
Some Recent Advances in Energetic Variational Approaches


Abstract
1. Introduction
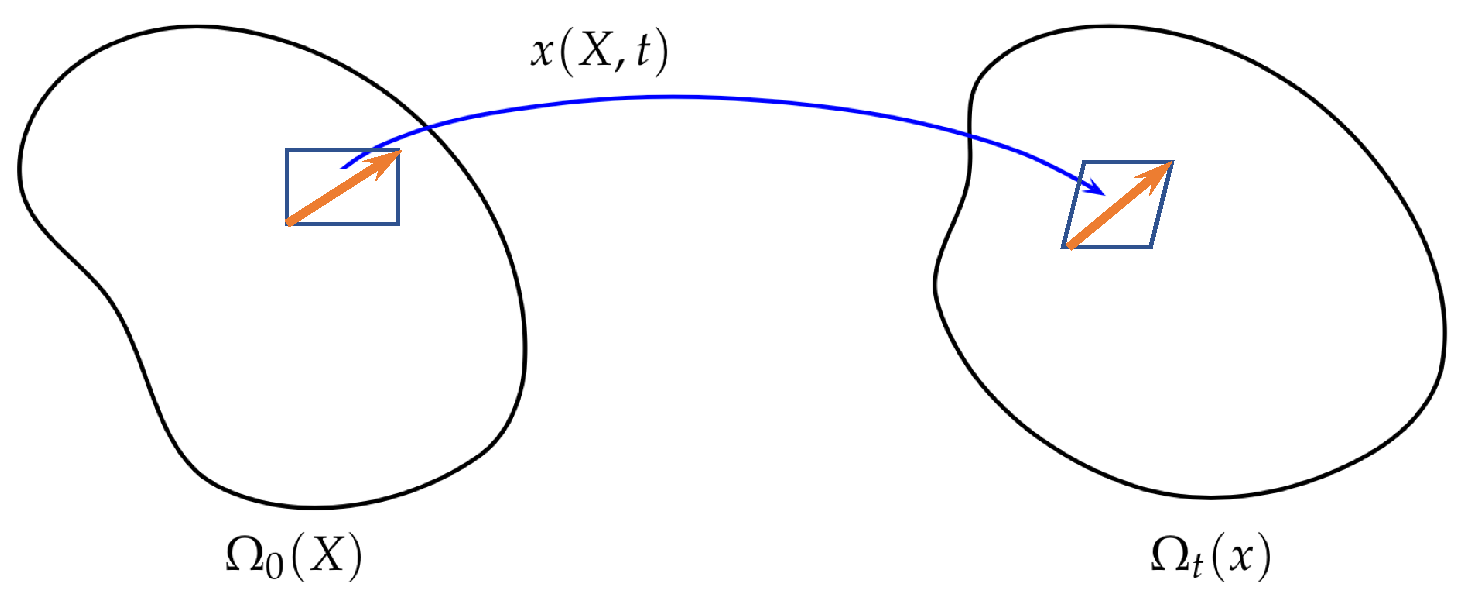
2. EnVarA in Continuum Mechanics
2.1. Generalized Diffusion
2.2. Micro-Macro Model for Polymeric Fluids
2.3. Kinetic Fokker–Planck Equation
3. EnVarA for Chemical Reactions
3.1. Dynamical Boundary Condition
3.2. Boltzmann Equation
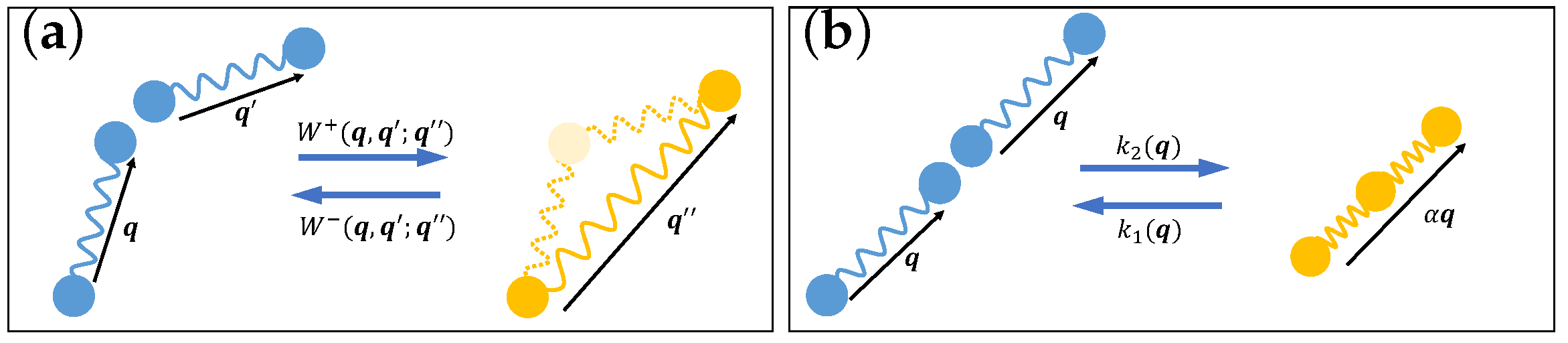
3.3. Reactive Fluids
4. EnVarA for Non-Isothermal Systems
5. Coarse-Graining and Numerical Realization
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beris, A.N.; Edwards, B.J. Thermodynamics of Flowing Systems: With Internal Microstructure; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Doi, M. Soft Matter Physics; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Rayleigh, L. Some General Theorems Relating to Vibrations. Proc. Lond. Math. Soc. 1873, 4, 357–368. [Google Scholar]
- Onsager, L. Reciprocal relations in irreversible processes. I. Phys. Rev. 1931, 37, 405. [Google Scholar] [CrossRef]
- Onsager, L. Reciprocal relations in irreversible processes. II. Phys. Rev. 1931, 38, 2265. [Google Scholar] [CrossRef]
- Liu, C. An introduction of elastic complex fluids: An energetic variational approach. In Multi-Scale Phenomena in Complex Fluids: Modeling, Analysis and Numerical Simulation; World Scientific: Singapore, 2009; pp. 286–337. [Google Scholar]
- Giga, M.H.; Kirshtein, A.; Liu, C. Variational modeling and complex fluids. In Handbook of Mathematical Analysis in Mechanics of Viscous Fluids; Springer: Berlin/Heidelberg, Germany, 2017; pp. 1–41. [Google Scholar]
- Lin, F.H.; Liu, C. Static and dynamic theories of liquid crystals. J. Partial Differ. Equ. 2001, 14, 289–330. [Google Scholar]
- Sun, H.; Liu, C. On energetic variational approaches in modeling the nematic liquid crystal flows. Discret. Contin. Dyn. Syst. 2009, 23, 455–475. [Google Scholar] [CrossRef]
- Lin, F.H.; Liu, C.; Zhang, P. On hydrodynamics of viscoelastic fluids. Commun. Pure Appl. Math. 2005, 58, 1437–1471. [Google Scholar] [CrossRef]
- Liu, C.; Shen, J. A phase field model for the mixture of two incompressible fluids and its approximation by a Fourier-spectral method. Phys. D Nonlinear Phenom. 2003, 179, 211–228. [Google Scholar] [CrossRef]
- Feng, J.J.; Liu, C.; Shen, J.; Yue, P. An energetic variational formulation with phase field methods for interfacial dynamics of complex fluids: Advantages and challenges. In Modeling of Soft Matter; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–26. [Google Scholar]
- Eisenberg, B.; Hyon, Y.; Liu, C. Energy variational analysis of ions in water and channels: Field theory for primitive models of complex ionic fluids. J. Chem. Phys. 2010, 133, 104104. [Google Scholar] [CrossRef]
- Ericksen, J.L. Introduction to the Thermodynamics of Solids; Applied Mathematical Sciences; Springer: Berlin/Heidelberg, Germany, 1998. [Google Scholar]
- Grmela, M.; Öttinger, H.C. Dynamics and thermodynamics of complex fluids. I. Development of a general formalism. Phys. Rev. E 1997, 56, 6620. [Google Scholar] [CrossRef]
