A Nonparametric Bayesian Approach to the Rare Type Match Problem

Abstract
1. Introduction
2. State of the Art
2.1. The Rare Type Match Problem
2.2. Evaluation of Matching Probabilities of Y-STR Data
3. The Model
3.1. Notation and Data
3.2. Model Assumptions
3.3. Prior
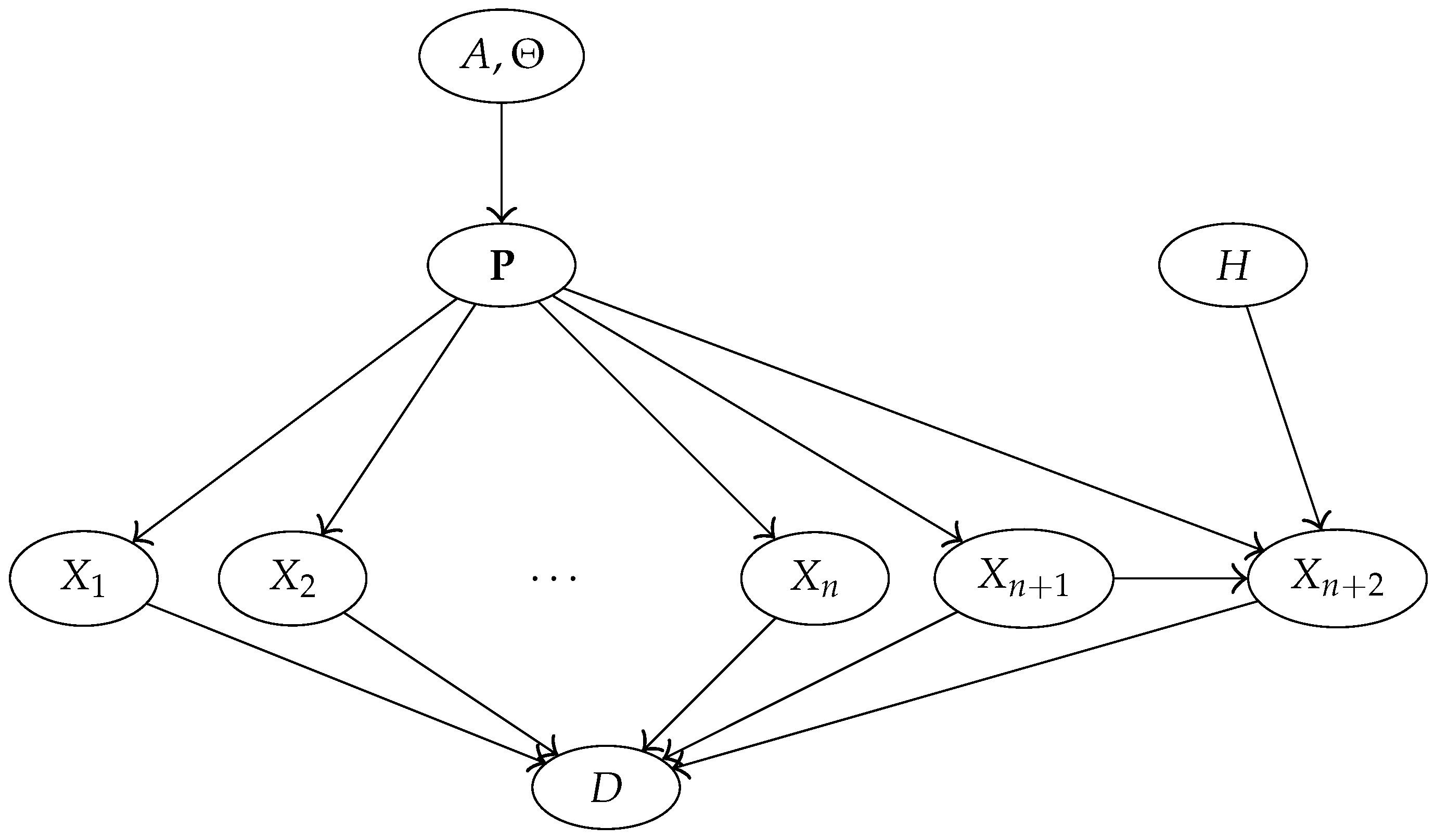
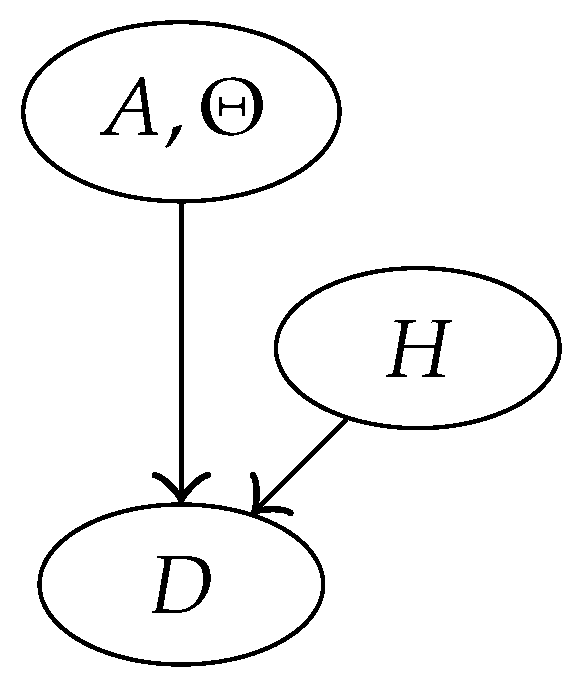
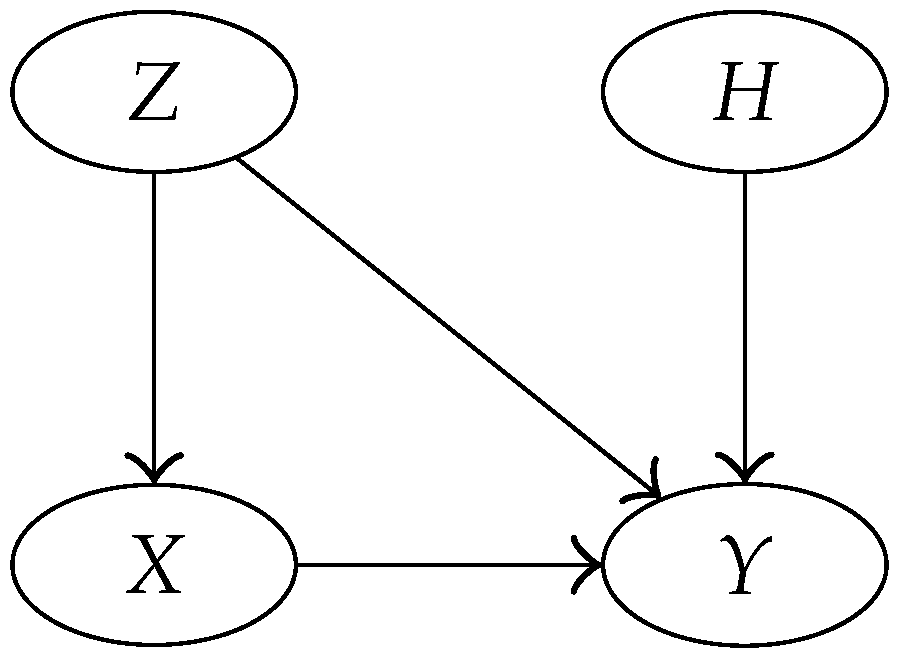
3.4. Bayesian Network Representation of the Model
- = suspect’s DNA type,
- = crime stain’s DNA type (matching the suspect’s type),
- B = a reference database of size n, which is treated here as a random sample of DNA types from the population of possible perpetrators.
- = The crime stain originated from the suspect,
- = The crime stain originated from someone else.
