytically in
Section 7.1, and it was shown that the universality class of the nearest-neighbour isotropic chain remains for all
[
31]. A recent study [
312] investigated the multicritical point using exact diagonalisation for comparably small system sizes. The critical points for
and
agree well with the exact limit and the pCUT+MC values. The study further confirms 2D-Ising universality up to
; however, in contrast to Ref. [
31], signatures of a different crossover criticality at
are claimed.
8. Long-Range Heisenberg Models
We review the results obtained from SSE and pCUT+MC calculations for Heisenberg models with long-range interactions. The starting point is the nearest-neighbour Heisenberg model, which can be written as
with the spin-1/2 operators
and
. Instead of a transverse field that induces quantum fluctuations in the (anti)ferromagnetic phase of the (LR)TFIM or (LR)TFAXYM, a magnetic field would immediately break the
-symmetry of the Heisenberg Hamiltonian. Here, we introduce a dimerisation limit. We consider two layers of lattices with Heisenberg spins on each site stacked on top of each other, where each spin couples to its nearest-neighbour. The nearest-neighbour coupling between the layers gives rise to interlayer dimers, also referred to as rungs. In one dimension, the layers are just two chains, resulting in a ladder system, while in two dimensions, we consider two square lattice layers, resulting in a square lattice bilayer model. Generalising this model for long-range interactions gives rise to intralayer, as well interlayer long-range couplings. The generic antiferromagnetic Hamiltonian is then given by
where the first term couples the nearest-neighbour spins between the layers with coupling strength
, the second term describes the long-range intralayer coupling with
, and the third term the long-range interlayer coupling of Heisenberg spins with
. The long-range interaction is given by
where
J is the coupling constant,
i and
j are the rung dimer indices, and
and
are the indices of the respective legs (layers) of the ladder (bilayer) model. In Equation (
319), we use the notation
and
. The long-range interaction introduced is similar to the one introduced in the sections before, however with the difference that the alternating sign in the numerator gives rise to staggered, non-frustrating antiferromagnetic long-range interactions, as we chose
. The exponent of the numerator is given by the one-norm
, while the usual geometric distance in the denominator given by
can be identified as the two-norm
. When increasing the overall interaction strength
J of intralayer and interlayer coupling and, likewise, making the long-range decay exponent
smaller, the Heisenberg spins will anti-align at odd distances and align at even distances, inducing a Néel-ordered antiferromagnetic phase. For
, the system becomes superextensive (analogous to the ferromagnetic LRTFIM in
Section 6.1), and for
, we recover the antiferromagnetic short-range ladder or bilayer model.
In order to employ the pCUT+MC method, we need an exactly soluble limit about which we can perform the series expansion. We introduce the perturbation parameter
rescaling the Hamiltonian by
and identify the first term of Equation (
319) as the unperturbed part
, which corresponds to a sum over Heisenberg dimers that can be easily diagonalised. For a single dimer, the lowest lying state with total spin
is the antisymmetric combination of
spins:
also called a rung singlet in our case with the associated energy
. The total spin
states:
are three-fold degenerate with the energy
and are called triplets. Alternatively, we can use an
-symmetric basis, where the singlet and triplet states read
There is also a convenient mapping from the spin operators to bosonic,
-symmetric operators creating and annihilating these states introduced in Ref. [
313], which can be readily adapted to hard-core bosonic operators for triplet excitations [
314]. We use the mapping
where
is the Levi-Civita symbol, and insert these expressions into Equation (
319). After some straightforward manipulations, we obtain
where
is the number of rungs,
, and
. The second and third line constitute the perturbation
associated with the physical perturbation parameter
. The
and
operators are absent, as terms contributing to these operators cancel out due to the reflection symmetry about the centre of the rung dimers.
For
, the ground state is given by a trivial product state of rung singlets, and for small, but finite
, the ground state is still adiabatically connected to this product state. We refer to this adiabatically connected ground state as the rung-singlet ground state. Elementary excitations in this phase are called triplons [
315], corresponding to dressed triplet excitations. For large
, we expect the non-frustrating antiferromagnetic long-range interaction to induce an antiferromagnetic phase giving rise to a quantum phase transition (QPT) between these two phases. In contrast to the transverse-field Ising model and the anisotropic XY model, where the ground state in the ordered phase spontaneously breaks the discrete
symmetry of the Hamiltonian, the Néel-ordered antiferromagnetic ground state for strong long-range couplings must break the continuous
symmetry of the Heisenberg Hamiltonian. Related to this observation, we present in the following two prominent theorems of great importance that apply to Hamiltonians with continuous symmetries:
First, there is the Hohenberg–Mermin–Wagner (HMW) theorem [
130,
131,
295], which rules out the spontaneous breaking of continuous symmetries in one- and two dimensional systems for
. From the quantum–classical correspondence (see Ref. [
113]), we can infer that, for one-dimensional systems, a QPT at
breaking such symmetry should be ruled out as well. Indeed, it was shown rigorously by Pitaevskii and Stringari in 1991 [
296] that it is prohibited in one dimension for
as well. However, there is the restriction that the interaction must be sufficiently short-ranged, i.e., the condition
for
must hold for the long-range decay exponent. A stronger condition was given by Ref. [
316] with
, where
d is the spatial dimension of the system. For the quantum case, it was shown in Ref. [
317] for a staggered antiferromagnetic long-range Heisenberg chain (
) that long-range order is absent for all
.
