A Quantum Chemical Investigation into the Molecular Mechanism of the Atmospheric Reactions of Chemi-Ions with Nitrogen and Nitrogen Oxides

 and
and
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
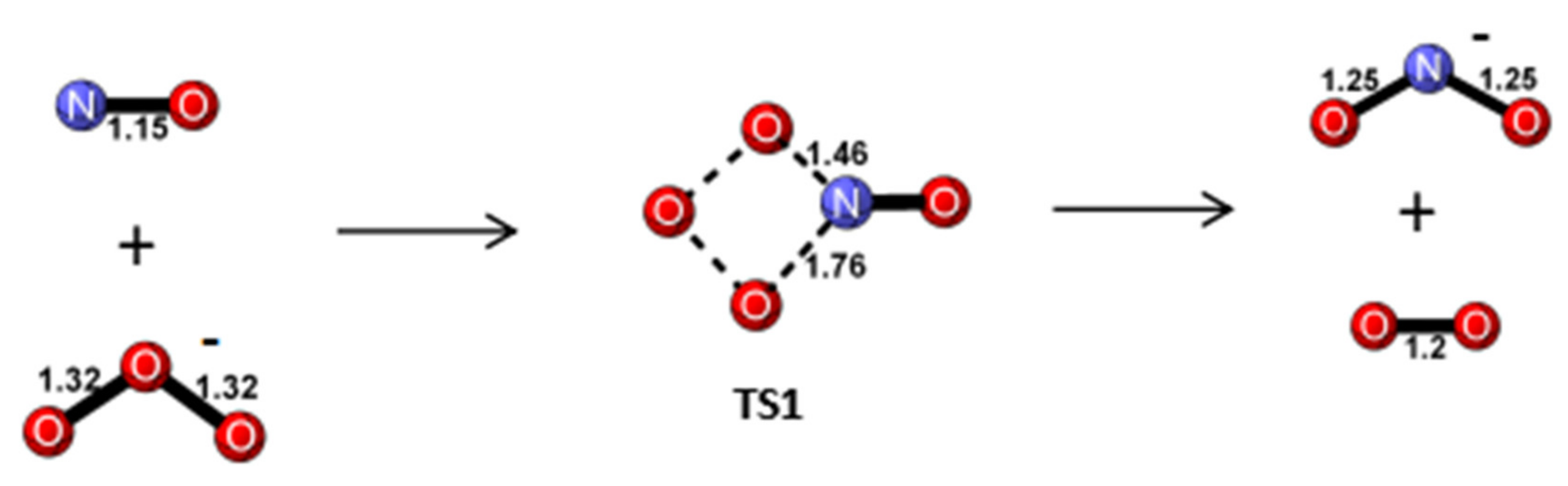
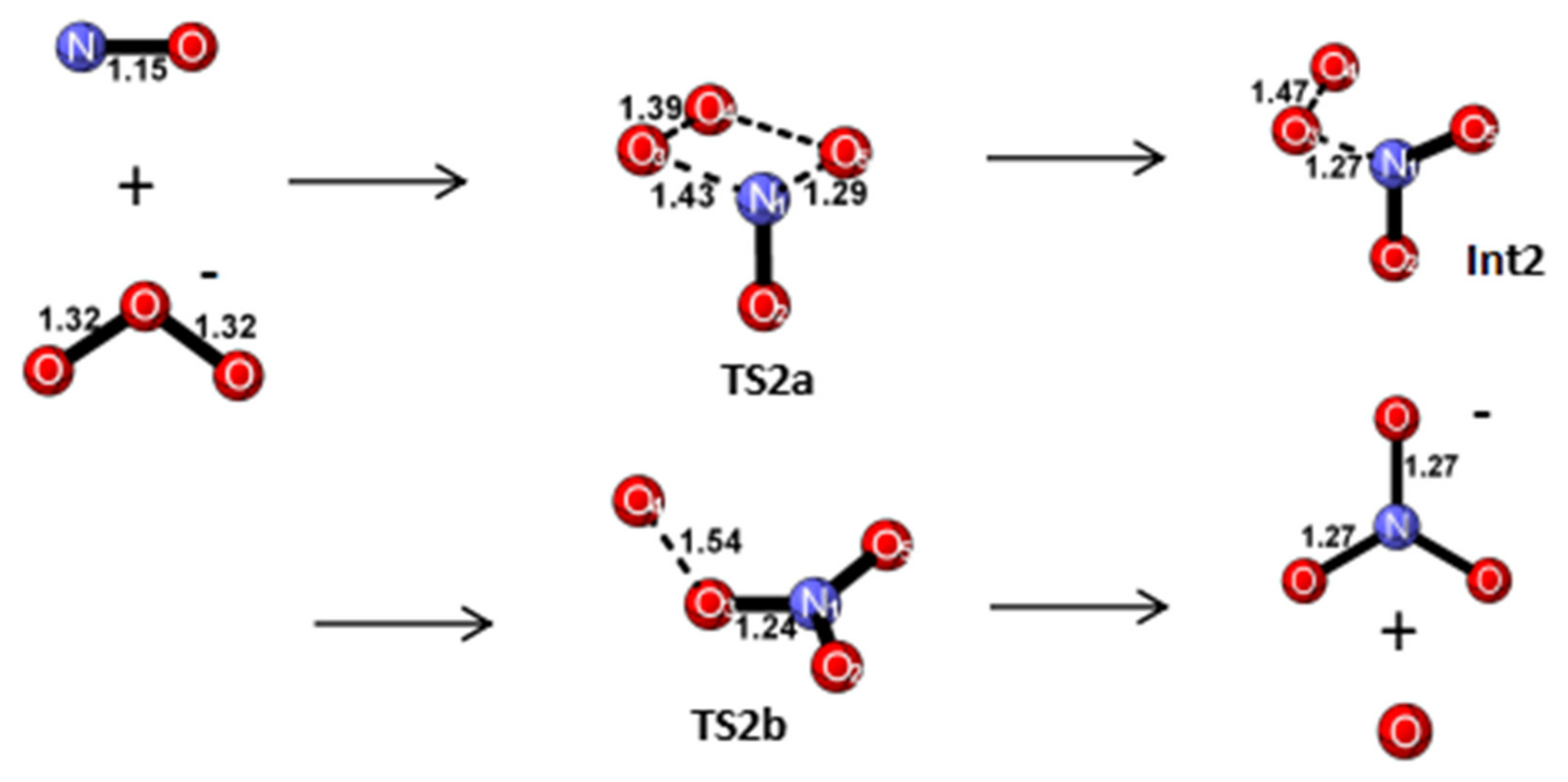
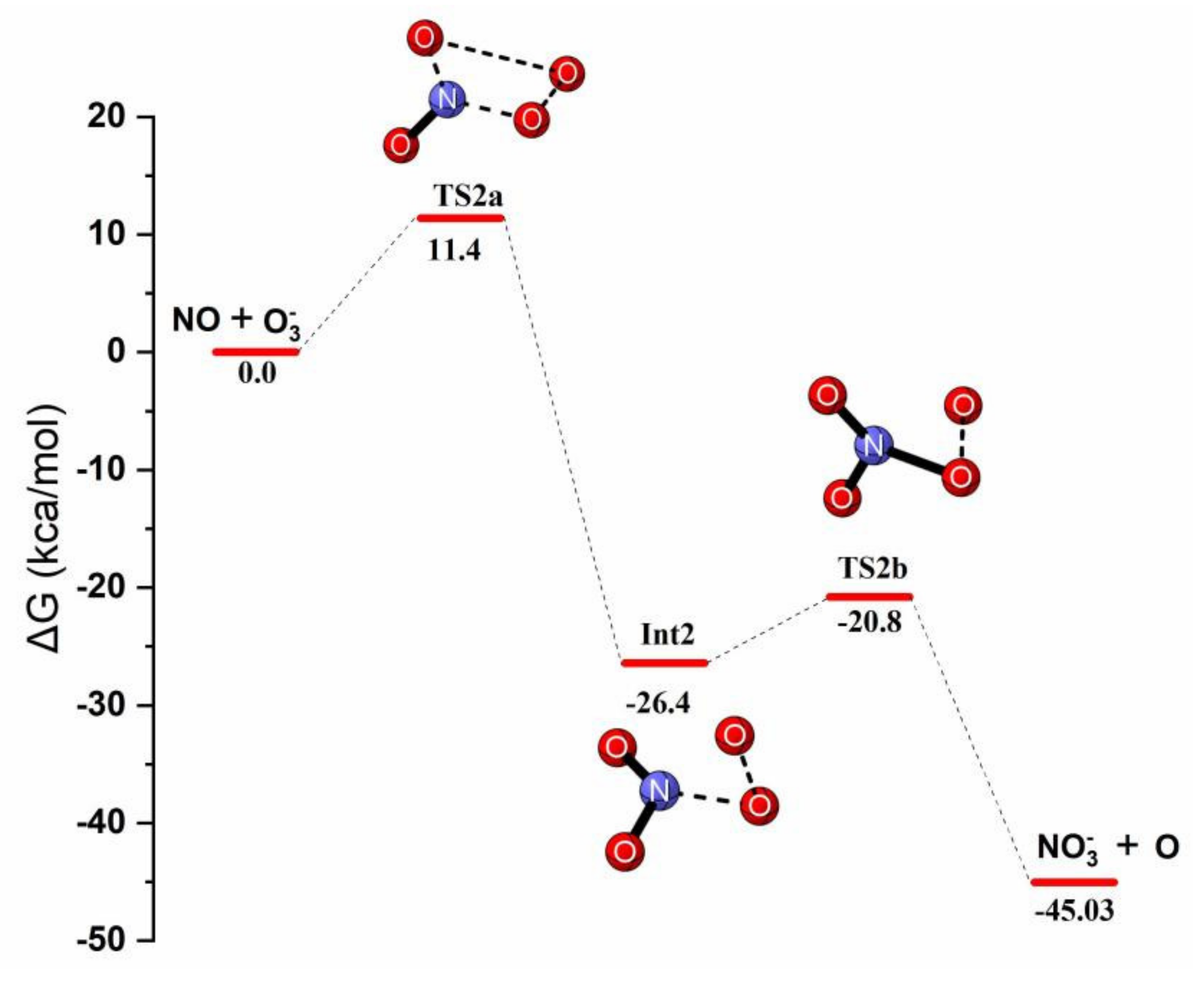
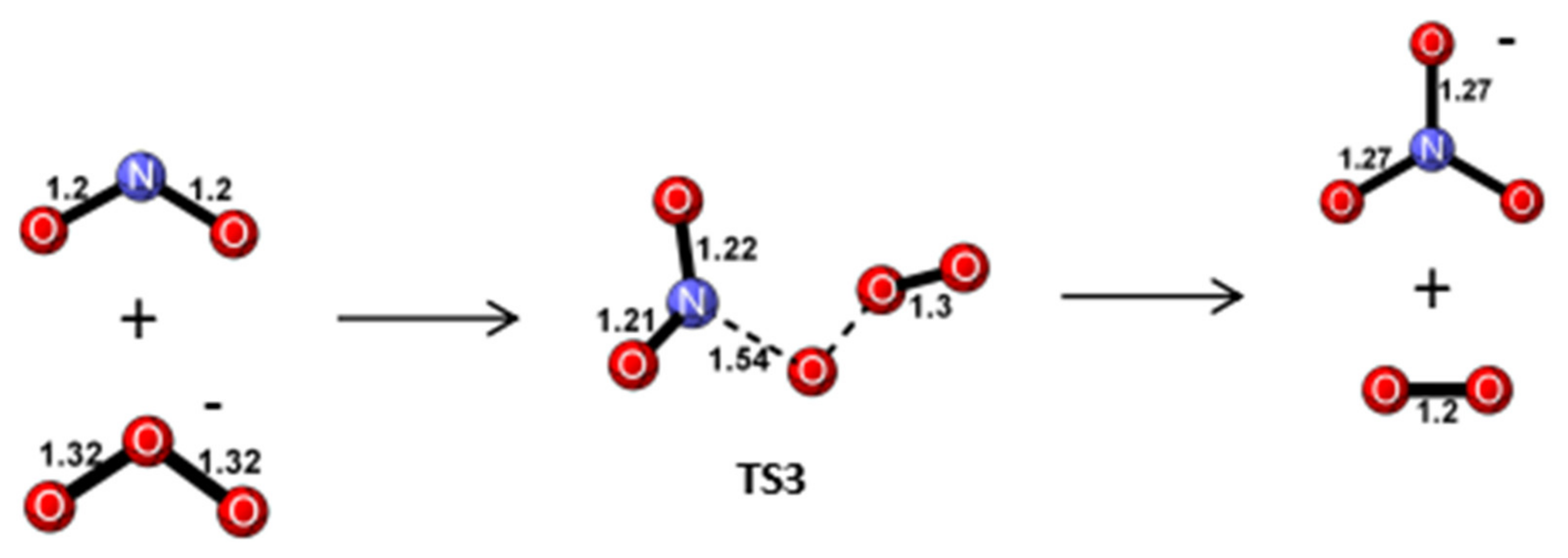
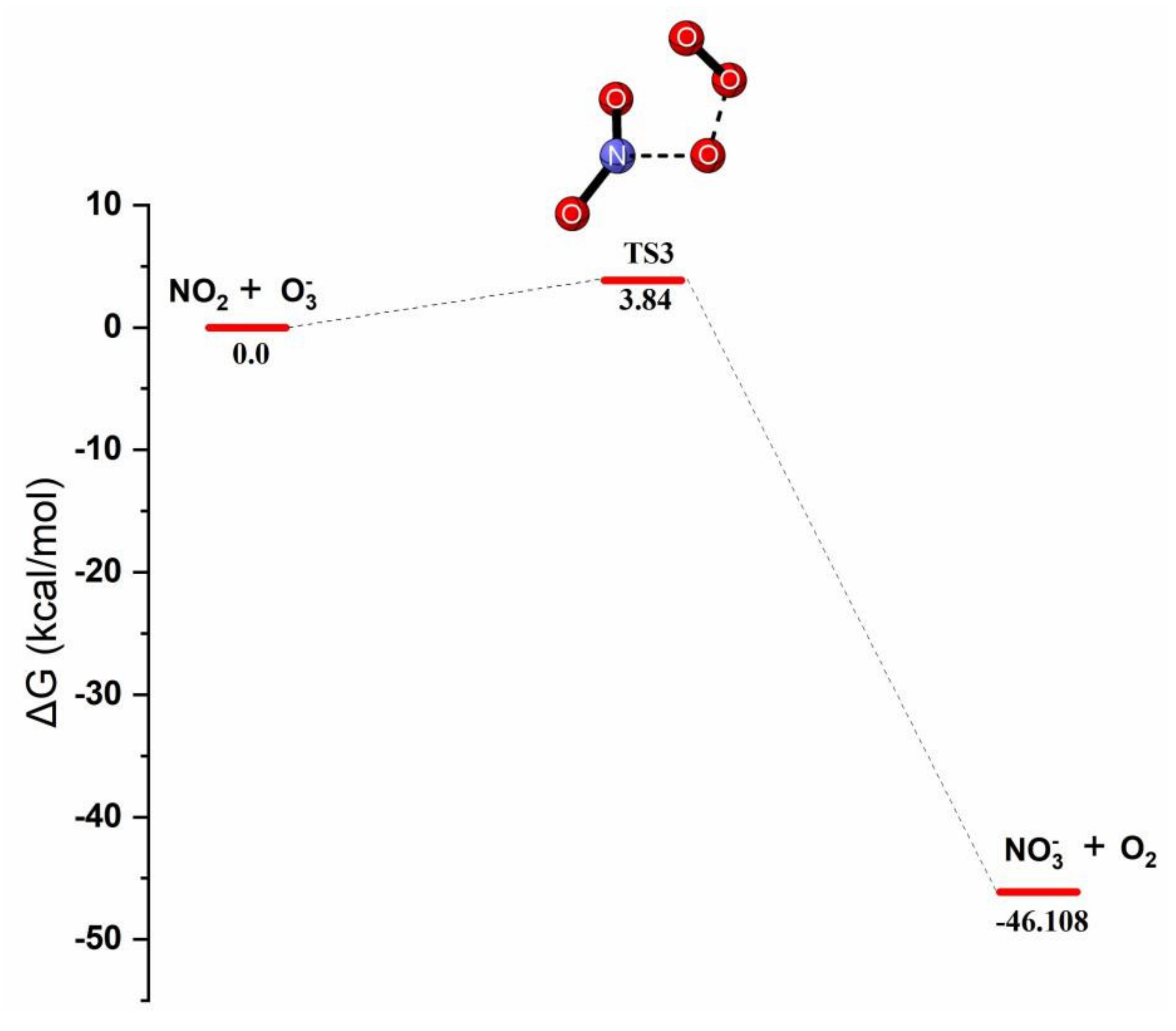
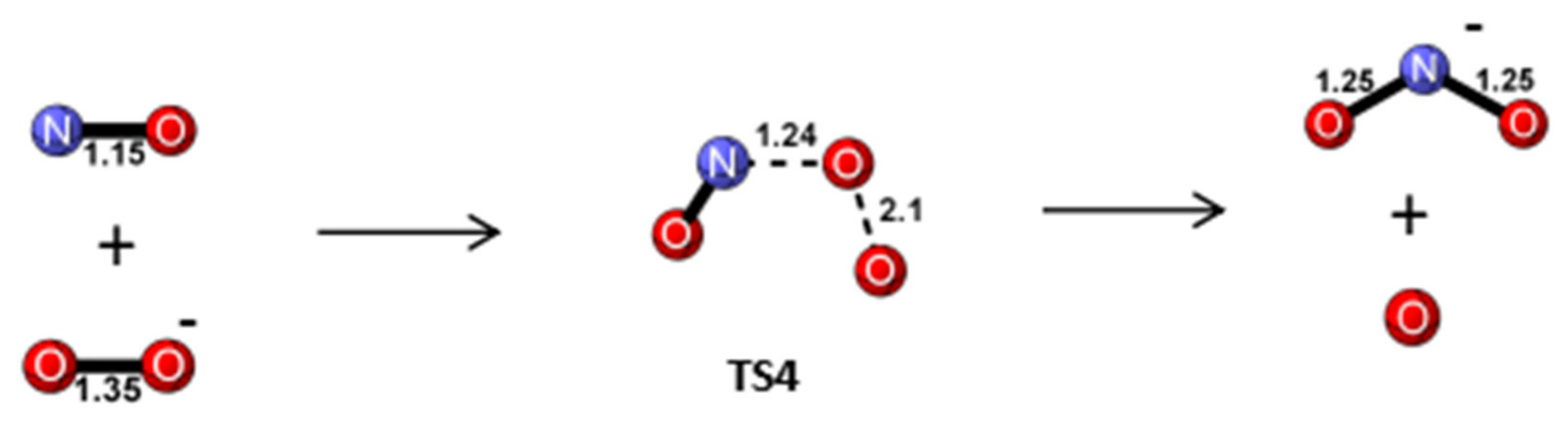
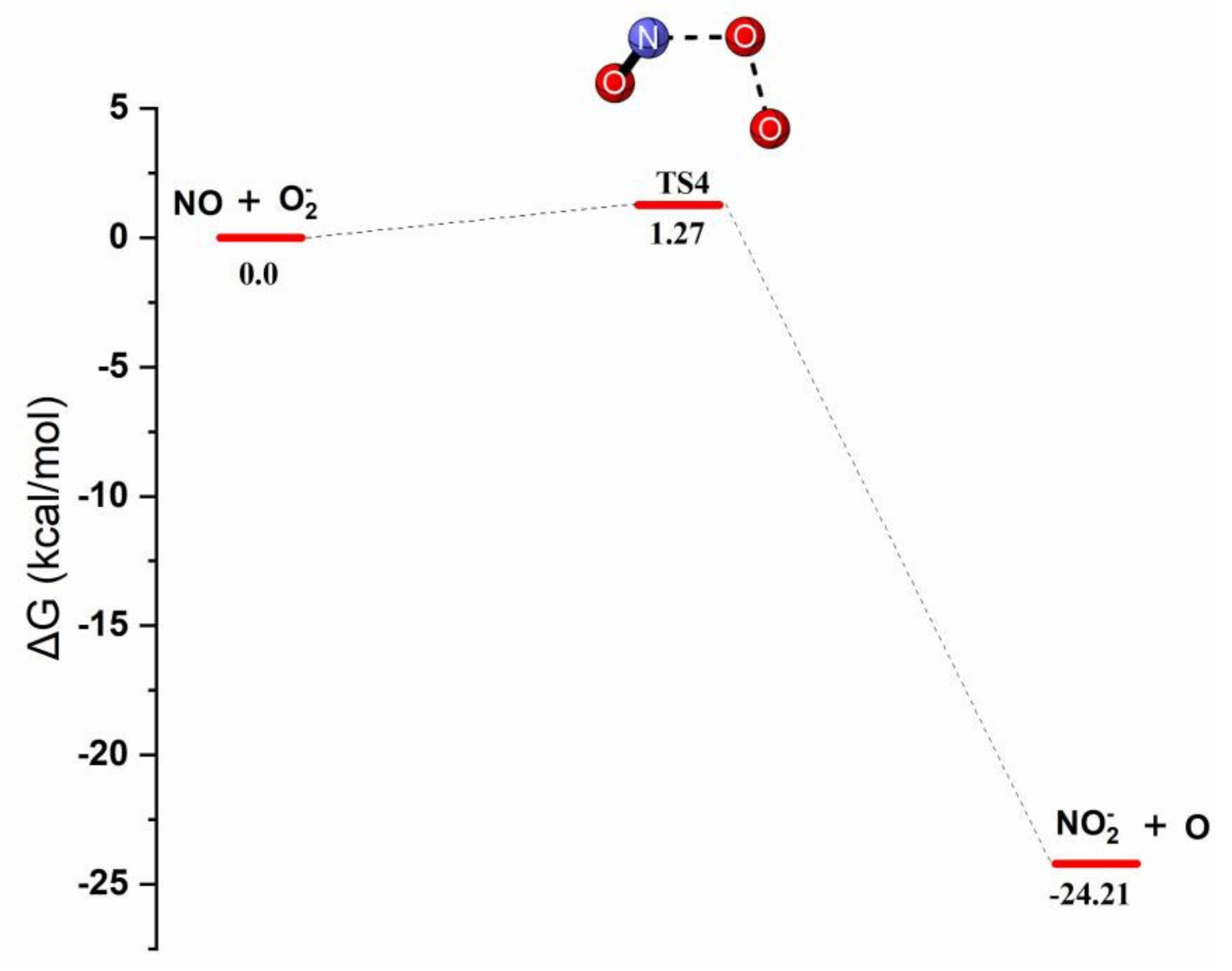
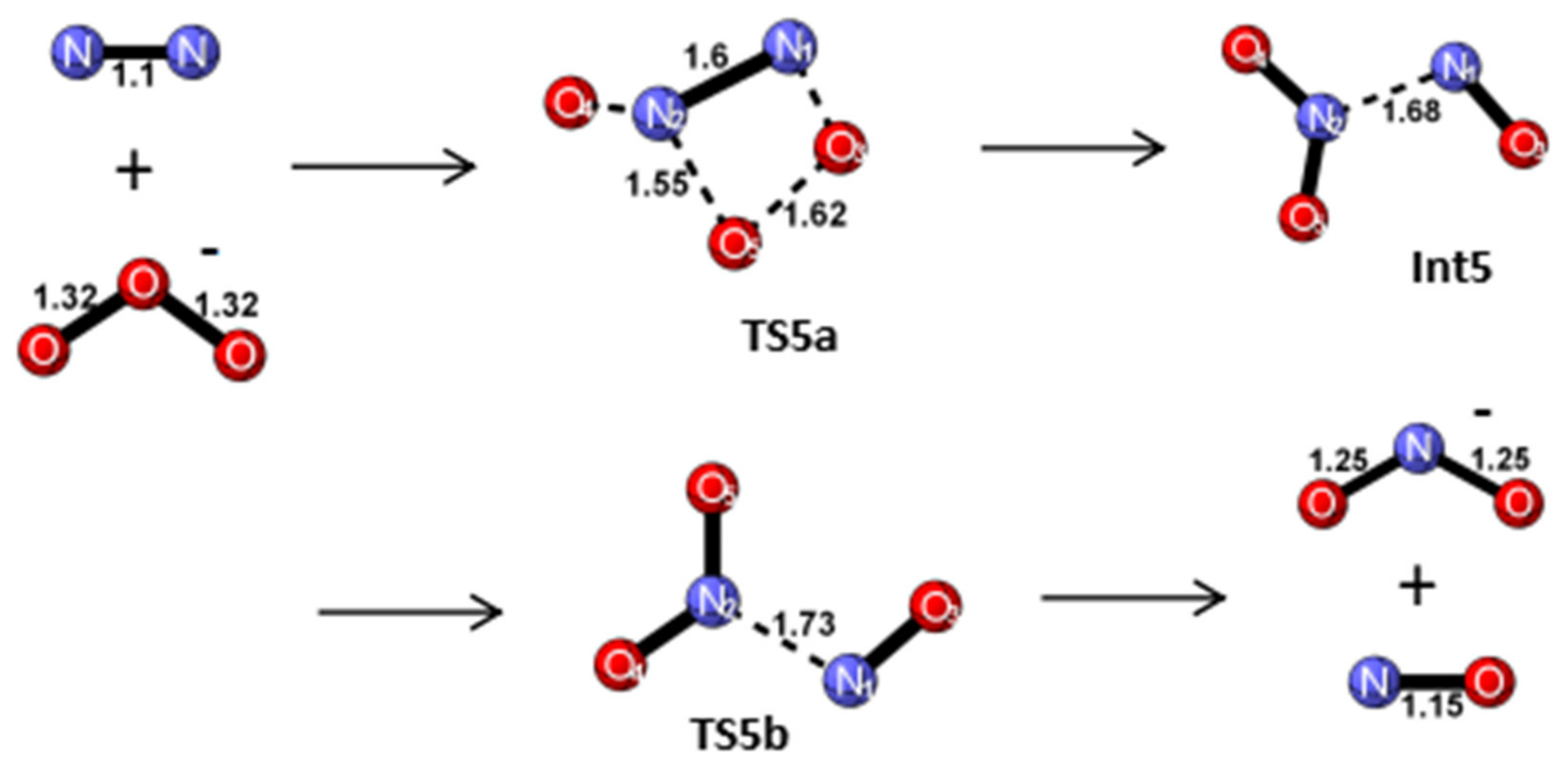
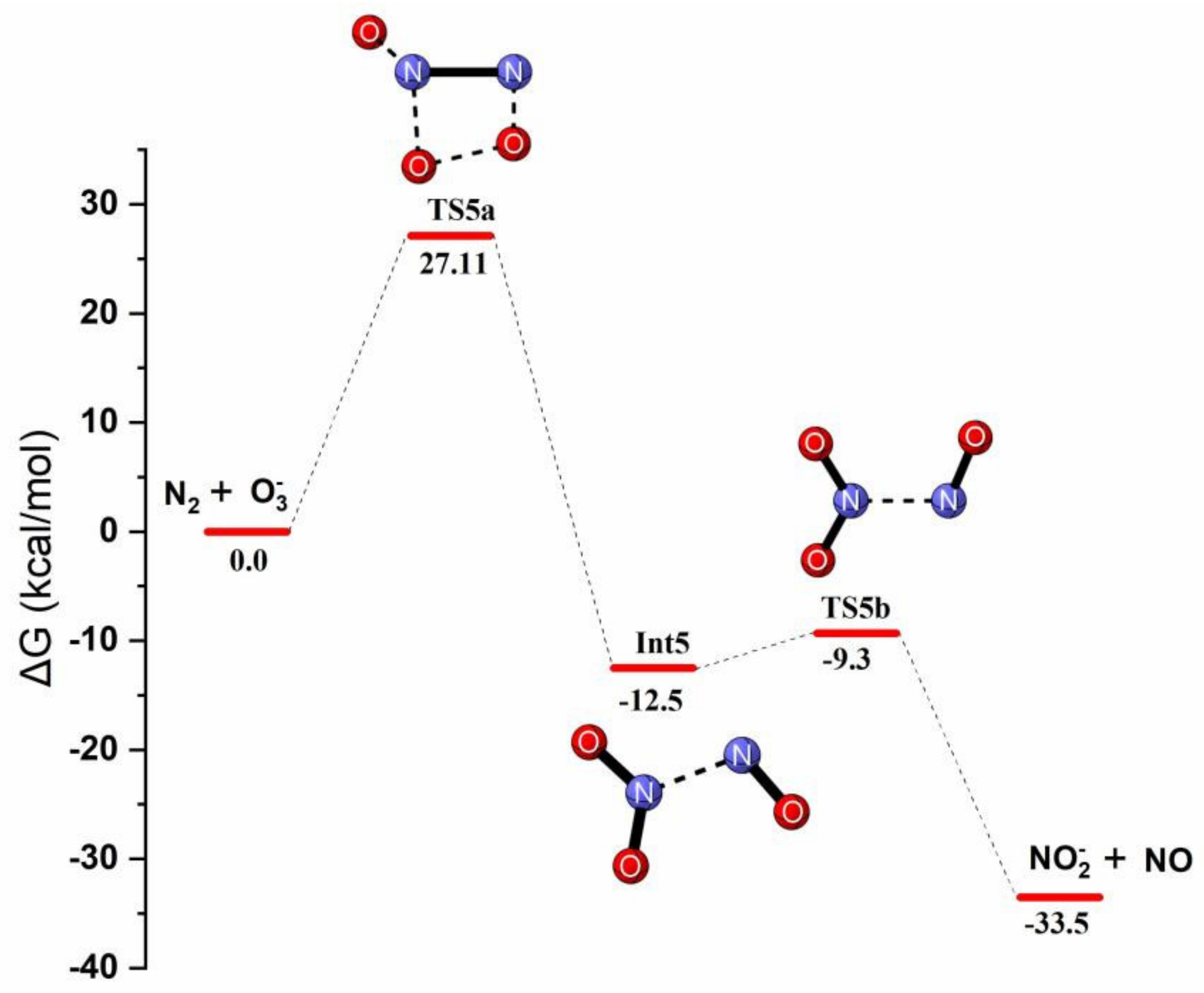
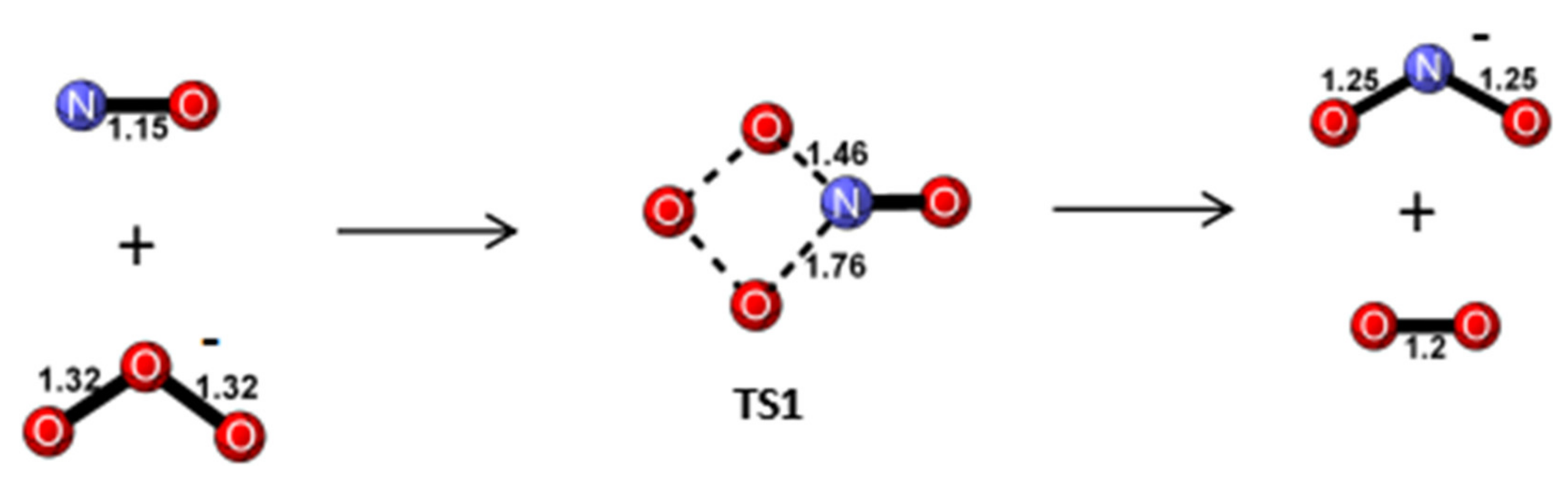
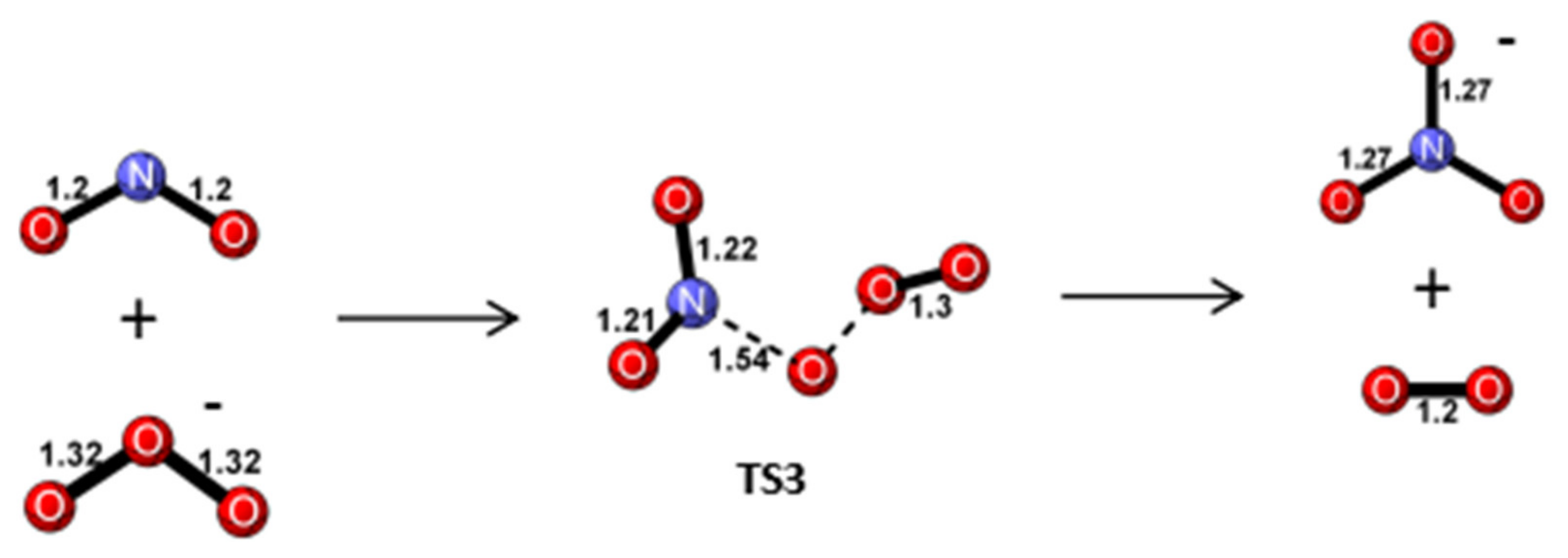
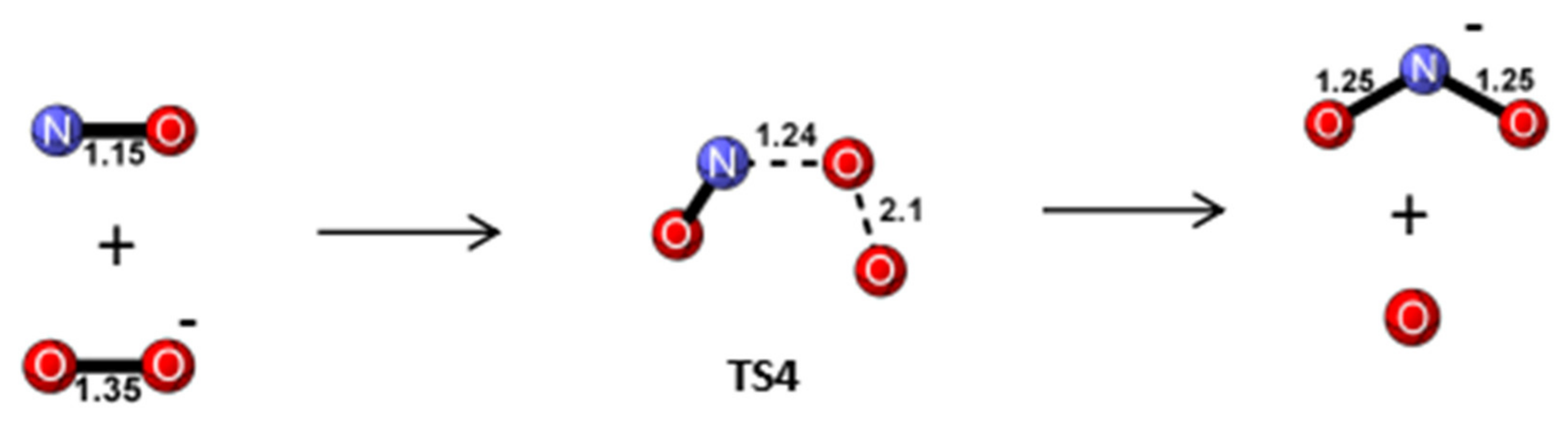
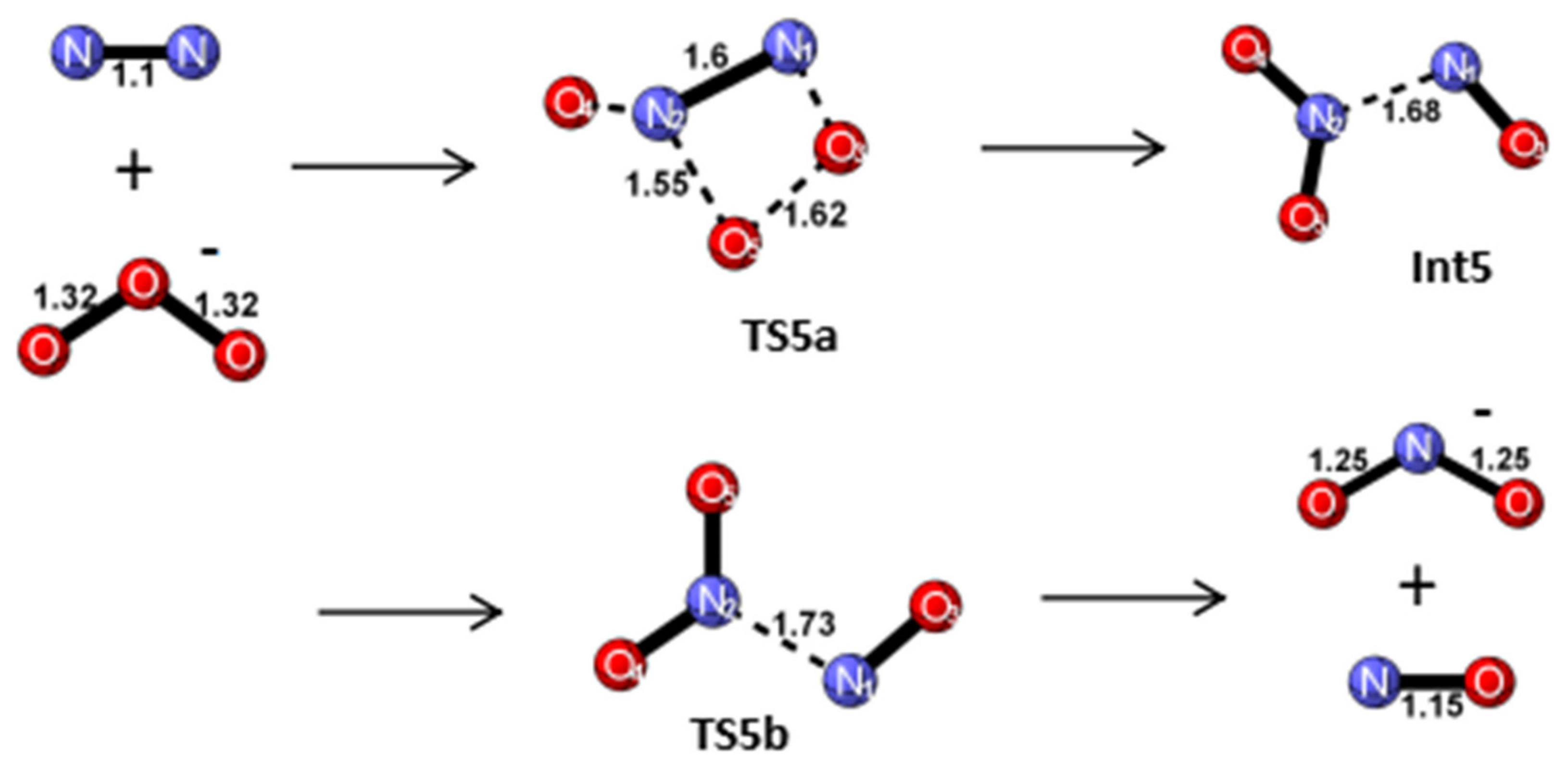
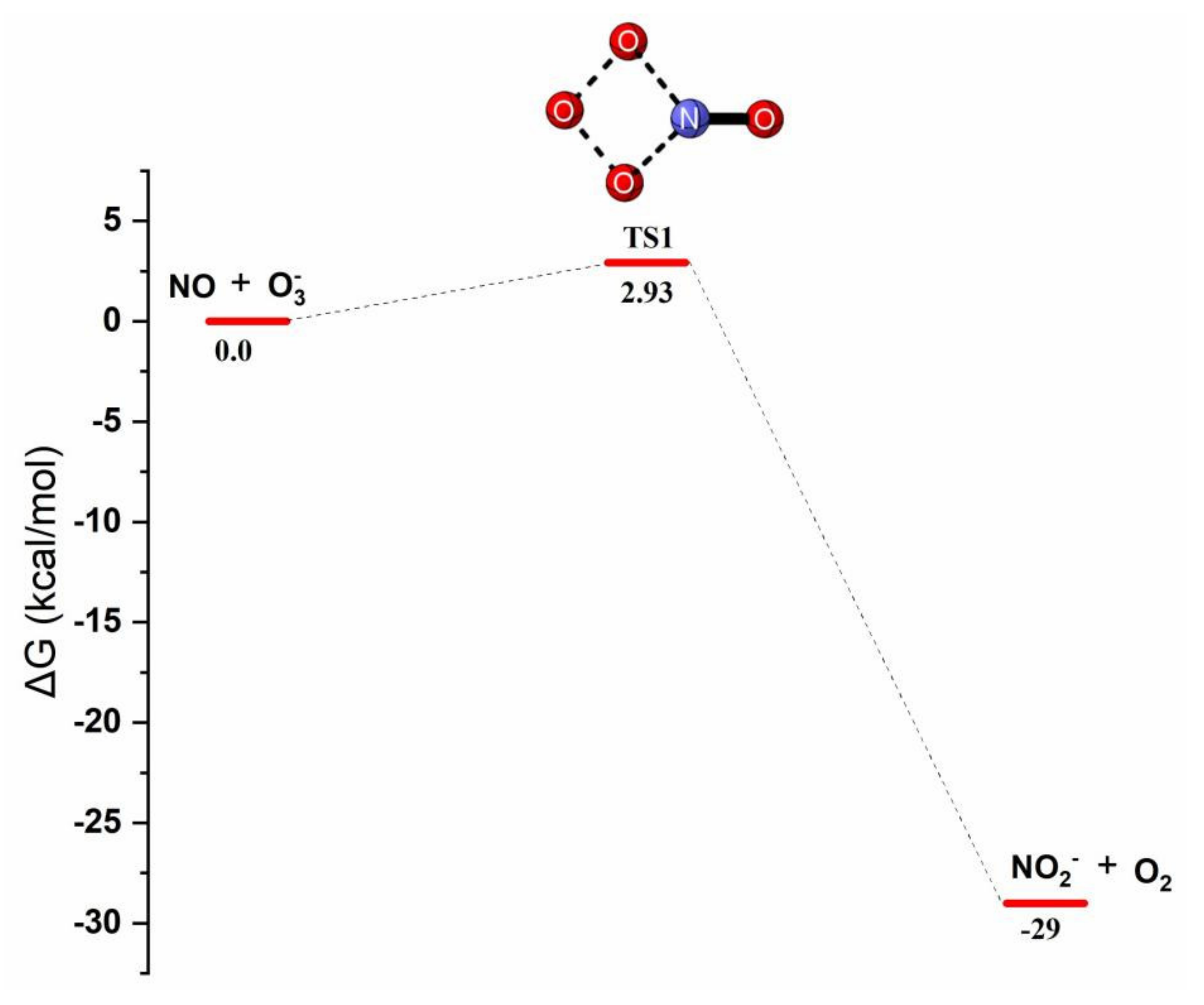
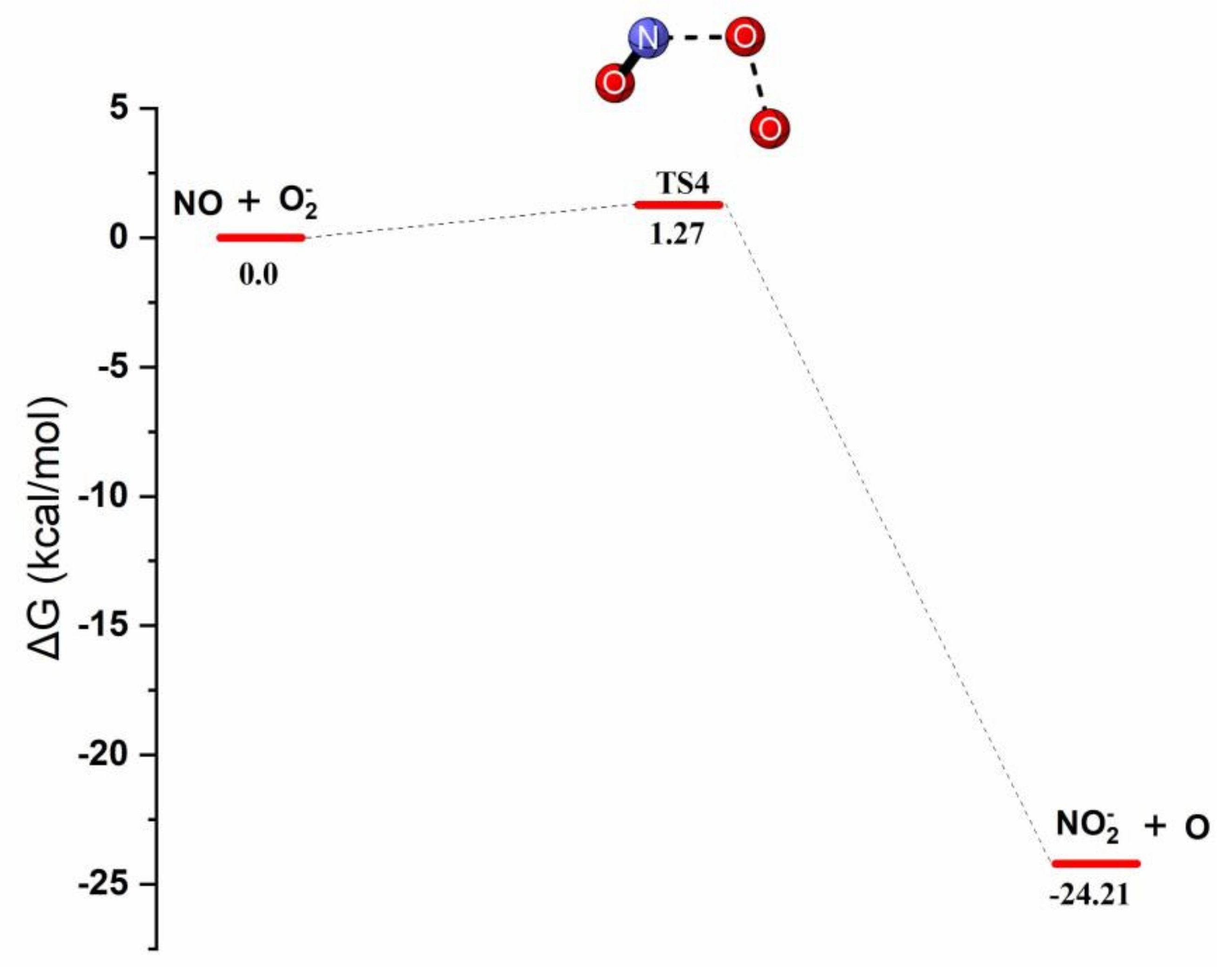
Reaction Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z. Energy and Air Pollution. In Comprehensive Energy Systems; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 9780128095973. [Google Scholar]
- Kampa, M.; Castanas, E. Human health effects of air pollution. Environ. Pollut. 2008, 151, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R. Atmospheric chemistry of VOCs and NO(x). Atmos. Environ. 2000, 34, 2063–2101. [Google Scholar] [CrossRef]
- Jenkin, M.E.; Clemitshaw, K.C. Ozone and other secondary photochemical pollutants: Chemical processes governing their formation in the planetary boundary layer. Atmos. Environ. 2000, 34, 2499–2527. [Google Scholar] [CrossRef]
- Johnston, H. Reduction of stratospheric ozone by nitrogen oxide catalysts from supersonic transport exhaust. Science 1971, 173, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.L.; Lee, D.S.; Raper, D.W.; De Foster, P.M.; Wilson, C.W.; Newton, P.J. The impacts of aviation on the atmosphere. Aeronaut. J. 2002, 106, 521–546. [Google Scholar]