3.5. Random Partitions and Database Partitions
- The random partitions defined through the random variables , …, and through the database are the same:
- Although , …, were not observable, the random partitions , and are observable.
3.6. Chinese Restaurant Representation
3.7. A Useful Lemma


4. The Likelihood Ratio
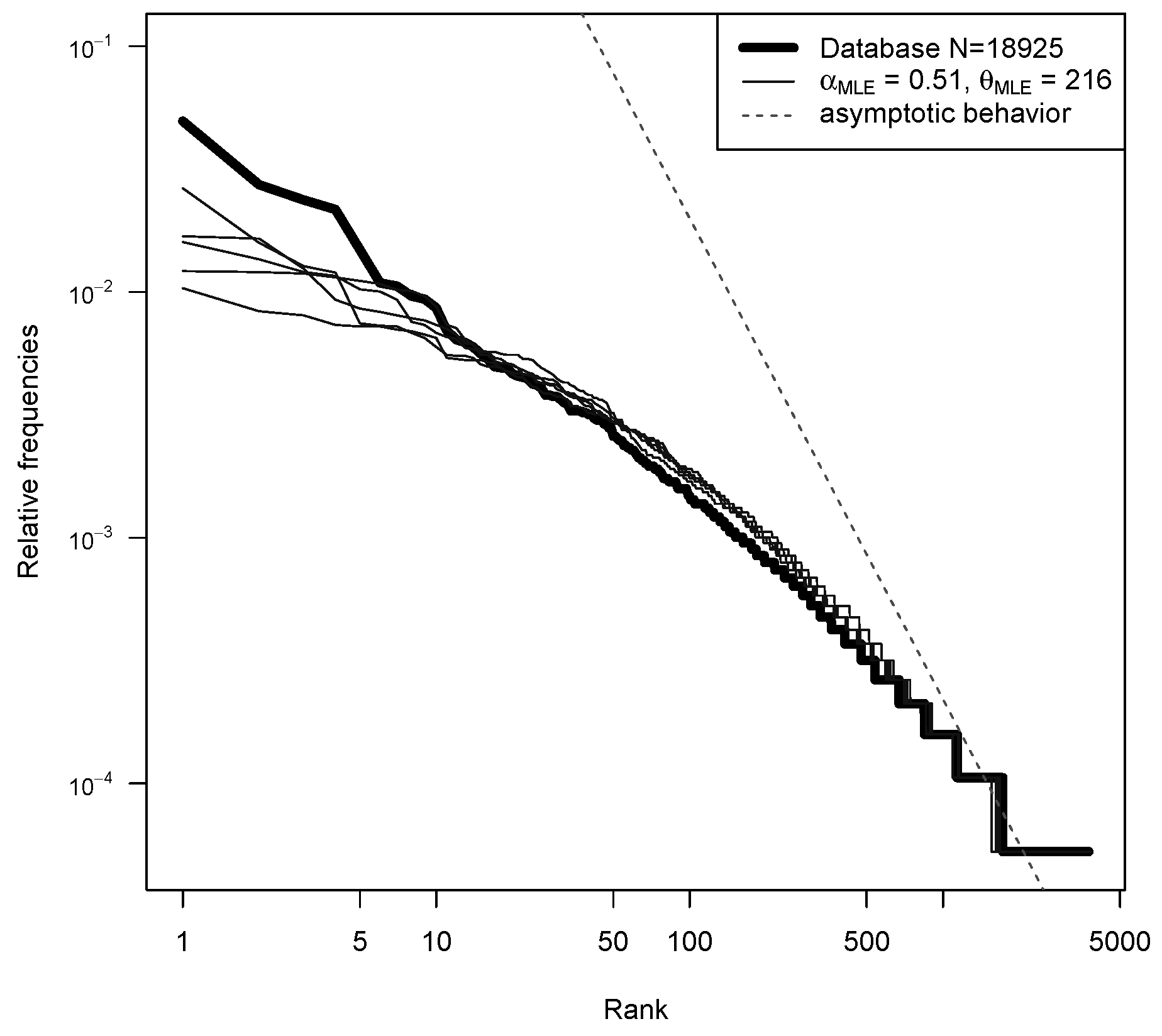
5. Analysis on the YHRD Database