- Öttinger, H.C.; Grmela, M. Dynamics and thermodynamics of complex fluids. II. Illustrations of a general formalism. Phys. Rev. E 1997, 56, 6633. [Google Scholar] [CrossRef]
- Doi, M. Onsager’s variational principle in soft matter. J. Phys. Condens. Matter 2011, 23, 284118. [Google Scholar] [CrossRef] [PubMed]
- Doi, M. A principle in dynamic coarse graining–Onsager principle and its applications. Eur. Phys. J. Spec. Top. 2016, 225, 1411–1421. [Google Scholar] [CrossRef]
- Doi, M.; Zhou, J.; Di, Y.; Xu, X. Application of the Onsager-Machlup integral in solving dynamic equations in nonequilibrium systems. Phys. Rev. E 2019, 99, 063303. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q. Generalized Onsager Principle and It Applications. In Frontiers and Progress of Current Soft Matter Research; Springer: Berlin/Heidelberg, Germany, 2021; pp. 101–132. [Google Scholar]
- Zhu, Y.; Hong, L.; Yang, Z.; Yong, W.A. Conservation-dissipation formalism of irreversible thermodynamics. J. Non-Equilib. Thermodyn. 2015, 40, 67–74. [Google Scholar] [CrossRef]
- Peng, L.; Hu, Y.; Hong, L. Conservation-Dissipation Formalism for soft matter physics: I. Augmentation to Doi’s variational approach. Eur. Phys. J. E 2019, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Arnold, V.I. Mathematical Methods of Classical Mechanics, 2nd ed.; Springer: New York, NY, USA, 1997. [Google Scholar]
- Berdichevsky, V. Variational Principles of Continuum Mechanics I. Fundamentals; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Gurtin, M.E. An Introduction to Continuum Mechanics, Volume 158, Mathematics in Science and Engineering; Academic Press: Cambridge, MA, USA, 1981. [Google Scholar]
- Evans, L.C.; Savin, O.; Gangbo, W. Diffeomorphisms and nonlinear heat flows. SIAM J. Math. Anal. 2005, 37, 737–751. [Google Scholar] [CrossRef]
- Carrillo, J.A.; Lisini, S. On the asymptotic behavior of the gradient flow of a polyconvex functional. Nonlinear Partial. Differ. Equ. Hyperbolic Wave Phenom. 2010, 526, 37–51. [Google Scholar]
- Liu, C.; Wang, Y. On Lagrangian schemes for porous medium type generalized diffusion equations: A discrete energetic variational approach. J. Comput. Phys. 2020, 417, 109566. [Google Scholar] [CrossRef]
- E, W.; Li, T.; Vanden-Eijnden, E. Applied Stochastic Analysis; American Mathematical Society: Providence, RI, USA, 2019; Volume 199. [Google Scholar]
- Kubo, R. The fluctuation-dissipation theorem. Rep. Prog. Phys. 1966, 29, 255. [Google Scholar] [CrossRef]
- Jordan, R.; Kinderlehrer, D.; Otto, F. The variational formulation of the Fokker–Planck equation. SIAM J. Math. Anal. 1998, 29, 1–17. [Google Scholar] [CrossRef]
- Benamou, J.D.; Brenier, Y. A computational fluid mechanics solution to the Monge-Kantorovich mass transfer problem. Numer. Math. 2000, 84, 375–393. [Google Scholar] [CrossRef]
- Benamou, J.D.; Carlier, G.; Laborde, M. An augmented Lagrangian approach to Wasserstein gradient flows and applications. ESAIM Proc. Surv. 2016, 54, 1–17. [Google Scholar] [CrossRef]