- Lee, D.S.; Pitari, G.; Grewe, V.; Gierens, K.; Penner, J.E.; Petzold, A.; Prather, M.J.; Schumann, U.; Bais, A.; Berntsen, T.; et al. Transport impacts on atmosphere and climate: Aviation. Atmos. Environ. 2010, 44, 4678–4734. [Google Scholar] [CrossRef] [PubMed]
- Crutzen, P.J. The role of NO and NO2 in the chemistry of the troposphere and stratosphere. Annu. Rev. Earth Planet. Sci. 1979, 7, 443–472. [Google Scholar] [CrossRef]
- Rowland, F.S. Stratospheric ozone depletion. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 769–790. [Google Scholar] [CrossRef]
- Sandhiya, L.; Kolandaivel, P.; Senthilkumar, K. Depletion of atmospheric ozone by nitrogen dioxide: A bifurcated reaction pathway. Theor. Chem. Acc. 2013, 132, 6–8. [Google Scholar] [CrossRef]
- Lammel, G.; Graßl, H. Greenhouse effect of NOX. Environ. Sci. Pollut. Res. 1995, 2, 40–45. [Google Scholar] [CrossRef]
- Sivasakthivel, T.; Reddy, K.K.S.K. Ozone Layer Depletion and Its Effects: A Review. Int. J. Environ. Sci. Dev. 2011, 2, 30. [Google Scholar] [CrossRef]
- Heal, M.R. Acid rain. Interdiscip. Sci. Rev. 1996, 21, 243–252. [Google Scholar] [CrossRef]
- Krug, E.C.; Frink, C.R. Acid rain on acid soil: A new perspective. Science 1983, 221, 520–525. [Google Scholar] [CrossRef]
- Singh, A.; Agrawal, M. Acid rain and its ecological consequences. J. Environ. Biol. 2008, 29, 15–24. [Google Scholar] [PubMed]
- Shevtsova, A.; Neuvonen, S. Responses of ground vegetation to prolonged simulated acid rain in sub-arctic pine-birch forest. New Phytol. 1997, 136, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S. Ecological consequences of The Acid rain. IOSR J. Appl. Chem. 2013, 5, 19–24. [Google Scholar] [CrossRef]
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef]