Second, Goldstone’s theorem [
318,
319,
320] applies. The theorem states that, when spontaneous breaking of a continuous symmetry occurs, it gives rise to massless excitations, also known as Nambu–Goldstone modes [
318,
319,
320]. For instance, magnons that are quantised spin–wave excitations are a manifestation of such gapless Nambu–Goldstone modes inside the ordered phase of Heisenberg systems. The recent findings in Ref. [
321] actually go beyond the conventional scenario of Goldstone modes due to long-range interactions. The authors identified three regimes depending on the decay exponent
: for ferromagnetic (antiferromagnetic) interactions, they found standard Goldstone modes for
(
) and an anomalous Goldstone regime with
and
(
) for
. Interestingly, a third regime for strong long-range interactions
(
) was found where the Goldstone modes become gapped via a generalised Higgs mechanism [
321]. A recent large-scale QMC study [
38] investigating the dynamic properties of the non-frustrating staggered antiferromagnetic square lattice Heisenberg model confirmed these three scenarios and found evidence that the Higgs regime already occurs in the regime
when the Hamiltonian is still extensive [
38]. Let us mention at this point that previous results from linear spin–wave theory [
22,
322] already indicated the existence of a sublinear dispersion in the staggered antiferromagnetic long-range Heisenberg chain a decade earlier. Remarkably, the existence of gapped Goldstone modes in the Heisenberg model with long-range interactions was already pointed out in the 1960s by Refs. [
323,
324].
The HMW theorem, as well as Nambu–Goldstone modes are a distinguishing feature of quantum systems with continuous symmetries like Heisenberg antiferromagnets. The quantum field theory describing dimerised Heisenberg antiferromagnets [
113] is given by the action
which is the same
n-component
-theory as the one describing the transverse-field Ising model. Here, the order-parameter field
is now a 3-component field instead of a 1-component one. We can readily include long-range interactions by adding the term
to the action (
326). For a more detailed discussion of the short-range
action, we refer to
Appendix A and to
Section 6.1.1, where the implications of long-range interactions were already discussed for
. The classical equivalent of the
action can describe the finite temperature phase transition of the ferromagnetic Heisenberg model [
1,
36,
113,
325]. Interestingly, the action of zero-temperature Heisenberg ferromagnets is not given by the above action [
113]. In fact, the quantum field theory describes a class of dimerised Heisenberg antiferromagnets, which can be appropriately described by Equation (
326) since their low-energy physics can be mapped onto the
quantum rotor model because pairwise antiferromagnetically coupled Heisenberg spins are an effective low-energy representation of quantum rotors [
113]. We can see this correspondence by looking at the Hamiltonian of the
quantum rotor model:
where the first part
with
and
the angular momentum operator gives the kinetic energy of the rotors. The second part
with
is the interaction between the three-component quantum rotors inducing parallel ordering of the rotors [
113]. The physical picture is that the kinetic energy is minimised when the orientation of the rotors is maximally uncertain and the energy of the interaction term is minimised when the rotors are aligned [
113]. The eigenvalues of the kinetic term of a single quantum rotor are given by
with
corresponding to a
-fold degenerate state. Now, given two spins
S interacting antiferromagnetically with coupling strength
forming a dimer, the total spin is
and the eigenenergies are given by
with a
-fold degeneracy. This gives a one-to-one correspondence to the kinetic energy of a rotor, but with an upper bound for the energy. Thus, this mapping is only valid when considering the low-energy properties of the models [
113] and holds certainly for large
. We just introduced a powerful mapping from the low-energy properties of the antiferromagnetic Heisenberg systems like ladder and bilayer models forming Heisenberg dimers on the rungs (see Equation (
319)) to an
quantum rotor model, which is described by the action (
326). It turns out that this mapping is not only valid for large
K/
J, but also in the ordered phase and at the quantum critical point [
113]. To include algebraically decaying long-range interactions, the term (
327) must be added to the action. The implications of long-range interactions for an
n-component order-parameter field in the
field theory was already extensively discussed in the 1970s [
1,
2,
3] for the classical action, but it took until 2001 for the quantum analogue to be studied by Ref. [
20]. More studies followed by N. Defenu et al. employing functional RG approaches [
21,
244,
326].
