
Phylogenetic Curved Optimal Regression for Adaptive Trait Evolution



Abstract
1. Introduction
2. Materials and Methods
2.1. Optimal Exponential Regression
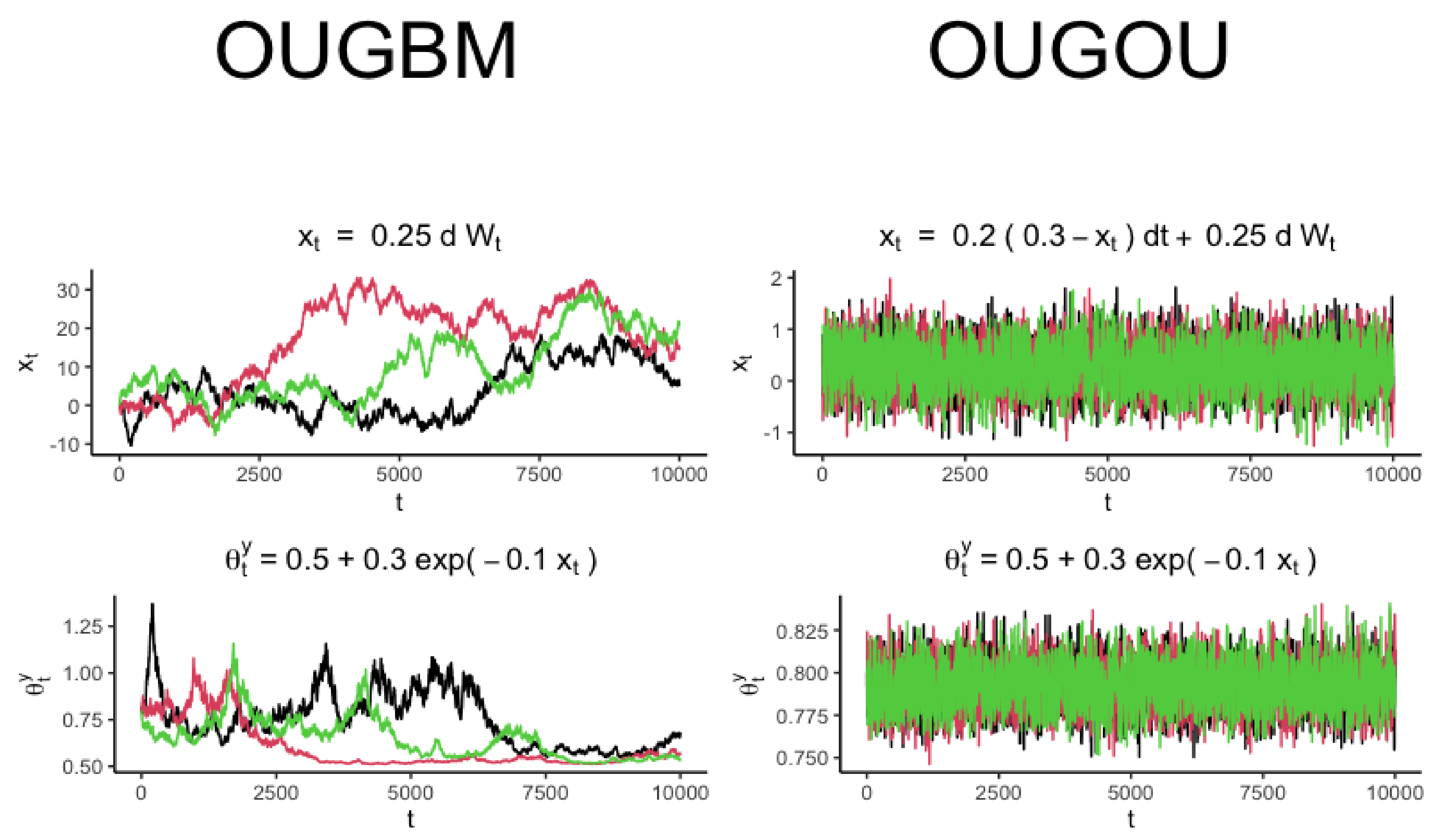
2.1.1. OUGBM Model
2.1.2. OUGOU Model
2.2. Optimal Linear Regression
2.2.1. OUBM Model
2.2.2. OUOU Model
2.3. Optimal Adaptive-Trait Evolution along Phylogenetic Tree
2.4. Approximate Bayesian Computation
| Algorithm 1: Approximate Bayesian computation for the models of adaptive trait evolution. |
|
2.5. Interpretation of Change of Optimum by Its Covariate
3. Results
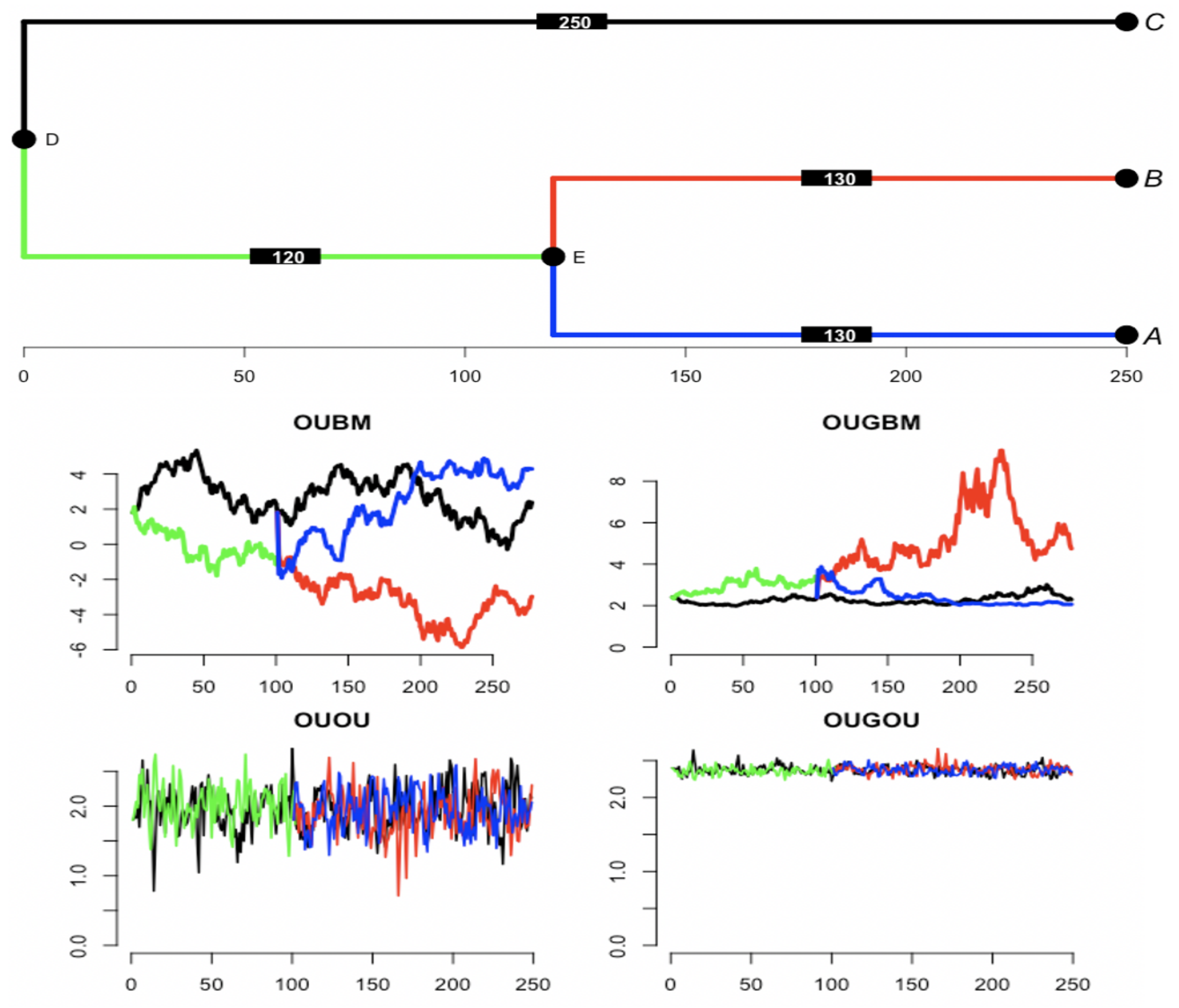
3.1. Simulation
3.1.1. Parameter Estimation
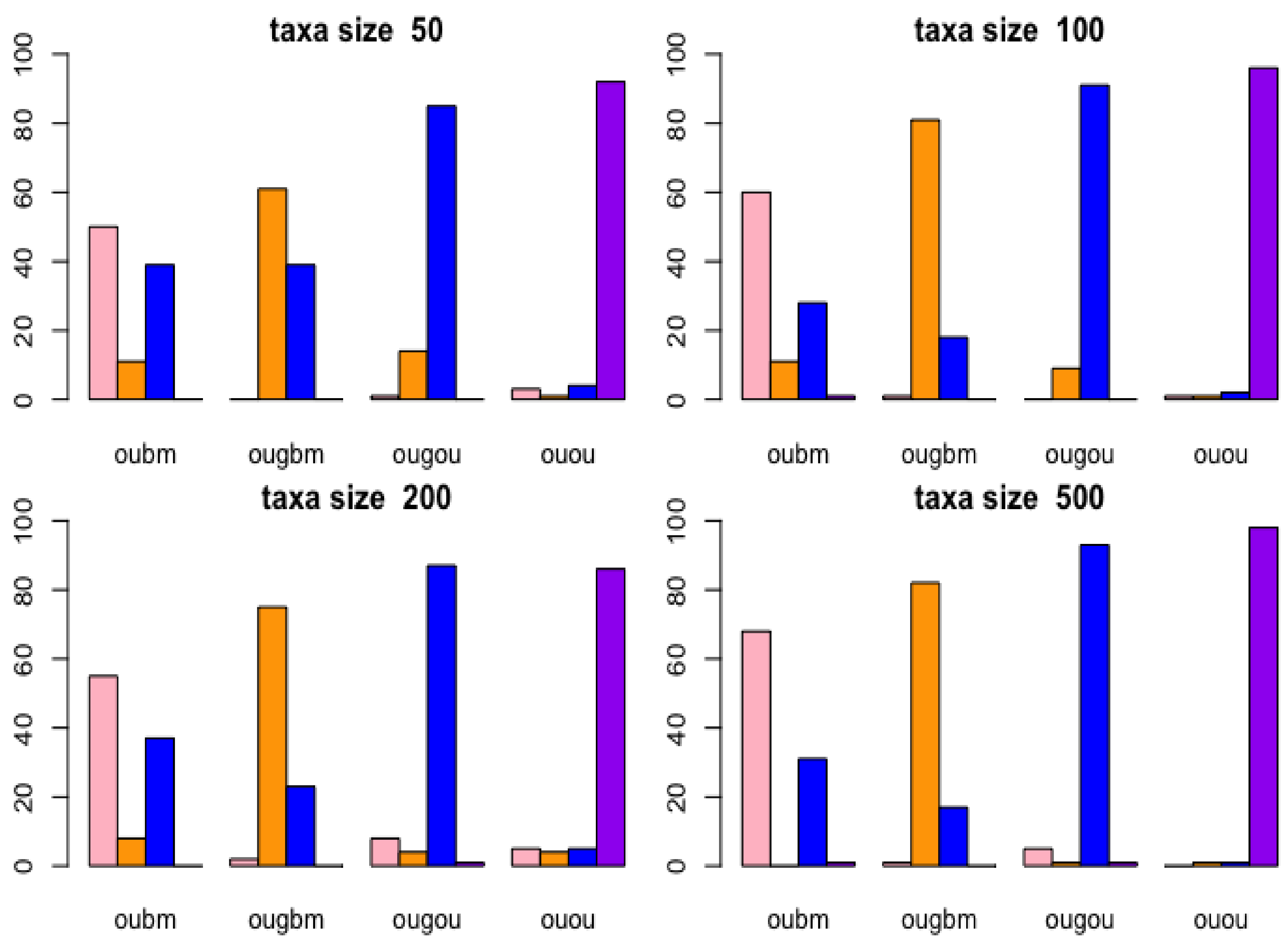
3.1.2. Cross-Validation
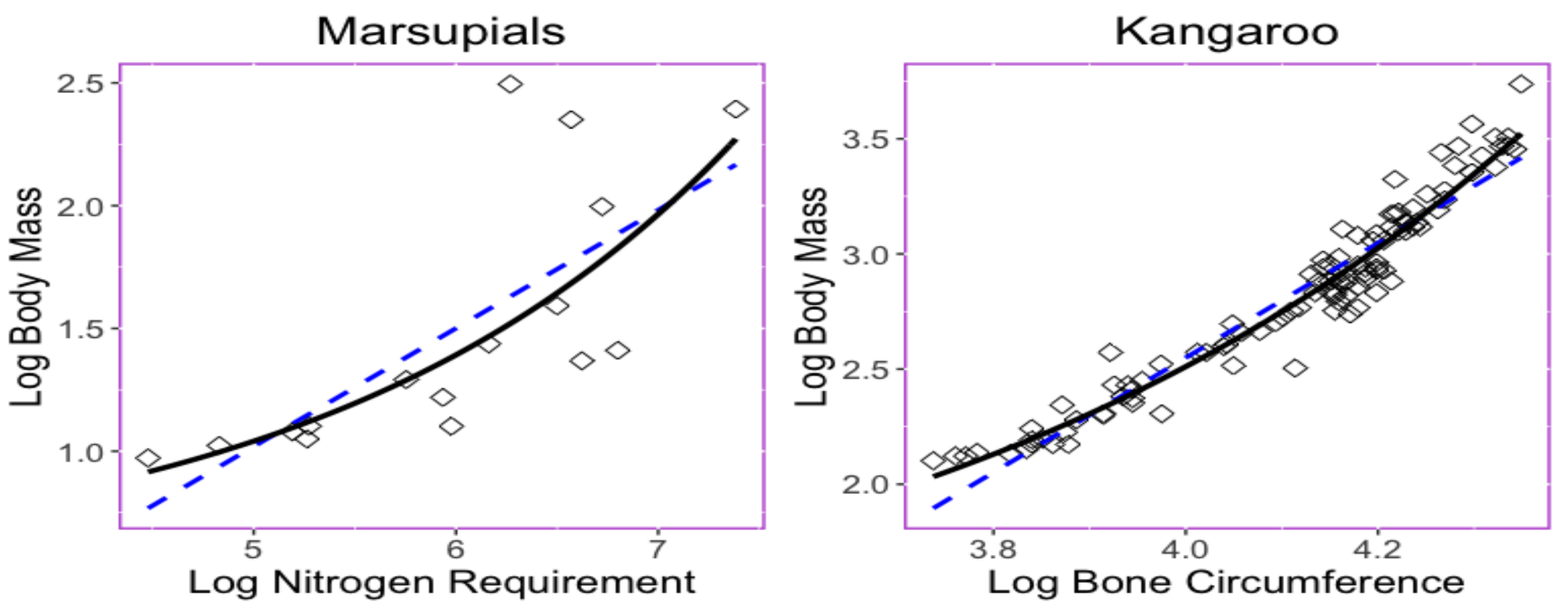
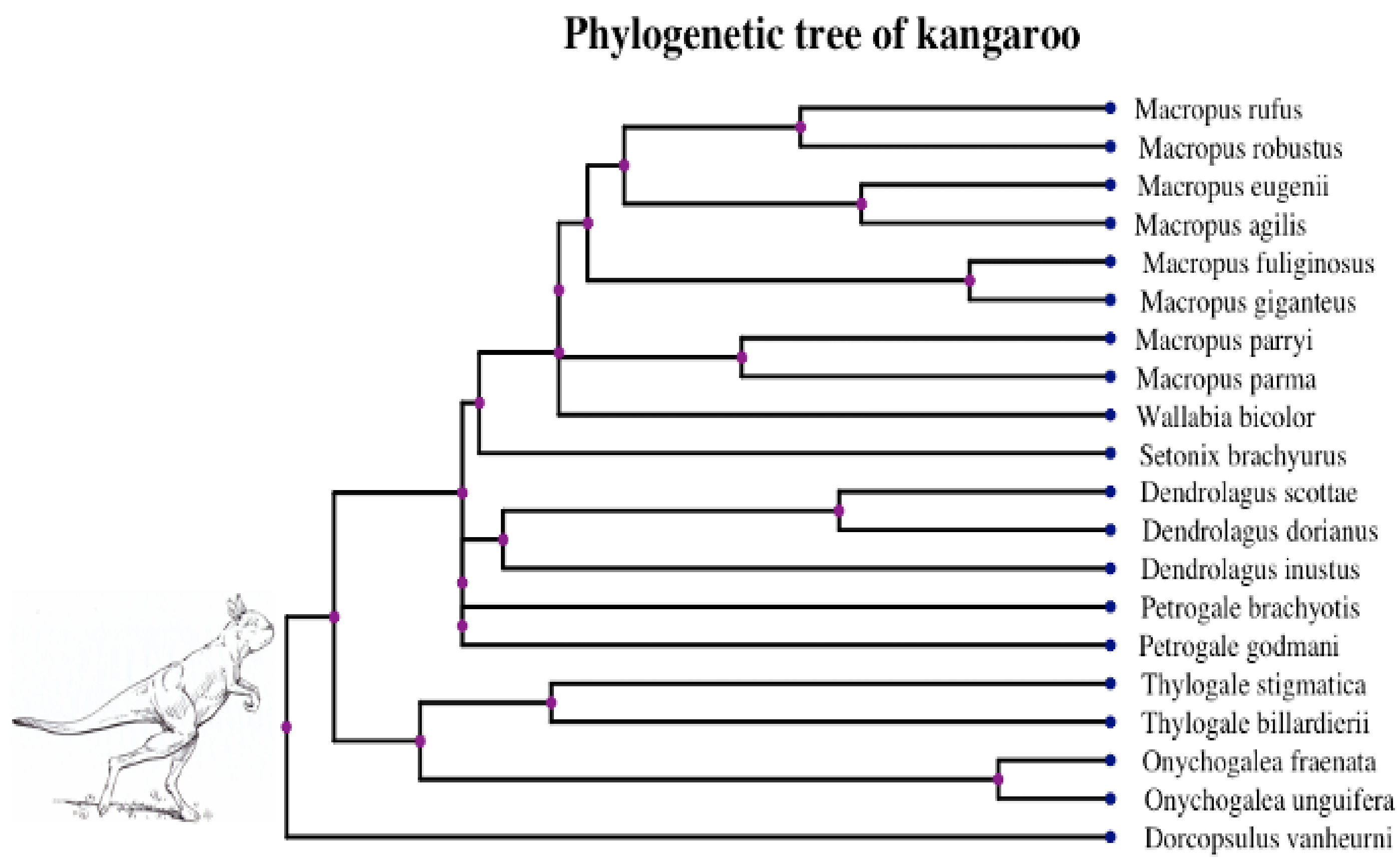
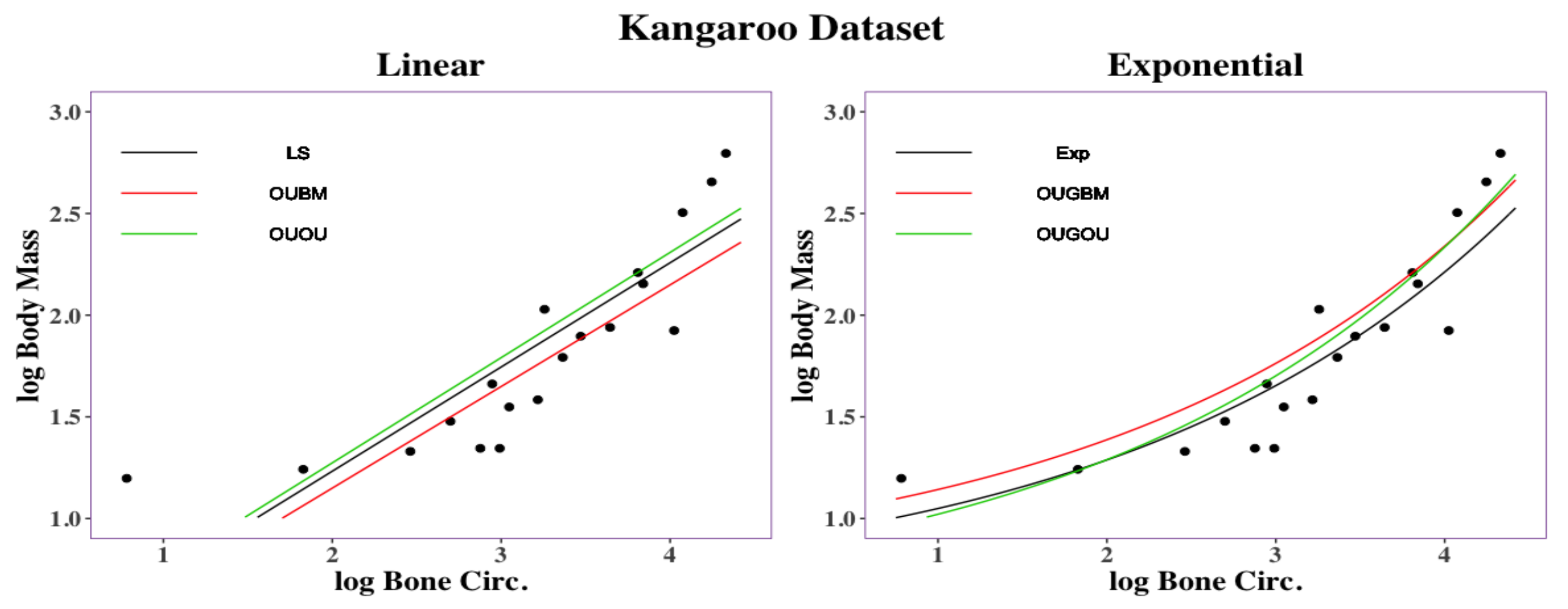
3.2. Empirical Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Felsenstein, J. Phylogenies and the comparative method. Am. Nat. 1985, 125, 1–15. [Google Scholar] [CrossRef]
- Lynch, M. Methods for the analysis of comparative data in evolutionary biology. Evolution 1991, 45, 1065–1080. [Google Scholar] [CrossRef]