- Jiménez, E.; Tapiador, F.J.; Sáez-Martínez, F.J. Atmospheric pollutants in a changing environment: Key issues in reactivity and monitoring, global warming, and health. Environ. Sci. Pollut. Res. 2015, 22, 4789–4792. [Google Scholar] [CrossRef] [PubMed]
- Fehsenfeld, F.C.; Schmeltekopf, A.L.; Schiff, H.I.; Ferguson, E.E. Laboratory measurements of negative ion reactions of atmospheric interest. Planet. Space Sci. 1967, 15, 373–379. [Google Scholar] [CrossRef]
- Nicolet, M.; Aikin, A.C. The formation of the D region of the ionosphere. J. Geophys. Res. 1960, 65, 1469–1483. [Google Scholar] [CrossRef]
- Mitra, A.P. The D-region of the ionosphere. Endeavour 1978, 2, 12–21. [Google Scholar] [CrossRef]
- Kovács, T.; Plane, J.M.C.; Feng, W.; Nagy, T.; Chipperfield, M.P.; Verronen, P.T.; Andersson, M.E.; Newnham, D.A.; Clilverd, M.A.; Marsh, D.R. D-region ion-neutral coupled chemistry (Sodankylä Ion Chemistry, SIC) within the Whole Atmosphere Community Climate Model (WACCM 4)—WACCM-SIC and WACCM-rSIC. Geosci. Model Dev. 2016, 9, 3123–3136. [Google Scholar] [CrossRef]
- Ferguson, E.E. Negative ion—Molecule reactions. Can. J. Chem. 1969, 47, 1815–1820. [Google Scholar] [CrossRef]
- Arnol, F.; Stilp, T.; Busen, R.; Schumann, U. Jet engine exhaust chemiion measurements. Atmos. Environ. 1998, 32, 3073–3077. [Google Scholar] [CrossRef]
- Kiendler, A.; Aberle, S.; Arnold, F. Negative chemiions formed in jet fuel combustion: New insights from jet engine and laboratory measurements using a quadrupole ion trap mass spectrometer apparatus. Atmos. Environ. 2000, 34, 2623–2632. [Google Scholar] [CrossRef]
- Haverkamp, H.; Wilhelm, S.; Sorokin, A.; Arnold, F. Positive and negative ion measurements in jet aircraft engine exhaust: Concentrations, sizes and implications for aerosol formation. Atmos. Environ. 2004, 38, 2879–2884. [Google Scholar] [CrossRef]