The predictions for long-range Heisenberg models are in large part the same as for the LRTFIM. We expect long-range mean-field behaviour with a Gaussian fixed point for
, a regime of nearest-neighbour criticality for
, and a non-trivial regime with continuously varying critical exponents in between [
20,
21,
244,
326]. For
, however, there is the major difference that a QPT associated with the spontaneous breaking of continuous symmetries is ruled out by the HMW theorem [
130,
131,
295,
296], at least for
[
21,
244,
317,
326]. The boundary is then called lower critical exponent
. For the Heisenberg model, the previously discussed action (
326) with long-range interactions (
327) holds, which together with the HMW theorem rules out the breaking of the order-parameter field for sufficiently short-range interactions and, therefore, a QPT in the regime
. For
, a QPT breaking the continuous symmetry is predicted [
20,
21,
244,
326]. Indeed, it has been long confirmed by several numerical studies [
22,
297,
298,
299,
300,
301,
302,
303,
304,
305,
306,
327] that the HMW theorem can be circumvented if the long-range interaction is sufficiently strong. Notably, in recent experiments with trapped ions [
328] and Rydberg atoms [
329] continuous symmetry breaking was realized in one and two dimensions which would be prohibited in the abscence of long-range interactions.
So far, most of the studies considered only Heisenberg Hamiltonians with long-range interactions in one-dimensional systems [
22,
35,
297,
298,
299,
300,
301,
302,
303,
304,
305,
306,
327], however often considering various modifications to the Hamiltonian. Some studies considered a one-dimensional Heisenberg chain with non-frustrating (staggered) antiferromagnetic long-range interactions [
22,
300,
305], while others included an anisotropy along the
z-components resulting in the
-model [
302,
303,
327]. An interesting example with frustrating long-range interactions is the exactly solvable Haldane–Shastry model [
330,
331] with
, which is known to show quasi-long-range order (QLRO) just like the conventional Heisenberg chain, but with vanishing logarithmic corrections [
332]. For the dimerisation transition between the QLRO and a valence bond solid (VBS), the frustrated long-range interaction seems to play only a minor role compared to the
-
model [
213]. Based on this, in Refs. [
213,
297,
298,
299,
333], a model combining the frustrated
-
model with additional non-frustrating staggered long-range interaction was investigated. The initial intention was to realise a one-dimensional analogue of deconfined criticality [
334,
335,
336,
337] between a Néel-order phase and a VBS. As the transition was found to be of first order, the authors in Ref. [
338] later turned to the one-dimensional long-range
J-
Q model, indeed finding evidence for a continuous deconfined QPT. Interestingly, a very recent paper [
327] also predicts a deconfined QPT in the antiferromagnetic long-range XXZ chain. There are also studies considering larger spin
. In Ref. [
304], a Heisenberg chain with single-ion anisotropy was studied, while in Ref. [
301], the XXZ chain was under scrutiny. In both cases, a rich phase diagram was found with intriguing quantum critical properties. For instance, continuous symmetry breaking due to long-range interactions gives rise to a new, possibly exotic, tricritical point in the XXZ chain with no analogue in short-range one-dimensional spin systems [
301].
In
Section 6 and
Section 7, we reviewed the results of various numerical studies investigating the quantum-critical properties of the LRTFM [
23,
24,
25,
26,
28,
29,
30,
32,
33,
34] and the LRTFAXYM [
31] with the focus on extracting the critical exponent as a function of the long-range decay
. In comparison to the LRTFIM, for long-range Heisenberg models, just recently, efforts have been made to extract the critical exponents associated with the QPT as a function of the long-range decay exponent [
22,
35,
37,
206]. Reference [
22], studying a one-dimensional long-range Heisenberg chain, is one notable exception in the sense that it was ahead of its time, performing large-scale SSE QMC simulations for long-range Heisenberg systems already in 2005, way before the rapid progress in implementing quantum simulators in quantum optics reignited the interest in long-range interacting systems.
In the following, we will first review the results from Refs. [
37,
206,
339] for the long-range square-lattice Heisenberg bilayer model in
Section 8.1. Then, we proceed with the discussion of the results from Ref. [
35] for long-range Heisenberg ladders in
Section 8.2. Finally, we conclude this section with the discussion of the long-range Heisenberg chain from Ref. [
22] in
Section 8.3.
8.1. Staggered Antiferromagnetic Long-Range Heisenberg Square Lattice Bilayer Model
In this section, we consider two square lattices stacked directly on top of each other, where the Heisenberg spins on each lattice site interact with their nearest neighbours. When the two spins on top of each other form a rung dimer interacting with coupling strength
, the Hamiltonian describing the bilayer system is given by Equation (
319). Here, we neglect the long-range interlayer interactions, which results in the Hamiltonian
We consider staggered non-frustrating long-range interactions along the layers
of the form (
320). The short-range model (
) was the subject of several studies from the 1990s onward [
162,
183,
184,
340,
341,
342,
343,
344,
345,
346] investigating its quantum-critical properties. For a critical coupling ratio
, the system undergoes a QPT breaking the continuous
symmetry of the Hamiltonian from a ground state adiabatically connected to the product singlet state towards an antiferromagnetic Néel ground state with gapless magnon excitations (Nambu–Goldstone modes). For long-range interactions, we expect that these Goldstone modes are altered [
38,
321] and that the criticality of the system changes as a function of the decay exponent
. We expect short-range criticality until
, a non-trivial regime of continuously varying criticality in
with
, and a long-range mean-field regime for
[
20,
21,
244,
326].