- Harvey, P.H.; Pagel, M.D. The Comparative Method in Evolutionary Biology; Oxford University Press: Oxford, UK, 1991; Volume 239. [Google Scholar]
- Felsenstein, J. Inferring Phylogenies; Sinauer Associates: Sunderland, MA, USA, 2004; Volume 2. [Google Scholar]
- O’Meara, B.C. Evolutionary inferences from phylogenies: A review of methods. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 267–285. [Google Scholar] [CrossRef]
- Hernández, C.E.; Rodríguez-Serrano, E.; Avaria-Llautureo, J.; Inostroza-Michael, O.; Morales-Pallero, B.; Boric-Bargetto, D.; Canales-Aguirre, C.B.; Marquet, P.A.; Meade, A. Using phylogenetic information and the comparative method to evaluate hypotheses in macroecology. Methods Ecol. Evol. 2013, 4, 401–415. [Google Scholar] [CrossRef]
- Pennell, M.W.; Harmon, L.J. An integrative view of phylogenetic comparative methods: Connections to population genetics, community ecology, and paleobiology. Ann. N. Y. Acad. Sci. 2013, 1289, 90–105. [Google Scholar] [CrossRef]
- Garamszegi, L.Z. Modern Phylogenetic Comparative Methods and Their Application in Evolutionary Biology. Concepts and Practice; Springer: London, UK, 2014. [Google Scholar]
- Grafen, A. The phylogenetic regression. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1989, 326, 119–157. [Google Scholar] [PubMed]
- Freckleton, R.P.; Harvey, P.H.; Pagel, M. Phylogenetic analysis and comparative data: A test and review of evidence. Am. Nat. 2002, 160, 712–726. [Google Scholar] [CrossRef]
- Xiao, X.; White, E.; Hooten, M.; Durham, S. On the use of log-transform vs. nonlinear regression for analyzing biological power laws. Ecology 2011, 92, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Harmon, L. Phylogenetic Comparative Methods: Learning from Trees; CreateSpace Independent Publishing Platform: Charleston, SC, USA, 2018. [Google Scholar]
- Ives, A.R.; Garland, T., Jr. Phylogenetic logistic regression for binary dependent variables. Syst. Biol. 2009, 59, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P.; Midford, P.E.; Otto, S.P. Estimating a binary character’s effect on speciation and extinction. Syst. Biol. 2007, 56, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Packard, G.C. On