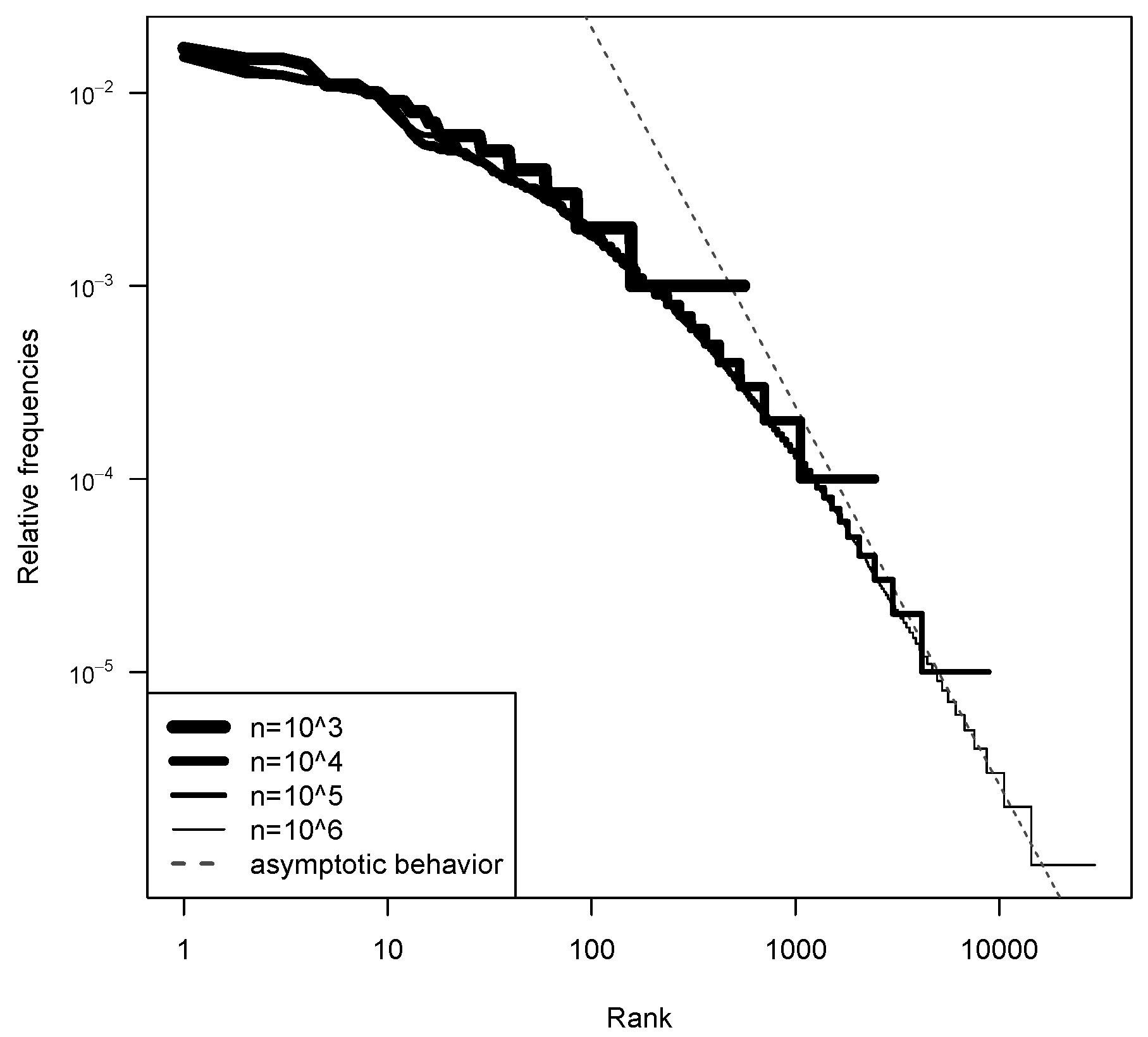
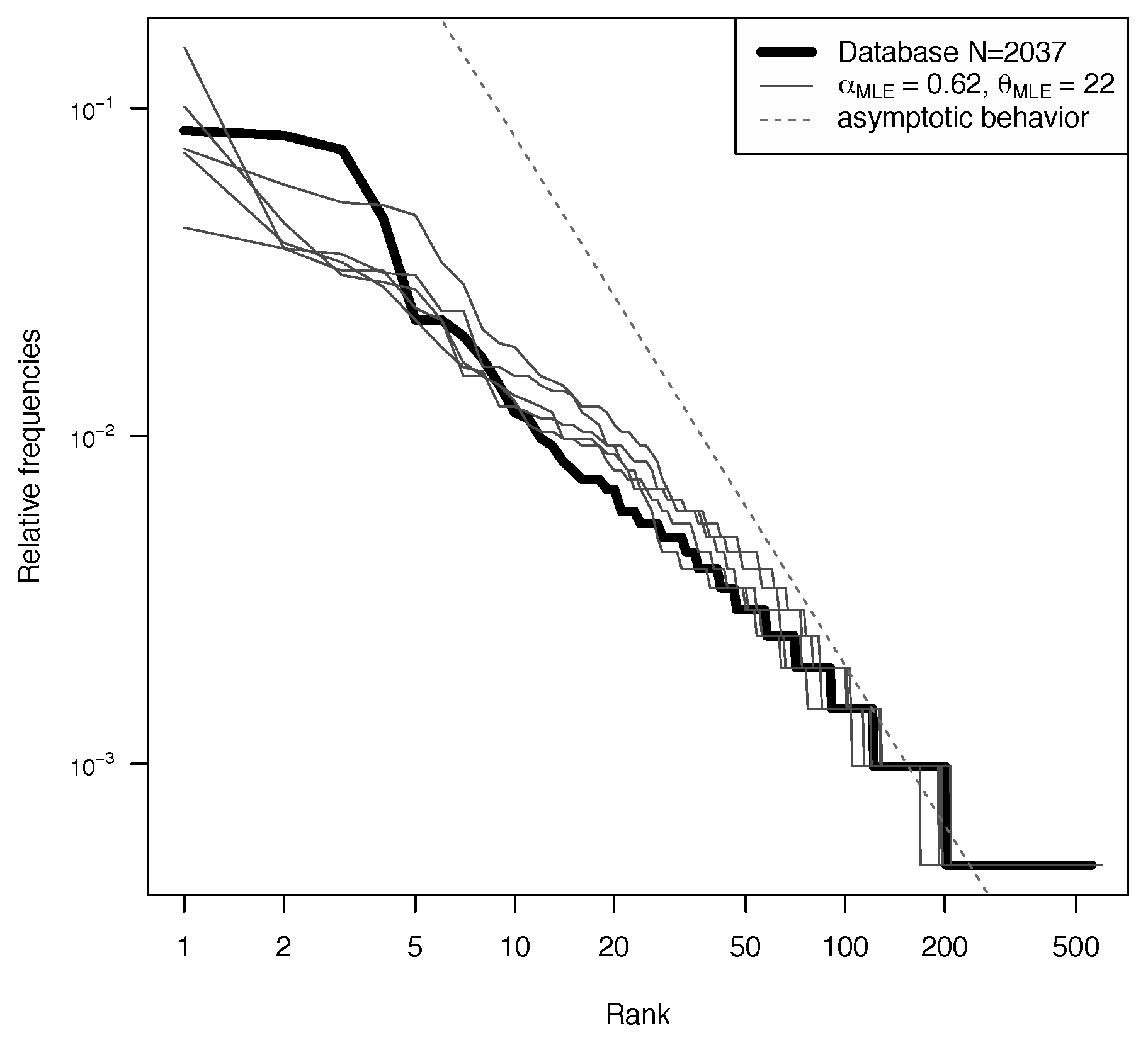
5.1. Model Fitting
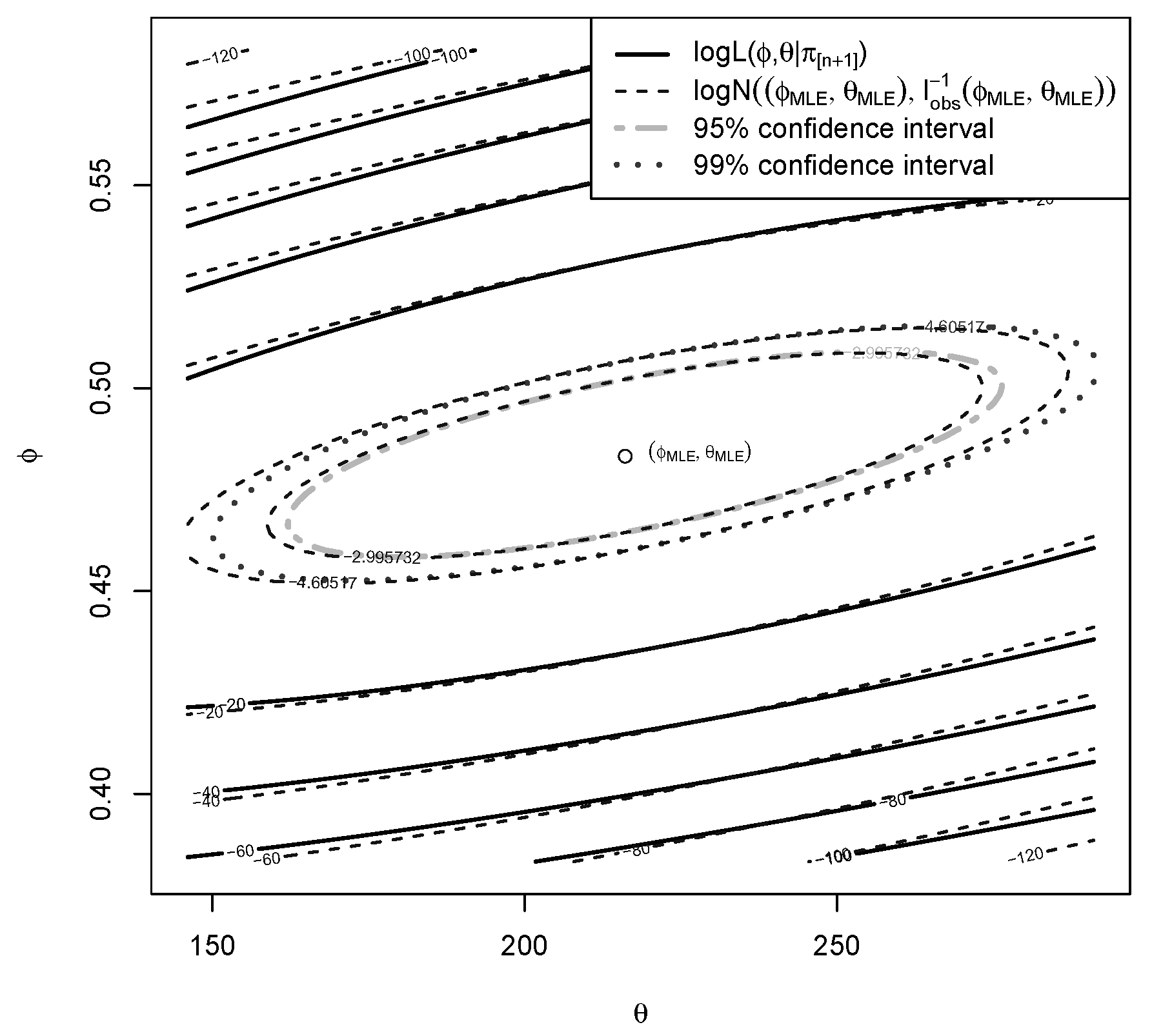
5.2. Log-Likelihood
5.3. True LR
Details of the Metropolis–Hastings Algorithm
5.4. Frequentist-Bayesian Analysis of the Error
- (loss due to choice of the Poisson Dirichlet model and approximation (13)),
- (overall loss).