Until recently, to the best of our knowledge, there was a complete absence of numerical confirmation of this scenario. The first data for the critical point and exponents as a function of
were published by Song et al. [
37] using the SSE QMC algorithm and is now supplemented by pCUT+MC data from Adelhardt and Schmidt [
206,
339].
In
Figure 33, the critical values from Refs. [
37,
206,
339] are plotted together with the results from functional renormalisation group (FRG) calculations from Ref. [
21] for the
quantum rotor model. The critical points
for both methods are in excellent agreement over the entire
-range. However, the critical exponents cannot be directly compared since the FSS of the magnetisation curves from SSE QMC simulations give
and
, while pCUT+MC calculations for the gap and spectral weight give
and
. While the QMC data show relatively large error bars in the long-range mean-field regime, the pCUT approach overestimates the critical exponents in the short-range regime. For the two-dimensional long-range Heisenberg bilayer, the deviation is larger than for the LRTFIM in two dimensions. This is no surprise, as a previous series expansion for the nearest-neighbour square-lattice Heisenberg bilayer model showed similar deviations [
347]. Also, the critical exponents from QMC are better at capturing the boundaries of the long-range mean-field and the short-range regime. In general, both approaches show good agreement with the FRG results from Ref. [
21] for the non-trivial intermediate regime (which apparently has its own shortcomings at the boundary to the short-range regime).
In conclusion, we summarised the results from SSE QMC and pCUT+MC studies for the square-lattice Heisenberg bilayer model with staggered antiferromagnetic interactions. Both approaches are in good agreement and confirm the three critical regimes [
1,
2,
3,
20,
21,
244,
326] from the two-dimensional
quantum rotor model within their limitations.
8.2. Staggered Antiferromagnetic Long-Range Heisenberg Ladder Models
After the discussion of the square-lattice bilayer model, we can imagine the Heisenberg ladder models as effectively reducing the dimension of the square-lattice bilayer model by one now considering two linear chains coupled by rung interactions. The long-range ladder Hamiltonian then reads
with long-range coupling
along the legs of the ladder and
between spins of different legs. Recalling the long-range interactions of Equation (
320) and the definitions of
and
, thereafter, we define two distinct Hamiltonians
and
, where the first one includes long-range interactions only along the legs, while the second one is the original Hamiltonian including long-range interactions both along and in between the legs.
A sketch of these ladders is provided in
Figure 34. As for the bilayer model, for small
(cf. Equation (
320)), the ground state is adiabatically connected to the product state of rung singlets (rung-singlet ground state), while for strong coupling ratios, the long-range interactions want to induce an antiferromagnetic ground state. However, in contrast to the bilayer model in
Section 8.1, there is no QPT for the nearest-neighbour Heisenberg ladder [
350,
351,
352], due to the HMW theorem ruling out continuous symmetry breaking for one-dimensional quantum models [
296]. Note that the HMW theorem only rules out a QPT with continuous symmetry breaking and not a QPT in general. For instance, the nearest-neighbour isotropic XY model in a transverse field exhibits a QPT without breaking the
symmetry [
284,
285,
286,
287,
288,
289,
290,
291,
292,
293], while in the nearest-neighbour Heisenberg ladder, there is no QPT at all [
350,
351,
352]. In fact, a QPT was ruled out until
in another one-dimensional model, namely the staggered antiferromagnetic long-range Heisenberg chain in Ref. [
317]. As there is a one-to-one correspondence between antiferromagnetic Heisenberg ladders and the low-energy properties of the one-dimensional
quantum rotor model [
113], we can expect a QPT predicted by the quantum field theory given by the action (
326) with Equation (
327) for
with
[
20,
21,
244]. The existence of a lower critical decay exponent
has the consequence that there are only two quantum-critical regimes. One is the long-range mean-field regime for
, and the other one is a regime of a continuously varying critical exponent with a non-trivial fixed point. The third regime in these models is non-critical.
The only study investigating the full parameter space of this model is Ref. [
35] using pCUT+MC and complementary linear spin–wave calculations. A previous study [
306] using QMC and DMRG investigated the
parameter line for
. There are also known limiting cases of decoupled staggered long-range Heisenberg chains at
, where a QPT from a QLRO phase towards the same Néel-ordered antiferromagnetic phase occurs. We can compare the critical points determined by pCUT+MC for
and the linear spin–wave results to the SSE QMC data from Ref. [
22] and also to the linear spin–wave calculations in Refs. [
22,
322] of the long-range Heisenberg chain. In fact, the linear spin–wave calculations for
can be seen as a generalisation of the ones for the Heisenberg chain and, therefore, exactly including them as a limiting case.