the use of log-transformation versus nonlinear regression for analyzing biological power laws. Biol. J. Linn. Soc. 2014, 113, 1167–1178. [Google Scholar] [CrossRef]
- Klaassen, M.; Nolet, B.A. Stoichiometry of endothermy: Shifting the quest from nitrogen to carbon. Ecol. Lett. 2008, 11, 785–792. [Google Scholar] [CrossRef]
- Hume, I.D. Marsupial Nutrition; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Helgen, K.M.; Wells, R.T.; Kear, B.P.; Gerdtz, W.R.; Flannery, T.F. Ecological and evolutionary significance of sizes of giant extinct kangaroos. Aust. J. Zool. 2006, 54, 293–303. [Google Scholar] [CrossRef]
- Jhwueng, D.C. Modeling rate of adaptive trait evolution using Cox–Ingersoll–Ross process: An Approximate Bayesian Computation approach. Comput. Stat. Data Anal. 2020, 145, 106924. [Google Scholar] [CrossRef]
- Hansen, T.F.; Pienaar, J.; Orzack, S.H. A comparative method for studying adaptation to a randomly evolving environment. Evolution 2008, 62, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Jhwueng, D.C.; Maroulas, V. Phylogenetic ornstein–uhlenbeck regression curves. Stat. Probab. Lett. 2014, 89, 110–117. [Google Scholar] [CrossRef][Green Version]