- Adams, S.; Dirr, N.; Peletier, M.; Zimmer, J. Large deviations and gradient flows. Philos. Trans. Royal Soc. A 2013, 371, 20120341. [Google Scholar] [CrossRef] [PubMed]
- Lisini, S.; Matthes, D.; Savaré, G. Cahn–Hilliard and thin film equations with nonlinear mobility as gradient flows in weighted-Wasserstein metrics. J. Differ. Equ. 2012, 253, 814–850. [Google Scholar] [CrossRef]
- Lin, F.H.; Liu, C.; Zhang, P. On a micro-macro model for polymeric fluids near equilibrium. Commun. Pure Appl. Math. 2007, 60, 838–866. [Google Scholar] [CrossRef]
- Li, T.; Zhang, P. Mathematical analysis of multi-scale models of complex fluids. Commun. Math. Sci. 2007, 5, 1–51. [Google Scholar] [CrossRef]
- Bird, R.B.; Ottinger, H.C. Transport properties of polymeric liquids. Annu. Rev. Phys. Chem. 1992, 43, 371–406. [Google Scholar] [CrossRef]
- Le Bris, C.; Lelievre, T. Micro-macro models for viscoelastic fluids: Modelling, mathematics and numerics. Sci. China Math. 2012, 55, 353–384. [Google Scholar] [CrossRef]
- Ma, L.; Li, X.; Liu, C. Fluctuation-dissipation theorem consistent approximation of the Langevin dynamics model. Commun. Math. Sci. 2017, 15, 1171–1181. [Google Scholar] [CrossRef]
- Espanol, P.; Warren, P. Statistical mechanics of dissipative particle dynamics. EPL (Europhys. Lett.) 1995, 30, 191. [Google Scholar] [CrossRef]
- Keener, J.P.; Sneyd, J. Mathematical Physiology; Springer: Berlin/Heidelberg, Germany, 1998; Volume 1. [Google Scholar]
- Coleman, B.D.; Gurtin, M.E. Thermodynamics with internal state variables. J. Chem. Phys. 1967, 47, 597–613. [Google Scholar] [CrossRef]
- Wei, J. Axiomatic treatment of chemical reaction systems. J. Chem. Phys. 1962, 36, 1578–1584. [Google Scholar] [CrossRef]
- Van Rysselberghe, P. General reciprocity relation between the rates and affinities of simultaneous chemical reactions. J. Chem. Phys. 1962, 36, 1329–1330. [Google Scholar] [CrossRef]
- Grmela, M. Thermodynamics of driven systems. Phys. Rev. E 1993, 48, 919. [Google Scholar] [CrossRef]
- Grmela, M. Fluctuations in extended mass-action-law dynamics. Phys. D Nonlinear Phenom. 2012, 241, 976–986. [Google Scholar] [CrossRef]
- Feinberg, M. Complex balancing in general kinetic systems. Arch. Ration. Mech. Anal. 1972, 49, 187–194. [Google Scholar] [CrossRef]
- Bataille, J.; Edelen, D.; Kestin, J. Nonequilibrium Thermodynamics of the Nonlinear Equations of Chemical Kinetics. J. Non-Equilib. Thermodyn. 1978, 3, 153–168. [Google Scholar] [CrossRef]
- Mielke, A. A gradient structure for reaction–diffusion systems and for energy-drift-diffusion systems. Nonlinearity 2011, 24, 1329. [Google Scholar] [CrossRef]
- Mielke, A.; Renger, D.M.; Peletier, M.A. A generalization of Onsager’s reciprocity relations to gradient flows with nonlinear mobility. J. Non-Equilib. Thermodyn. 2016, 41, 141–149. [Google Scholar] [CrossRef]