We can find a plot in
Figure 35 showing a ground-state phase diagram for both
and
from Refs. [
35,
353]. The figure also includes other known values from Refs. [
22,
306], which fit into the overall picture of the pCUT+MC results. We also show the critical exponents
,
, and
determined by the pCUT+MC approach. It is easy to identify the two critical regimes of long-range mean-field behaviour and continuously varying critical exponents as predicted by quantum field theory [
20,
21,
244]. In the non-trivial regime, the exponents shown here diverge when approaching the lower critical dimension
, which is in reasonable agreement with FRG results in Refs. [
21,
244]. The largest deviation can be seen for
, which is the hardest exponent to extract from DlogPadé extrapolations. Using the (hyper)scaling relations (
3)–(
9), the remaining critical exponents can be determined. See
Figure A2 in
Appendix D showing all critical exponents for the Heisenberg ladders. It should be noted that there are three main difficulties discussed in Ref. [
35]. First, as just stated, the
exponent is difficult to determine. Second, it becomes increasingly hard to extrapolate the perturbative series in the regions
(
) for
(
). Third, the presence of logarithmic corrections to the dominant power-law behaviour close to the critical point spoils the exponents around the upper critical dimension at
. In the end, all factors play a role when determining all critical exponents due to error propagation. We can observe in
Figure A2 that several exponents from the pCUT+MC approach deviate from the FRG exponents significantly in the non-trivial regime. For instance, the
exponent seems to approach a constant value
for
for pCUT+MC, while the FRG predicts a diverging exponent. Another important finding is that the lower critical decay exponent
is apparently not universal in these two models and considerably smaller than the predicted value
from quantum field theory [
21,
244]. This claim is made in Ref. [
35] due to the known limiting case of the long-range Heisenberg chain [
22] from SSE QMC simulations and due to linear spin–wave calculations [
35].
Beyond these interesting discrepancies, there was speculation about a possible deconfined quantum critical point along the
parameter line for
in Ref. [
306]. The reason for this was the fact that the staggered long-range Heisenberg ladder undergoes a QPT from a disordered phase with a non-local string order parameter towards a Néel-ordered phase with conventional order. Also, they found a sharp peak and a gap in the dynamic structure factor at the ordering momentum
in the ordered phase, which could be indicative of deconfined excitations in terms of spinons [
306,
354]. Usually, when there is a QPT between two competing ordered phases, the system undergoes a first-order phase transition or there must be a coexistence phase. There is also a much more exotic scenario beyond the Landau–Ginzburg–Wilson theory of phase transitions [
334]. A deconfined quantum critical point [
334,
335,
336,
337] is a second-order QPT that is not described by a “confining” order parameter, but by an emergent
-symmetric gauge field with “deconfined” degrees of freedom accompanied by a fractionalisation of the order parameters [
334]. A paradigmatic example is the deconfined QPT between a Néel-ordered antiferromagnetic ground state and a VBS state on a two-dimensional lattice [
334,
335,
337,
355,
356,
357,
358,
359,
360]. While deconfined criticality was originally proposed in two dimensions, similarities in terms of a conventional Luttinger Liquid theory description have been drawn in one dimension [
361,
362,
363,
364,
365,
366,
367,
368]. Interestingly, a very close analogy to a two-dimensional deconfined critical point was found in Ref. [
338] using a toy model with six-spin Heisenberg interactions and long-range two-spin interactions inducing a continuous phase transition between a VBS phase and an antiferromagnetic phase. A scenario between a conventional order and non-local string order as proposed by Ref. [
306] would go even beyond the one-dimensional scenarios found so far. However, it was argued in Ref. [
35] that there should be no such deconfined critical point in the above long-range Heisenberg ladders due to the critical exponents found and the fact that the rung-singlet phase is adiabatically connected to the trivial product state of rung singlets not falling into the category of symmetry-protected topological phases despite the presence of a non-local string order parameter [
35,
369]. Another possible interpretation of the finding in Ref. [
306] is probably along the lines of Refs. [
38,
321]. The observed gap in the dynamic structure factor is in agreement with strong finite-size artefacts arising from the altered dispersion
of sublinear behaviour
in the anomalous Goldstone regime
.