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Robertson, B.; Vignaux, G.A. Interpreting Evidence: Evaluating Forensic Science in the Courtroom; John Wiley & Sons: Chichester, UK, 1995. [Google Scholar]
- Evett, I.; Weir, B. Interpreting DNA Evidence: Statistical Genetics for Forensic Scientists; Sinauer Associates: Sunderland, UK, 1998. [Google Scholar]
- Aitken, C.; Taroni, F. Statistics and the Evaluation of Evidence for Forensics Scientists; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Balding, D. Weight-of-Evidence for Forensic DNA Profiles; John Wiley & Sons: Chichester, UK, 2005. [Google Scholar]
- Taroni, F.; Aitken, C.; Garbolino, P.; Biedermann, A. Bayesian Networks and Probabilistic Inference in Forensic Science; John Wiley & Sons: Chichester, UK, 2006. [Google Scholar]
- Cereda, G. Bayesian approach to LR in case of rare type match. Stat. Neerl. 2017, 71, 141–164. [Google Scholar] [CrossRef]
- Cereda, G. Impact of model choice on LR assessment in case of rare haplotype match (frequentist approach). Scand. J. Stat. 2017, 44, 230–248. [Google Scholar] [CrossRef]
- Brenner, C.H. Fundamental problem of forensic mathematics—The evidential value of a rare haplotype. Forensic Sci. Int. Genet. 2010, 4, 281–291. [Google Scholar] [CrossRef]
- Cereda, G.; Biedermann, A.; Hall, D.; Taroni, F. An investigation of the potential of DIP-STR markers for DNA mixture analyses. Forensic Sci. Int. Genet. 2014, 11, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Laplace, P. Essai Philosophique sur les Probabilites; Mme. Ve Courcier: Paris, France, 1814. [Google Scholar]
- Krichevsky, R.; Trofimov, V. The performance of universal coding. IEEE Trans. Inf. Theory 1981, 27, 199–207. [Google Scholar] [CrossRef]
- Gale, W.A.; Church, K.W. What’s wrong with adding one? In Corpus-Based Research into Language; Rodolpi: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Good, I. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Orlitsky, A.; Santhanam, N.P.; Viswanathan, K.; Zhang, J. On Modeling Profiles Instead of Values. In Proceedings of the 20th Conference on Uncertainty in Artificial Intelligence (UAI ’04), Banff, AB, Canada, 7–11 July 2004; pp. 426–435. [Google Scholar]
- Anevski, D.; Gill, R.D.; Zohren, S. Estimating a probability mass function with unknown labels. Ann. Stat. 2017, 45, 2708–2735. [Google Scholar] [CrossRef]
- Tiwari, R.C.; Tripathi, R.C. Nonparametric Bayes estimation of the probability of discovering a new species. Commun. Stat. Theory Methods 1989, A18, 877–895. [Google Scholar] [CrossRef]
- Lijoi, A.; Mena, R.H.; Pruenster, I. Bayesian nonparametric estimation of the probability of discovering new species. Biometrika 2007, 94, 769–786. [Google Scholar] [CrossRef]
- De Blasi, P.; Favaro, S.; Lijoi, A.; Mena, R.H.; Pruenster, I.; Ruggiero, M. Are Gibbs-Type Priors the Most Natural Generalization of the Dirichlet Process. IEEE Trans. Patterns Anal. Ans Mach. Intell. 2015, 37, 212–229. [Google Scholar] [CrossRef]
- Favaro, S.; Lijoi, A.; Mena, R.H.; Pruenster, I. Bayesian nonparametric inference for species variety with a two parameter Poisson-Dirichlet process prior. J. R. Stat. Soc. Ser. (Methodol.) 2009, 71, 993–1008. [Google Scholar] [CrossRef]