- Kondepudi, D.; Prigogine, I. Modern Thermodynamics: From Heat Engines to Dissipative Structures; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Réti, P.; Ropolyi, L. Analogies between point mechanics and chemical reaction kinetics. React. Kinet. Catal. Lett. 1984, 25, 109–113. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Liu, P.; Eisenberg, B. Field theory of reaction-diffusion: Law of mass action with an energetic variational approach. Phys. Rev. E 2020, 102, 062147. [Google Scholar] [CrossRef] [PubMed]
- Oster, G.F.; Perelson, A.S. Chemical reaction dynamics. Arch. Ration. Mech. Anal. 1974, 55, 230–274. [Google Scholar] [CrossRef]
- De Groot, S.R.; Mazur, P. Non-Equilibrium Thermodynamics; Courier Corporation: North Chelmsford, MA, USA, 2013. [Google Scholar]
- Liu, C.; Wang, C.; Wang, Y. A structure-preserving, operator splitting scheme for reaction-diffusion equations with detailed balance. J. Comput. Phys. 2021, 436, 110253. [Google Scholar] [CrossRef]
- Salazar, D.S.; Landi, G.T. Nonlinear Onsager relations for Gaussian quantum maps. Phys. Rev. Res. 2020, 2, 033090. [Google Scholar] [CrossRef]
- Giordano, S. Entropy production and Onsager reciprocal relations describing the relaxation to equilibrium in stochastic thermodynamics. Phys. Rev. E 2021, 103, 052116. [Google Scholar] [CrossRef]
- Chizat, L.; Peyré, G.; Schmitzer, B.; Vialard, F.X. Scaling algorithms for unbalanced optimal transport problems. Math. Comput. 2018, 87, 2563–2609. [Google Scholar] [CrossRef]
- Mroueh, Y.; Rigotti, M. Unbalanced Sobolev Descent. arXiv 2020, arXiv:2009.14148. [Google Scholar]
- Yang, K.D.; Uhler, C. Scalable unbalanced optimal transport using generative adversarial networks. arXiv 2018, arXiv:1810.11447. [Google Scholar]
- Feydy, J.; Séjourné, T.; Vialard, F.X.; Amari, S.i.; Trouvé, A.; Peyré, G. Interpolating between optimal transport and MMD using Sinkhorn divergences. In Proceedings of the 22nd International Conference on Artificial Intelligence and Statistics—PMLR, Naha, Japan, 16–18 April 2019; pp. 2681–2690. [Google Scholar]
- Pham, K.; Le, K.; Ho, N.; Pham, T.; Bui, H. On unbalanced optimal transport: An analysis of Sinkhorn algorithm. In Proceedings of the International Conference on Machine Learning—PMLR, online, 13–18 July 2020; pp. 7673–7682. [Google Scholar]
- Gallouët, T.O.; Monsaingeon, L. A JKO Splitting Scheme for Kantorovich–Fisher–Rao Gradient Flows. SIAM J. Math. Anal. 2017, 49, 1100–1130. [Google Scholar] [CrossRef]
- Liu, C.; Wu, H. An energetic variational approach for the Cahn–Hilliard equation with dynamic boundary condition: Model derivation and mathematical analysis. Arch. Ration. Mech. Anal. 2019, 233, 167–247. [Google Scholar] [CrossRef]
- Knopf, P.; Lam, K.F.; Liu, C.; Metzger, S. Phase-field dynamics with transfer of materials: The Cahn–Hillard equation with reaction rate dependent dynamic boundary conditions. ESAIM Math. Model. Numer. Anal. 2021, 55, 229–282. [Google Scholar] [CrossRef]
- Wang, X.P.; Qian, T.; Sheng, P. Moving contact line on chemically patterned surfaces. J. Fluid Mech. 2008, 605, 59–78. [Google Scholar] [CrossRef]