In this subsection, we have seen that the antiferromagnetic Heisenberg ladders with staggered long-range interactions show two quantum-critical regimes: one regime with long-range mean-field behaviour and a second non-trivial regime with continuously varying critical exponents. There is also a third regime that does not show any QPT because continuous symmetry breaking is ruled out by the HMW theorem. Despite the numerical confirmation of the two critical regimes predicted from quantum field theory [
20,
21,
244,
326], a discrepancy between the upper bounds of the Néel-ordered phase and the predicted lower critical exponent
was identified by Ref. [
35]. On the other hand, the data in Refs. [
35,
353] are in agreement with other literature for the limiting case of long-range Heisenberg spin chains [
22,
322]. We also discussed briefly the possibility of a deconfined QPT and the presence of anomalous Goldstone modes in one dimension for the above ladder models. To put it briefly, the Hamiltonian (
330) hosts some intriguing physics with several aspects that need further clarification.
8.3. Staggered Antiferromagnetic Long-Range Heisenberg Chain
Lastly, we consider another one-dimensional system, the Heisenberg chain with staggered non-frustrating antiferromagnetic long-range interactions:
In the previous two models (bilayer and ladders), the unperturbed part at
consisted of uncoupled dimers with a trivial product singlet state as its ground state and local triplet excitations above. Here, the unperturbed part is the nearest-neighbour Heisenberg chain with a ground state exhibiting quasi-long-range order (QLRO) and fractionalised elementary excitations. These excitations are referred to as spinons and can be seen as propagating domain walls carrying
degrees of freedom. The perturbation consists of long-range interactions that couple sites beyond the nearest-neighbours. This interaction is of the same algebraic form as Equation (
320). Because of the non-frustrating nature of the antiferromagnetic long-range interactions, it induces a Néel-ordered antiferromagnetic phase upon increasing its coupling strength. Again, a QPT breaking the
-symmetry of the Hamiltonian is only allowed when the long-range decay exponent satisfies
due to the HMW theorem [
22,
317], and thus, such a transition can be ruled out for larger decay exponents. In this model, a QPT from a QLRO towards an ordered phase is expected to occur, and therefore, the
theory of Equation (
326) does not apply. The
Wess–Zumino–Witten non-linear
model [
370,
371] is known to describe the low-energy physics of Heisenberg chains and includes topological coupling to account for the presence of QLRO in the lattice model [
22,
213]. As for the
theory, a long-range coupling analogous to Equation (
327) can be added to describe the Hamiltonian (
331) [
22].
Reference [
22] is a comprehensive study of the Heisenberg chain (
331) using large-scale SSE QMC simulations to extract the critical properties of the QPT. The results for the critical point, as well as the critical exponents
and
z can be found in
Figure 36. The overall behaviour of the critical point as a function of the decay exponent
is very similar to the one found in the previous subsection for the Heisenberg ladder. Here, the critical point diverges at about
when approaching the lower critical exponent
. The hard boundary for a QPT is again given by the HMW theorem. Yet, there is a significant difference. While, for the Heisenberg ladders, the disordered rung-singlet phase exists for any
, for the Heisenberg chain, the QLRO phase only exists for
. Thus, for the Heisenberg chain, the critical line terminates in a marginal point at
and
. For any
, the perturbation parameter
becomes irrelevant, and the system is always in the antiferromagnetic phase [
22]. The long-range Heisenberg chain (
331) was also studied in Refs. [
213,
297,
333] in the context of a Heisenberg chain with frustrated next-nearest-neighbour and non-frustrating long-range interactions, i.e., the
chain with staggered long-range interactions. In both models, the QLRO-Néel QPT can be identified by a level crossing between a triplet
and quintuplet
excitations (The transition was initially misidentified as a level crossing between two
states in Refs. [
213,
297] until it was later clarified to be a level crossing between the
and
states [
333]). The critical point from finite-size scaling of the level crossing from the ED [
297] and DMRG [
333] is in very good agreement with the QMC results at
(see
Figure 36). Proceeding with the critical exponents, we can see that the critical exponent
matches well with the field-theoretical expectations
(linear in at least leading order, but also a simple scaling argument predicts the linear behaviour [
22]) in the range
. For
, the
from SSE QMC simulations starts to deviate from the linear behaviour. One interesting observation was pointed out in Ref. [
35]. In both the Heisenberg ladders and the Heisenberg chain, the linear behaviour
is expected from the underlying quantum field theory, yet the data from pCUT+MC and SSE QMC studies indicate a deviation from this with
for the Heisenberg ladders and
for the Heisenberg chain. It should be noted, however, that the exponent from the pCUT+MC approach is determined using scaling relations and, therefore, can suffer from unfavourable error propagation, especially when the
exponent is involved. The dynamical exponent
z of the Heisenberg chain was extracted from SSE QMC simulations as well. The exponent
z is one at the marginal point
and then quickly drops to
, where it seems to be constant within the error bars up to
. This finding is also in contrast to the RG prediction, where
in leading order, and the exponent is expected to be constant even in higher orders [
22]. In Ref. [
213], for
, the dynamic exponent
z was determined in excellent agreement with the QMC results (see
Figure 36). Also, the results for the
model with non-frustrating long-range interactions, where the same QLRO-Néel transition is realised, the exponent is in agreement with
[
213,
297]. Further, the RG analysis in Ref. [
22] gave also a prediction for
. However, this could not be compared with QMC as it was not possible to obtain accurate estimates of
[
22]. Note, also, that neither the long-range transverse-field XY chain nor the Heisenberg chain show long-range mean-field behaviour.