- Yu, F.; Turco, R.P. The role of ions in the formation and evolution of particles in aircraft plumes. Geophys. Res. Lett. 1997, 24, 1927–1930. [Google Scholar] [CrossRef]
- Yu, F.; Turco, R.P. The formation and evolution of aerosols in stratospheric aircraft plumes: Numerical simulations and comparisons with observations. J. Geophys. Res. Atmos. 1998, 103, 25915–25934. [Google Scholar] [CrossRef]
- Starik, A.M.; Savel’ev, A.M.; Titova, N.S.; Schumann, U. Modeling of sulfur gases and chemiions in aircraft engines. Aerosp. Sci. Technol. 2002, 6, 63–81. [Google Scholar] [CrossRef]
- Sorokin, A.; Vancassel, X.; Mirabel, P. Emission of ions and charged soot particles by aircraft engines. Atmos. Chem. Phys. 2003, 3, 325–334. [Google Scholar] [CrossRef]
- Calcote, H.F.; Keil, D.G. The role of ions in soot formation. Pure Appl. Chem. 1990, 62, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, A.; Arnold, F. Organic positive ions in aircraft gas-turbine engine exhaust. Atmos. Environ. 2006, 40, 6077–6087. [Google Scholar] [CrossRef]
- Manzoor, S.; Kulshrestha, U. Atmospheric Aerosols: Air Quality and Climate Change Perspectives. Curr. World Environ. 2015, 10, 738–746. [Google Scholar] [CrossRef]
- Andreae, M.O.; Crutzen, P.J. Atmospheric aerosols: Biogeochemical sources and role in atmospheric chemistry. Science 1997, 276, 1052–1058. [Google Scholar] [CrossRef]
- Arnold, F. Multi-ion complexes in the stratosphere—Implications for trace gases and aerosol. Nature 1980, 284, 610–611. [Google Scholar] [CrossRef]
- Guo, X.; Nadykto, A.B.; Xu, Y.; Zhang, Q.; Hu, J. Ab intio investigation of the thermochemistry and kinetics of the SO2 + O3− → SO3− + O2 reaction in aircraft engines and the environment. Entropy 2014, 16, 6300–6312. [Google Scholar] [CrossRef]
- Francisco, J.S. A new frontier in atmospheric chemistry: Computational atmospheric chemistry. Comput. Theor. Chem. 2011, 965, 248. [Google Scholar] [CrossRef]