- Dreyer, W.; Guhlke, C.; Müller, R. A new perspective on the electron transfer: Recovering the Butler–Volmer equation in non-equilibrium thermodynamics. Phys. Chem. Chem. Phys. 2016, 18, 24966–24983. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Xu, J. Structure-preserving finite element methods for stationary MHD models. Math. Comput. 2019, 88, 553–581. [Google Scholar] [CrossRef]
- Bou-Rabee, N.; Holmes-Cerfon, M.C. Sticky Brownian motion and its numerical solution. SIAM Rev. 2020, 62, 164–195. [Google Scholar] [CrossRef]
- Goldstein, G.R.; Miranville, A.; Schimperna, G. A Cahn–Hilliard model in a domain with non-permeable walls. Phys. D Nonlinear Phenom. 2011, 240, 754–766. [Google Scholar] [CrossRef]
- Erbar, M. A gradient flow approach to the Boltzmann equation. arXiv 2016, arXiv:1603.00540. [Google Scholar]
- Öttinger, H.C. GENERIC formulation of Boltzmann’s kinetic equation. J. Non-Equilib. Thermodyn 1997, 222, 386–391. [Google Scholar] [CrossRef]
- Gorban, A.; Yablonsky, G. Three waves of chemical dynamics. Math. Model. Nat. Phenom. 2015, 10, 1–5. [Google Scholar] [CrossRef]
- Doostmohammadi, A.; Ignés-Mullol, J.; Yeomans, J.M.; Sagués, F. Active nematics. Nat. Commun. 2018, 9, 3246. [Google Scholar] [CrossRef]
- Marchetti, M.C.; Joanny, J.F.; Ramaswamy, S.; Liverpool, T.B.; Prost, J.; Rao, M.; Simha, R.A. Hydrodynamics of soft active matter. Rev. Mod. Phys. 2013, 85, 1143. [Google Scholar] [CrossRef]
- Needleman, D.; Dogic, Z. Active matter at the interface between materials science and cell biology. Nat. Rev. Mater. 2017, 2, 17048. [Google Scholar] [CrossRef]
- Prost, J.; Jülicher, F.; Joanny, J.F. Active gel physics. Nat. Phys. 2015, 11, 111–117. [Google Scholar] [CrossRef]
- Wang, H.; Qian, T.; Xu, X. Onsager’s variational principle in active soft matter. arXiv 2020, arXiv:2011.10821. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, J.; Forest, M.G.; Wang, Q. Hydrodynamic theories for flows of active liquid crystals and the generalized onsager principle. Entropy 2016, 18, 202. [Google Scholar] [CrossRef]
- Cates, M. Reptation of living polymers: Dynamics of entangled polymers in the presence of reversible chain-scission reactions. Macromolecules 1987, 20, 2289–2296. [Google Scholar] [CrossRef]
- Adams, J.; Fielding, S.M.; Olmsted, P.D. Transient shear banding in entangled polymers: A study using the Rolie-Poly model. J. Rheol. 2011, 55, 1007–1032. [Google Scholar] [CrossRef]
- Grmela, M.; Chinesta, F.; Ammar, A. Mesoscopic tube model of fluids composed of worm-like micelles. Rheol. Acta 2010, 49, 495–506. [Google Scholar] [CrossRef]
- Olmsted, P.; Radulescu, O.; Lu, C.Y. Johnson–Segalman model with a diffusion term in cylindrical Couette flow. J. Rheol. 2000, 44, 257–275. [Google Scholar] [CrossRef][Green Version]
- Vasquez, P.A.; McKinley, G.H.; Cook, L.P. A network scission model for wormlike micellar solutions: I. Model formulation and viscometric flow predictions. J. Non-Newton. Fluid Mech. 2007, 144, 122–139. [Google Scholar] [CrossRef]