The Heisenberg chain with staggered long-range interactions is another prime example of long-range models hosting intriguing critical behaviour. The remaining discrepancy between the numerical SSE QMC results and the underlying field-theoretical description shows that the critical properties are not yet fully settled and further exploration of the model is necessary.
9. Summary and Outlook
In this review, we gave an overview of recent advances in the investigation of the quantum-critical properties of quantum magnets with long-range interactions focusing on two techniques, both based on Monte Carlo integration, but complementary in spirit. On the one hand, we described pCUT+MC, where classical Monte Carlo integration is decisive in the embedding scheme of white graphs. This allows extracting series expansions of relevant physical quantities directly in the thermodynamic limit. On the other hand, SSE QMC enables calculations on large finite systems where finite-size scaling can be used to determine the physical properties of the infinite system. Both quantitative and unbiased approaches take the full long-range interaction into account and can be used a priori in any spatial dimension for any geometry.
In recent years, both techniques, alongside other methods, have been applied successfully to one- and two-dimensional quantum magnets involving long-range Ising, XY, and Heisenberg interactions on various bipartite and non-bipartite lattices. In this work, we have summarised the obtained quantum-critical properties including quantum phase diagrams and the (full sets of) critical exponents for all these systems coherently. Further, we reviewed how long-range interactions are used to study quantum phase transitions above the upper critical dimension and how the scaling techniques are extended to extract these quantum critical properties from the numerical calculations. This is indeed generically the case for all unfrustrated systems in this review, with the exception of the one-dimensional isotropic XY and Heisenberg chain. For frustrated systems, one can apply both MC techniques successfully for Ising interactions, while in general, only the pCUT+MC method is applicable (if an appropriate perturbative limit exists) due to the sign problem of SSE QMC. Nevertheless, in all frustrated cases, the small- regime of long-range interactions is challenging for both approaches, and further technical developments are desirable.
In the future, several extensions and research directions are interesting. As mentioned in
Section 6.3 of this review, the interplay between long-range interactions and geometric frustration is a vibrant research field at the moment. It has been demonstrated numerically and experimentally that this interplay provides a great resource to engineer exotic phases of matter [
54,
55,
109,
283,
372] with the most spectacular example being the
quantum spin liquid on the Ruby lattice [
55,
109,
372]. We expect further rapid development in the field since many promising theoretical proposals can be realised in analogue quantum simulation platforms (e.g., programmable Rydberg atom quantum simulators [
52,
55]).
In terms of methods, pCUT+MC is yet to be extended to arbitrary unit cells, larger spin values, and multi-spin interactions. The access to larger unit cells will enable the investigation of the interplay between long-range interactions and frustration on even more relevant lattice structures, e.g., the Kagome or Ruby lattice. There are systems with multi-spin interactions hosting deconfined quantum criticality [
338,
355,
356,
357,
358,
359], and an introduction of long-range interactions to this type of systems seems to be an interesting research topic [
338]. We hope to spark further interest in the development and application of pCUT+MC by other users.
The SSE QMC approach is a widely used numerical tool for the calculation of unbiased thermal averages of observables. A large variety of distinct QPTs is potentially accessible by these QMC simulations. We envision that, for all systems that do not suffer from a sign problem, SSE QMC, in combination with appropriate zero-temperature protocols and finite-size scaling, can be used to study how long-range interactions affect QPTs beyond the standard
-symmetry. The SSE QMC method has also been extended to tackle frustrated systems in a more efficient way [
261,
262,
373]. However, an efficient treatment of both the long-range interaction and frustration has not been introduced yet [
32,
40]. Reference [
374] developed an SSE QMC approach to access the toric code quantum spin liquid regime [
55,
109,
372]. An application of the SSE QMC approach to extended long-range interacting Bose–Hubbard models (see
Section 5.5) along the lines of directed loop updates [
215,
216,
217] would also be a natural development. This would enable numerically calculating observables with SSE QMC for ultracold gas experiments with optical lattices. A possible application could be the study of complex crystalline phases and their breakdown in frustrated Bose–Hubbard systems with long-range interactions [
64,
73,
111,
230].