- Vereecken, L.; Francisco, J.S. Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem. Soc. Rev. 2012, 41, 6259–6293. [Google Scholar] [CrossRef]
- Peiró-García, J.; Nebot-Gil, I. Ab initio study of the mechanism of the atmospheric reaction: NO2 + O3 → NO3 + O2. J. Comput. Chem. 2003, 24, 1657–1663. [Google Scholar] [CrossRef]
- Jaroszyńska-Wolińska, J. The reaction mechanism of ozone with the NO and NO2 oxides. J. Mol. Struct. THEOCHEM 2010, 952, 74–83. [Google Scholar] [CrossRef]
- Savarino, J.; Bhattacharya, S.K.; Morin, S.; Baroni, M.; Doussin, J.F. The NO+ O3 reaction: A triple oxygen isotope perspective on the reaction dynamics and atmospheric implications for the transfer of the ozone isotope anomaly. J. Chem. Phys. 2008, 128, 194303. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.M.; McCartney, H.A. Some measurements of ambient air pollution arising from the manufacture of nitric acid and ammonium nitrate fertiliser. Atmos. Environ. 1979, 13, 1105–1120. [Google Scholar] [CrossRef]
- Laaksonen, A.; Hienola, J.; Kulmala, M.; Arnold, F.F. Supercooled cirrus cloud formation modified by nitric acid pollution of the upper troposphere. Geophys. Res. Lett. 1997, 24, 3009–3012. [Google Scholar] [CrossRef]
- Cisneros, R.; Bytnerowicz, A.; Schweizer, D.; Zhong, S.; Traina, S.; Bennett, D.H. Ozone, nitric acid, and ammonia air pollution is unhealthy for people and ecosystems in southern Sierra Nevada, California. Environ. Pollut. 2010, 158, 3261–3271. [Google Scholar] [CrossRef]
- Chirik, P.J. Nitrogen fixation: One electron at a time. Nat. Chem. 2009, 1, 520–522. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Y.; Jia, R.; Zhang, C.; Zhang, B. Electrochemical synthesis of nitric acid from air and ammonia through waste utilization. Natl. Sci. Rev. 2019, 6, 730–738. [Google Scholar] [CrossRef]