- Germann, N.; Cook, L.; Beris, A.N. Nonequilibrium thermodynamic modeling of the structure and rheology of concentrated wormlike micellar solutions. J. Non-Newton. Fluid Mech. 2013, 196, 51–57. [Google Scholar] [CrossRef]
- Pipe, C.; Kim, N.; Vasquez, P.; Cook, L.; McKinley, G. Wormlike micellar solutions: II. Comparison between experimental data and scission model predictions. J. Rheol. 2010, 54, 881–913. [Google Scholar] [CrossRef]
- Zhou, L.; Cook, L.P.; McKinley, G.H. Multiple shear-banding transitions for a model of wormlike micellar solutions. SIAM J. Appl. Math. 2012, 72, 1192–1212. [Google Scholar] [CrossRef]
- Germann, N.; Cook, L.; Beris, A. Investigation of the inhomogeneous shear flow of a wormlike micellar solution using a thermodynamically consistent model. J. Non-Newton. Fluid Mech. 2014, 207, 21–31. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.F.; Liu, C. A two species micro–macro model of wormlike micellar solutions and its maximum entropy closure approximations: An energetic variational approach. J. Non-Newton. Fluid Mech. 2021, 293, 104559. [Google Scholar] [CrossRef]
- Liu, C.; Wang, Y.; Zhang, T.F. Global existence of classical solutions for a reactive polymeric fluid near equilibrium. Calc. Var. Partial Differ. Equ. 2022, 61, 117. [Google Scholar] [CrossRef]
- De Anna, F.; Liu, C. Non-isothermal general Ericksen–Leslie system: Derivation, analysis and thermodynamic consistency. Arch. Ration. Mech. Anal. 2019, 231, 637–717. [Google Scholar] [CrossRef]
- Liu, P.; Wu, S.; Liu, C. Non-isothermal electrokinetics: Energetic variational approach. Commun. Math. Sci. 2018, 16, 1451–1463. [Google Scholar] [CrossRef]
- Liu, C.; Sulzbach, J.E. The Brinkman-Fourier System with Ideal Gas Equilibrium. arXiv 2020, arXiv:2007.07304. [Google Scholar] [CrossRef]
- Liu, C.; Sulzbach, J.E. Well-Posedness for the Reaction-Diffusion Equation with Temperature in a critical Besov Space. arXiv 2021, arXiv:2101.10419. [Google Scholar] [CrossRef]
- Hieber, M.; Prüss, J.W. Modeling and analysis of the Ericksen-Leslie equations for nematic liquid crystal flows. In Handbook of Mathematical Analysis in Mechanics of Viscous Fluids; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1075–1134. [Google Scholar]
- De Anna, F.; Liu, C.; Schlömerkemper, A.; Sulzbach, J.E. Temperature dependent extensions of the Cahn-Hilliard equation. arXiv 2021, arXiv:2112.14665. [Google Scholar]
- Qian, H.; Kjelstrup, S.; Kolomeisky, A.B.; Bedeaux, D. Entropy production in mesoscopic stochastic thermodynamics: Nonequilibrium kinetic cycles driven by chemical potentials, temperatures, and mechanical forces. J. Phys. Condens. Matter 2016, 28, 153004. [Google Scholar] [CrossRef] [PubMed]
- Prigogine, I. Etude thermodynamique des phenomenes irreversible. Bull. Acad. Roy. Blg. Cl. Sci. 1945, 31, 600–606. [Google Scholar]
- Liu, C.; Wang, Y. A Variational Lagrangian Scheme for a Phase-Field Model: A Discrete Energetic Variational Approach. SIAM J. Sci. Comput. 2020, 42, B1541–B1569. [Google Scholar] [CrossRef]