- Jhwueng, D.C.; Maroulas, V. Adaptive trait evolution in random environment. J. Appl. Stat. 2016, 43, 2310–2324. [Google Scholar] [CrossRef]
- Bartoszek, K.; Pienaar, J.; Mostad, P.; Andersson, S.; Hansen, T.F. A phylogenetic comparative method for studying multivariate adaptation. J. Theor. Biol. 2012, 314, 204–215. [Google Scholar] [CrossRef]
- Cressler, C.E.; Butler, M.A.; King, A.A. Detecting adaptive evolution in phylogenetic comparative analysis using the Ornstein–Uhlenbeck model. Syst. Biol. 2015, 64, 953–968. [Google Scholar] [CrossRef]
- Marass, F.; Mouliere, F.; Yuan, K.; Rosenfeld, N.; Markowetz, F. A phylogenetic latent feature model for clonal deconvolution. Ann. Appl. Stat. 2016, 10, 2377–2404. [Google Scholar] [CrossRef]
- Oksendal, B. Stochastic Differential Equations: An Introduction with Applications; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Ksendal, B. Stochastic differential equations. In Stochastic Differential Equations; Springer: Berlin, Germany, 2003; pp. 65–84. [Google Scholar]
- Vega, C.A.M. Calibration of the exponential Ornstein–Uhlenbeck process when spot prices are visible through the maximum log-likelihood method. Example with gold prices. Adv. Differ. Equations 2018, 2018, 269. [Google Scholar] [CrossRef]
- Lyasoff, A. Another look at the integral of exponential Brownian motion and the pricing of Asian options. Financ. Stochastics 2016, 20, 1061–1096. [Google Scholar] [CrossRef]
- Dufresne, D. The distribution of a perpetuity, with applications to risk theory and pension funding. Scand. Actuar. J. 1990, 1990, 39–79. [Google Scholar] [CrossRef]
- Dufresne, D. The integral of geometric Brownian motion. Adv. Appl. Probab. 2001, 33, 223–241. [Google Scholar] [CrossRef]
- Yor, M. On some exponential functionals of Brownian motion. Adv. Appl. Probab. 1992, 24, 509–531. [Google Scholar] [CrossRef]
- Burden, R.L.; Faires, J.D. Numerical Analysis, 9th ed.; Brooks Cole Publishing: Monterey, CA, USA, 2010. [Google Scholar]
- Borchers, H.W. Pracma: Practical Numerical Math Functions. R Package Version 2.2.9. 2019. Available online: https://CRAN.R-project.org/package=pracma (accessed on 12 September 2020).