- Brasseur, G.P.; Cox, R.A.; Hauglustaine, D.; Isaksen, I.; Lelieveld, J.; Lister, D.H.; Sausen, R.; Schumann, U.; Wahner, A.; Wiesen, P. European scientific assessment of the atmospheric effects of aircraft emissions. Atmos. Environ. 1998, 32, 2329–2418. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, R. Chapter 10 Theoretical Investigation of Atmospheric Oxidation of Biogenic Hydrocarbons: A Critical Review. Adv. Quantum Chem. 2008, 55, 177–213. [Google Scholar] [CrossRef]
- Pulay, P.; Saebø, S. Orbital-invariant formulation and second-order gradient evaluation in Møller-Plesset perturbation theory. Theor. Chim. Acta 1986, 69, 357–368. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Li, Y.; Iwata, S. Potential energy surfaces of the ground and low-lying states of HCCS and NCS: CASSCF, MRCI and CCSD(T) studies. Chem. Phys. Lett. 1997, 273, 91–97. [Google Scholar] [CrossRef]
- Gonzalez, C.; Bernhard Schlegel, H. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lewars, E.G.; Lewars, E.G. The Concept of the Potential Energy Surface. In Computational Chemistry; Springer: Cham, Switzerland, 2011. [Google Scholar]
- Hotokka, M.; Pyykkö, P. An ab initio study of bonding trends in the series BO33-, CO32-, NO3- and O4(D3h). Chem. Phys. Lett. 1989, 157, 415–418. [Google Scholar] [CrossRef]
- Stirling, A.; Pápai, I.; Mink, J.; Salahub, D.R. Density functional study of nitrogen oxides. J. Chem. Phys. 1994, 100, 2910–2923. [Google Scholar] [CrossRef]
- Creighton, J.A.; Lippincott, E.R. Vibrational frequency and dissociation energy of the superoxide ion. J. Chem. Phys. 1964, 40, 1779–1780. [Google Scholar] [CrossRef]
- Dupuis, M.; Fitzgerald, G.; Hammond, B.; Lester, W.A.; Schaefer, H.F. Theoretical study of the H+O3↔OH+O2↔O+HO2 system. J. Chem. Phys. 1985, 84, 2691–2697. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Parameters | CCSD/ 6-31g(d) | CCSD/ 6-311g(d) | CCSD/ 6-311+g(d) | * Exp 1,2,3 |