- Feng, K.; Qin, M. Symplectic Geometric Algorithms for Hamiltonian Systems; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Noh, J.; Wang, Y.; Liang, H.L.; Jampani, V.S.R.; Majumdar, A.; Lagerwall, J.P. Dynamic tuning of the director field in liquid crystal shells using block copolymers. Phys. Rev. Res. 2020, 2, 033160. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Liu, C.; Kang, L. Particle-based energetic variational inference. Stat. Comput. 2021, 31, 34. [Google Scholar] [CrossRef]
- Furihata, D.; Matsuo, T. Discrete Variational Derivative Method: A Structure-Preserving Numerical Method for Partial Differential Equations; Chapman and Hall/CRC: Boca Raton, FL, USA, 2010. [Google Scholar]
- Christiansen, S.H.; Munthe-Kaas, H.Z.; Owren, B. Topics in structure-preserving discretization. Acta Numer. 2011, 20, 1–119. [Google Scholar] [CrossRef]
- Carrillo, J.A.; Moll, J.S. Numerical simulation of diffusive and aggregation phenomena in nonlinear continuity equations by evolving diffeomorphisms. SIAM J. Sci. Comput. 2009, 31, 4305–4329. [Google Scholar] [CrossRef]
- Celledoni, E.; Grimm, V.; McLachlan, R.I.; McLaren, D.; O’Neale, D.; Owren, B.; Quispel, G. Preserving energy resp. dissipation in numerical PDEs using the “Average Vector Field” method. J. Comput. Phys. 2012, 231, 6770–6789. [Google Scholar] [CrossRef]
- Carrillo, J.A.; Ranetbauer, H.; Wolfram, M.T. Numerical simulation of nonlinear continuity equations by evolving diffeomorphisms. J. Comput. Phys. 2016, 327, 186–202. [Google Scholar] [CrossRef]
- Xu, X.; Di, Y.; Doi, M. Variational method for liquids moving on a substrate. Phys. Fluids 2016, 28, 087101. [Google Scholar] [CrossRef]
- Marsden, J.E.; West, M. Discrete mechanics and variational integrators. Acta Numer. 2001, 10, 357–514. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y.; Wu, S.; Bousquet, A. On the stability and accuracy of partially and fully implicit schemes for phase field modeling. Comput. Methods Appl. Mech. Eng. 2019, 345, 826–853. [Google Scholar] [CrossRef]
- Du, Q.; Feng, X. The phase field method for geometric moving interfaces and their numerical approximations. In Handbook of Numerical Analysis; Elsevier: Amsterdam, The Netherlands, 2020; Volume 21, pp. 425–508. [Google Scholar]
- Braides, A. Local Minimization, Variational Evolution and Γ-Convergence; Springer: Berlin/Heidelberg, Germany, 2014; Volume 2094. [Google Scholar]
- Esedog Lu, S.; Otto, F. Threshold dynamics for networks with arbitrary surface tensions. Commun. Pure Appl. Math. 2015, 68, 808–864. [Google Scholar] [CrossRef]
- Carrillo, J.A.; Düring, B.; Matthes, D.; McCormick, D.S. A Lagrangian scheme for the solution of nonlinear diffusion equations using moving simplex meshes. J. Sci. Comput. 2018, 75, 1463–1499. [Google Scholar] [CrossRef]
- Matthes, D.; Plazotta, S. A variational formulation of the BDF2 method for metric gradient flows. ESAIM Math. Model. Numer. Anal. 2019, 53, 145–172. [Google Scholar] [CrossRef]
- Elliott, C.M.; Stuart, A. The global dynamics of discrete semilinear parabolic equations. SIAM J. Numer. Anal. 1993, 30, 1622–1663. [Google Scholar] [CrossRef]