- Baum, D. Trait evolution on a phylogenetic tree: Relatedness. Nat. Educ. 2008, 1, 191. [Google Scholar]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Csillery, K.; Francois, O.; Blum, M.G.B. abc: An R package for approximate Bayesian computation (ABC). Methods Ecol. Evol. 2012. [Google Scholar] [CrossRef]
- Blomberg, S.P.; Garland, T., Jr.; Ives, A.R. Testing for phylogenetic signal in comparative data: Behavioral traits are more labile. Evolution 2003, 57, 717–745. [Google Scholar] [CrossRef] [PubMed]
- Pagel, M. Inferring thetheirtorical patterns of biological evolution. Nature 1999, 401, 877–884. [Google Scholar] [CrossRef]
- Adams, D.C.; Felice, R.N. Assessing trait covariation and morphological integration on phylogenies using evolutionary covariance matrices. PLoS ONE 2014, 9, e94335. [Google Scholar] [CrossRef]
- Bartoszek, K.; Liò, P. Modelling trait dependent speciation with Approximate Bayesian Computation. arXiv 2018, arXiv:1812.03715. [Google Scholar] [CrossRef]
- Lepers, C.; Billiard, S.; Porte, M.; Méléard, S.; Tran, V.C. Inference with selection, varying population size and evolving population structure: Application of ABC to a forward-backward coalescent process with interactions. arXiv 2019, arXiv:1910.10201. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, J.C.; Harmon, L.J. A novel Bayesian method for inferring and interpreting the dynamics of adaptive landscapes from phylogenetic comparative data. Syst. Biol. 2014, 63, 902–918. [Google Scholar] [CrossRef]
- Bastide, P.; Ho, L.S.T.; Baele, G.; Lemey, P.; Suchard, M.A. Efficient Bayesian Inference of General Gaussian Models on Large Phylogenetic Trees. arXiv 2020, arXiv:2003.10336. [Google Scholar]
- Stadler, T. TreeSim: Simulating Phylogenetic Trees. R package version 2.4. 2019. Available online: https://CRAN.R-project.org/package=TreeSim (accessed on 28 August 2020).
- Bo, L.; Wang, L.; Jiao, L. Feature scaling for kernel fisher discriminant analysis using leave-one-out cross validation. Neural Comput. 2006, 18, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Janis, C.M.; Buttrill, K.; Figueirido, B. Locomotion in extinct giant kangaroos: Were sthenurines hop-less monsters? PLoS ONE 2014, 9, e109888. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.; Thomas, G.H.; Venditti, C.; Meade, A.; Freckleton, R.P. A cautionary note on the use of Ornstein Uhlenbeck models in macroevolutionary studies. Biol. J. Linn. Soc. 2016, 118, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, W.; Nakagawa, S. Phylogenetic comparative methods. Curr. Biol. 2017, 27, R333–R336. [Google Scholar] [CrossRef] [PubMed]
- Jhwueng, D.C. Building an adaptive trait simulator package to infer parametric diffusion model along phylogenetic tree. MethodsX 2020, 7, 100978. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Par | True 1 | Prior 1 | True 2 | Prior 2 |
|---|---|---|---|---|
| 0.50 | 0.20 | (rate = 5) | ||
| 0.125 | 0.125 | (rate = 8) | ||
| 0.00 | 1.00 | (mean = 1, sd = 1) | ||
| 2.50 | 0.5 | (sh = 2,sc = 0.5) | ||
| 1.00 | 0.5 | (sh = 2, sc = 0.5) | ||
| 0.00 | 0.00 | |||
| 1.00 | −2.00 | |||
| −0.50 | −0.5 |
| Model | Taxa | |||||
|---|---|---|---|---|---|---|
| True Value | ||||||
| OUGBM | 16 | 0.52 (0.06, 0.96) | 2.16 (0.26, 4.59) | 0.89 (0.16, 1.83) | ||
| 32 | 0.53 (0.08, 0.95) | 1.83 (0.25, 4.3) | 0.95 (0.2, 1.82) | |||
| 64 | 0.54 (0.09, 0.95) | 1.66 (0.2, 4.1) | 0.93 (0.2, 1.78) | |||
| 128 | 0.52 (0.08, 0.95) | 1.65 (0.21, 4.07) | 0.91 (0.2, 1.78) | |||
| OUGOU | 16 | 0.44 (0.04, 0.95) | 0.12 (0.01, 0.24) | −1.14 (−4.49, 2.81) | 2.25 (0.65, 4.28) | 1.16 (0.38, 1.88) |