|---|---|---|---|---|---|
| NO | R(N-O) | 1.166 | 1.144 | 1.151 | 1.151 |
| O2 | R(O-O) | 1.220 | 1.200 | 1.204 | 1.207 |
| N2 | R(N-N) | 1.102 | 1.102 | 1.103 | 1.098 |
| O2− | R(O-O) | 1.357 | 1.341 | 1.349 | 1.350 |
| NO2 | R(N-O) | 1.204 | 1.190 | 1.198 | 1.207 |
| <O-N-O | 134.3 | 134.5 | 134.1 | 134.1 | |
| NO2− | R(N-O) | 1.271 | 1.254 | 1.253 | 1.257 |
| <O-N-O | 115.7 | 116.1 | 116.1 | 116.0 | |
| NO3− | R(N-O) | 1.261 | 1.251 | 1.268 | 1.270 |
| <O-N-O | 120.0 | 120 | 120.0 | 120.0 | |
| O3− | R(O-O) | 1.36 | 1.342 | 1.319 | 1.314 |
| <O-O-O | 115.3 | 115.4 | 114.6 | 114.6 |
| Molecule | CCSD/6-31g(d) | CCSD/ 6-311g(d) | CCSD/ 6-311+g(d) | Exp 1,2,3,4 |
|---|---|---|---|---|
| NO | 1951.7 | 1971.9 | 1963.8 | 1904.1 |
| O2 | 1650.1 | 1678.9 | 1667.0 | 1580.4 |
| N2 | 2411.12 | 2411.1 | 2408.0 | 2330.0 |
| O2− | 1208.0 | 1214.5 | 1183.5 | 1074.0 |
| NO2 | 770.1 | 713.7 | 784.0 | 756.8 |
| 1352.4 | 1226.4 | 1375.0 | 1355.9 | |
| 1383.3 | 2181.6 | 2106.1 | 1663.5 | |
| NO2− | 801.9 | 822.1 | 814.9 | 776.0 |
| 1386.8 | 1385.1 | 1353.9 | 1241.5 | |
| 1402.8 | 1414.4 | 1381.2 | 1284.0 | |
| NO3− | 715.3 | 735.8 | 725.0 | 720.0 |
| 860.1 | 875.8 | 852.0 | 830.0 | |
| 1099.9 | 1109.0 | 1101.4 | 1050.0 | |
| 1503.8 | 1493.6 | 1427.0 | 1390.0 | |
| O3− | 590.3 | 617.2 | 671.6 | - |
| 1006.7 | 984.7 | 1102.9 | - | |
| 1006.8 | 1089.3 | 1321.1 | - |
| Molecule | Parameters | CCSD/ 6-31G(d) | CCSD/ 6-311G(d) | CCSD/ 6-311+G(d) |
|---|---|---|---|---|
| NO | Energy | −129.584286 | −129.641792 | −129.647432 |
| Z.P.E | 0.004446 | 0.004492 | 0.004474 | |
| O2 | Energy | −149.967988 | −150.041012 | −150.047750 |
| Z.P.E | 0.003759 | 0.003825 | 0.003798 | |
| N2 | Energy | −109.308796 | −109.308790 | −109.313030 |
| Z.P.E | 0.005493 | 0.005493 | 0.005487 | |