- Eyre, D.J. Unconditionally gradient stable time marching the Cahn-Hilliard equation. In Materials Research Society Symposium Proceedings; Materials Research Society: Warrendale, PA, USA, 1998; Volume 529, pp. 39–46. [Google Scholar]
- Shen, J.; Wang, C.; Wang, X.; Wise, S.M. Second-order convex splitting schemes for gradient flows with Ehrlich–Schwoebel type energy: Application to thin film epitaxy. SIAM J. Numer. Anal. 2012, 50, 105–125. [Google Scholar] [CrossRef]
- Jiang, K.; Si, W.; Bao, C. An efficient method for computing stationary states of phase field crystal models. arXiv 2019, arXiv:1909.00305. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, J.; Wang, Q. Numerical approximations for the molecular beam epitaxial growth model based on the invariant energy quadratization method. J. Comput. Phys. 2017, 333, 104–127. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Q.; Yang, X. Numerical approximations for a phase field dendritic crystal growth model based on the invariant energy quadratization approach. Int. J. Numer. Methods Eng. 2017, 110, 279–300. [Google Scholar] [CrossRef]
- Shen, J.; Xu, J.; Yang, J. The scalar auxiliary variable (SAV) approach for gradient flows. J. Comput. Phys. 2018, 353, 407–416. [Google Scholar] [CrossRef]
- Shen, J.; Xu, J.; Yang, J. A new class of efficient and robust energy stable schemes for gradient flows. SIAM Rev. 2019, 61, 474–506. [Google Scholar] [CrossRef]
- Du, Q.; Nicolaides, R.A. Numerical analysis of a continuum model of phase transition. SIAM J. Numer. Anal. 1991, 28, 1310–1322. [Google Scholar] [CrossRef]
- Gottlieb, S.; Shu, C.W.; Tadmor, E. Strong stability-preserving high-order time discretization methods. SIAM Rev. 2001, 43, 89–112. [Google Scholar] [CrossRef]
- Canevari, G.; Harris, J.; Majumdar, A.; Wang, Y. The well order reconstruction solution for three-dimensional wells, in the Landau–de Gennes theory. Int. J. Non-Linear Mech. 2020, 119, 103342. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P.; Chen, J.Z.Y. Topological defects in an unconfined nematic fluid induced by single and double spherical colloidal particles. Phys. Rev. E 2017, 96, 042702. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P.; Chen, J.Z.Y. Formation of three-dimensional colloidal crystals in a nematic liquid crystal. Soft Matter 2018, 14, 6756–6766. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, C. Some Recent Advances in Energetic Variational Approaches. Entropy 2022, 24, 721. https://doi.org/10.3390/e24050721
Wang Y, Liu C. Some Recent Advances in Energetic Variational Approaches. Entropy. 2022; 24(5):721. https://doi.org/10.3390/e24050721
Chicago/Turabian StyleWang, Yiwei, and Chun Liu. 2022. "Some Recent Advances in Energetic Variational Approaches" Entropy 24, no. 5: 721. https://doi.org/10.3390/e24050721
APA StyleWang, Y., & Liu, C. (2022). Some Recent Advances in Energetic Variational Approaches. Entropy, 24(5), 721. https://doi.org/10.3390/e24050721