| 32 | 0.47 (0.04, 0.95) | 0.12 (0.01, 0.24) | −1.22 (−4.59, 2.75) | 2.52 (0.82, 4.56) | 0.99 (0.19, 1.83) | |
| 64 | 0.48 (0.04, 0.95) | 0.12 (0.01, 0.24) | −1.16 (−4.58, 2.93) | 2.61 (0.87, 4.59) | 0.95 (0.18, 1.81) | |
| 128 | 0.49 (0.04, 0.95) | 0.12 (0.01, 0.24) | −1.16 (−4.58, 2.88) | 2.57 (0.79, 4.57) | 0.9 (0.16, 1.78) | |
| OUBM | 16 | 0.5 (0.05, 0.95) | 2.14 (0.59, 4.22) | 1.13 (0.11, 1.92) | ||
| 32 | 0.56 (0.07, 0.96) | 2.05 (0.55, 4.21) | 1.05 (0.1, 1.92) | |||
| 64 | 0.52 (0.06, 0.96) | 1.95 (0.48, 4.12) | 1.07 (0.11, 1.92) | |||
| 128 | 0.54 (0.06, 0.96) | 1.92 (0.51, 4.05) | 1.06 (0.11, 1.91) | |||
| OUOU | 16 | 0.53 (0.05, 0.95) | 0.12 (0.01, 0.24) | 0.63 (−4.15, 4.49) | 2.13 (0.64, 4.11) | 1.08 (0.12, 1.92) |
| 32 | 0.55 (0.05, 0.95) | 0.12 (0.01, 0.24) | 0.94 (−4.15, 4.56) | 1.9 (0.42, 4) | 1.06 (0.13, 1.9) | |
| 64 | 0.53 (0.05, 0.94) | 0.12 (0.01, 0.24) | 0.79 (−4.18, 4.54) | 1.85 (0.42, 3.96) | 1.06 (0.12, 1.91) | |
| 128 | 0.55 (0.05, 0.95) | 0.12 (0.01, 0.24) | 0.79 (−4.26, 4.54) | 1.81 (0.44, 3.92) | 1.05 (0.11, 1.9) |
| Model | Taxa | |||
|---|---|---|---|---|
| True Value | ||||
| OUGBM | 16 | −0.08 (−0.91, 0.84) | 1.01 (0.14, 1.89) | −0.46 (−0.94, −0.05) |
| 32 | −0.02 (−0.9, 0.87) | 0.95 (0.12, 1.87) | −0.47 (−0.95, −0.05) | |
| 64 | −0.02 (−0.91, 0.89) | 0.96 (0.14, 1.86) | −0.48 (−0.95, −0.05) | |
| 128 | 0.01 (−0.9, 0.9) | 0.97 (0.14, 1.86) | −0.48 (−0.95, −0.04) | |
| OUGOU | 16 | −0.01 (−0.92, 0.88) | 0.88 (0.06, 1.89) | −0.47 (−0.92, −0.05) |
| 32 | −0.03 (−0.92, 0.88) | 0.89 (0.07, 1.89) | −0.48 (−0.94, −0.05) | |
| 64 | −0.05 (−0.92, 0.88) | 0.88 (0.07, 1.89) | −0.48 (−0.93, −0.05) | |
| 128 | −0.05 (−0.92, 0.88) | 0.91 (0.07, 1.89) | −0.49 (−0.94, −0.05) | |
| OUBM | 16 | −0.03 (−0.88, 0.89) | 0.8 (0.11, 1.81) | |
| 32 | 0.01 (−0.89, 0.9) | 0.78 (0.09, 1.82) | ||
| 64 | −0.01 (−0.9, 0.89) | 0.79 (0.09, 1.83) | ||
| 128 | −0.02 (−0.9, 0.89) | 0.8 (0.09, 1.83) | ||
| OUOU | 16 | −0.11 (−0.9, 0.88) | 0.86 (0.11, 1.81) | |
| 32 | −0.11 (−0.9, 0.88) | 0.81 (0.09, 1.83) | ||
| 64 | −0.1 (−0.89, 0.88) | 0.85 (0.1, 1.85) | ||
| 128 | −0.09 (−0.89, 0.88) | 0.82 (0.1, 1.84) |
| Model | Parameter | |||||||
|---|---|---|---|---|---|---|---|---|
| EXP | 0.5987 | 0.2946 | 0.4251 | |||||
| OUGBM | 0.0016 | 1.3420 | 0.7888 | 0.6848 | 0.2985 | 0.4281 | ||
| OUGOU | 0.0014 | 0.0015 | 0.8034 | 0.5480 | −1.2113 | 0.5208 | 0.3258 | 0.4293 |
| LS | 0.2078 | 0.5125 | ||||||
| OUBM | 0.0015 | 1.4413 | 0.7392 | 0.1504 | 0.4996 | |||
| OUOU | 0.0014 | 0.0014 | 0.9952 | 0.6931 | −0.5732 | 0.2392 | 0.5713 | |
| 0.3360 | 0.2810 | 0.2240 | 0.1590 | ||
|---|---|---|---|---|---|
| Rank | Model | OUGBM | OUGOU | OUBM | OUOU |
| 1st | OUGBM | 1.0000 | 1.1957 | 1.5000 | 2.1132 |
| 2nd | OUGOU | 0.8363 | 1.0000 | 1.2545 | 1.7673 |
| 3rd | OUBM | 0.6667 | 0.7972 | 1.0000 | 1.4088 |
| 4th | OUOU | 0.4732 | 0.5658 | 0.7098 | 1.0000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jhwueng, D.-C.; Wang, C.-P. Phylogenetic Curved Optimal Regression for Adaptive Trait Evolution. Entropy 2021, 23, 218. https://doi.org/10.3390/e23020218
Jhwueng D-C, Wang C-P. Phylogenetic Curved Optimal Regression for Adaptive Trait Evolution. Entropy. 2021; 23(2):218. https://doi.org/10.3390/e23020218
Chicago/Turabian StyleJhwueng, Dwueng-Chwuan, and Chih-Ping Wang. 2021. "Phylogenetic Curved Optimal Regression for Adaptive Trait Evolution" Entropy 23, no. 2: 218. https://doi.org/10.3390/e23020218
APA StyleJhwueng, D.-C., & Wang, C.-P. (2021). Phylogenetic Curved Optimal Regression for Adaptive Trait Evolution. Entropy, 23(2), 218. https://doi.org/10.3390/e23020218