| O2− | Energy | −149.936820 | −150.010960 | −150.048378 |
| Z.P.E | 0.002752 | 0.002764 | 0.002696 | |
| NO2 | Energy | −204.565748 | −204.638166 | −204.664268 |
| Z.P.E | 0.007987 | 0.009390 | 0.013591 | |
| NO2− | Energy | −204.661106 | −204.704836 | −204.744319 |
| Z.P.E | 0.008182 | 0.008251 | 0.008088 | |
| NO3− | Energy | −279.628433 | −279.759869 | −279.78294 |
| Z.P.E | 0.014577 | 0.014680 | 0.014261 | |
| O3− | Energy | −224.912758 | −225.020595 | −225.060074 |
| Z.P.E | 0.006069 | 0.006131 | 0.005914 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulay, R.; Krishnan, A.; Muralikrishna, B.; Devadas, S.; Rajalakshmi, C.; Mathew, J.; Thomas, V.I. A Quantum Chemical Investigation into the Molecular Mechanism of the Atmospheric Reactions of Chemi-Ions with Nitrogen and Nitrogen Oxides. Entropy 2022, 24, 1257. https://doi.org/10.3390/e24091257
Sulay R, Krishnan A, Muralikrishna B, Devadas S, Rajalakshmi C, Mathew J, Thomas VI. A Quantum Chemical Investigation into the Molecular Mechanism of the Atmospheric Reactions of Chemi-Ions with Nitrogen and Nitrogen Oxides. Entropy. 2022; 24(9):1257. https://doi.org/10.3390/e24091257
Chicago/Turabian StyleSulay, Rehin, Anandhu Krishnan, Balasubramoniam Muralikrishna, Sudheesh Devadas, Chandralekha Rajalakshmi, Jintumol Mathew, and Vibin Ipe Thomas. 2022. "A Quantum Chemical Investigation into the Molecular Mechanism of the Atmospheric Reactions of Chemi-Ions with Nitrogen and Nitrogen Oxides" Entropy 24, no. 9: 1257. https://doi.org/10.3390/e24091257
APA StyleSulay, R., Krishnan, A., Muralikrishna, B., Devadas, S., Rajalakshmi, C., Mathew, J., & Thomas, V. I. (2022). A Quantum Chemical Investigation into the Molecular Mechanism of the Atmospheric Reactions of Chemi-Ions with Nitrogen and Nitrogen Oxides. Entropy, 24(9), 1257. https://doi.org/10.3390/e24091257