Finally, in the context of the long-range mean-field regime above the upper critical dimension, much research has been conducted regarding finite-size scaling in classical systems [
12,
13,
14,
15,
16,
17,
18,
19,
41,
125,
128,
142,
145], including the study of multiplicative logarithmic corrections for the characteristic length scale at the upper critical dimension [
17] and the investigation of the role of Fourier modes and boundary conditions [
19]. On the contrary, its quantum counterpart has only been treated successfully in recent years [
32,
34]. In addition to the transfer of established concepts from classical to quantum Q-FSS, one interesting open question is a detailed understanding of the crossover regime between classical and quantum Q-FSS for small temperatures. Even though we focused on the quantum version of the Q-FSS of Ref. [
34] due to the quantum nature of the models analysed in this review, the ground work has been conducted by the inventors of classical Q-FSS (Refs. [
15,
16,
17,
18,
19,
41,
125]) and, in general, many other researchers who provided valuable insight into the scaling above the upper critical dimension, e.g., Refs. [
10,
12,
13,
14,
127,
128,
142]. Overall, it is exciting that the abstract concept of dangerous irrelevant variables and the physics above the upper critical dimension are accessible in quantum–optical platforms realising long-range interactions.











































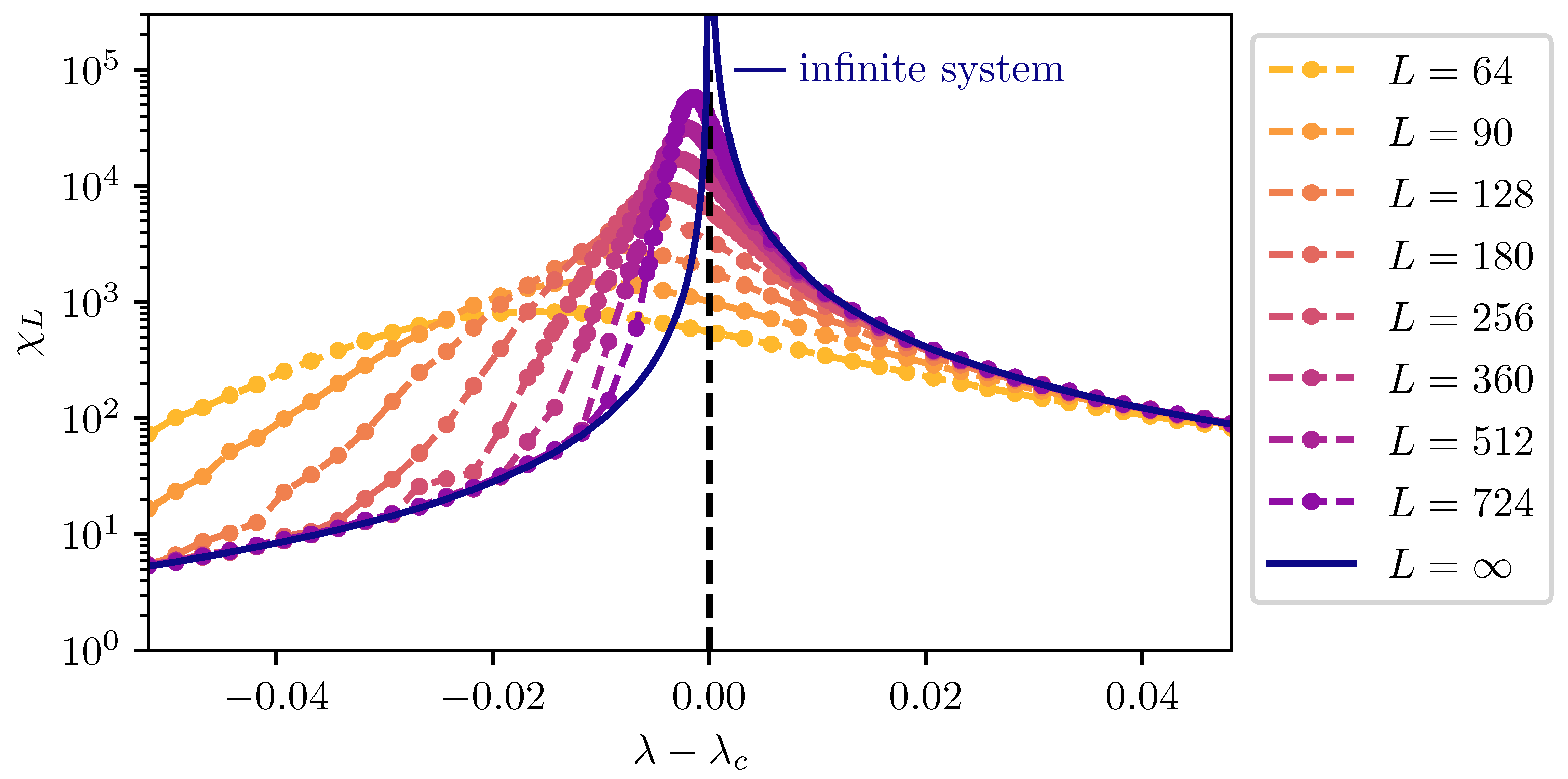{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}