- Arbel, J.; Favaro, S.; Nipoti, B.; Teh, Y.W. Bayesian nonparametric inference for discovery probabilities: credible intervals and large sample asymptotics. Stat. Sin. 2017, 27, 839–859. [Google Scholar] [CrossRef]
- Favaro, S.; Nipoti, B.; Teh, Y.W. Rediscovery of Good-Turing estimators via Bayesian nonparametrics. Biometrics 2016, 72, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Teh, Y.W.; Jordan, M.I.; Beal, M.J.; Blei, D.M. Hierarchical Dirichlet processes. J. Am. Stat. Assoc. 2006, 101, 1566–1581. [Google Scholar] [CrossRef]
- Newman, M. Power laws, Pareto distributions and Zipf’s law. Contemp. Phys. 2005, 46, 323–351. [Google Scholar] [CrossRef]
- Caliebe, A.; Jochens, A.; Willuweit, S.; Roewer, L.; Krawczak, M. No shortcut solutions to the problem of Y-STR match probability calculation. Forensic Sci. Int. Genet. 2015, 15, 69–75. [Google Scholar] [CrossRef]
- Andersen, M.M.; Curran, J.M.; de Zoete, J.; Taylor, D.; Buckleton, J. Modelling the dependence structure of Y-STR haplotypes using graphical models. Forensic Sci. Int. Genet. 2018, 37, 29–36. [Google Scholar] [CrossRef]
- Andersen, M.M.; Caliebe, A.; Kirkeby, K.; Knudsen, M.; Vihrsa, N.; Curran, J.M. Estimation of Y haplotype frequencies with lower order dependencies. Forensic Sci. Int. Genet. 2019, 46, 102214. [Google Scholar] [CrossRef]
- Balding, D.J.; Nichols, R.A. DNA profile match probability calculation: How to allow for population stratification, relatedness, database selection and single bands. Forensic Sci. Int. 1994, 64, 125–140. [Google Scholar] [CrossRef]
- Andersen, M.M.; Balding, D.J. How convincing is a matching Y-chromosome profile? PLoS Genet. 2017, 13, e1007028. [Google Scholar] [CrossRef]
- Andersen, M.M.; Balding, D.J. Y-profile evidence: Close paternal relatives and mixtures. Forensic Sci. Int. Genet. 2019, 38, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Egeland, T.; Salas, A. Estimating Haplotype Frequency and Coverage of Databases. PLoS ONE 2008, 3, e3988. [Google Scholar] [CrossRef] [PubMed]
- Roewer, L. Y chromosome STR typing in crime casework. Forensic Sci. Med. Pathol. 2009, 5, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Buckleton, J.; Krawczak, M.; Weir, B. The interpretation of lineage markers in forensic DNA testing. Forensic Sci. Int. Genet. 2011, 5, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Willuweit, S.; Caliebe, A.; Andersen, M.M.; Roewer, L. Y-STR Frequency Surveying Method: A critical reappraisal. Forensic Sci. Int. Genet. 2011, 5, 84–90. [Google Scholar] [CrossRef]
- Wilson, I.J.; Weale, M.E.; Balding, D.J. Inferences from DNA data: Population histories, evolutionary processes and forensic match probabilities. J. R. Stat. Soc. Ser. (Stat. Soc.) 2003, 166, 155–188. [Google Scholar] [CrossRef]
- Andersen, M.M.; Eriksen, P.S.; Morling, N. The discrete Laplace exponential family and estimation of Y-STR haplotype frequencies. J. Theor. Biol. 2013, 329, 39–51. [Google Scholar] [CrossRef]
- Willuweit, S.; Roewer, L. Y chromosome haplotype reference database (YHRD): Update. Forensic Sci. Int. Genet. 2007, 1, 83–87. [Google Scholar] [CrossRef]
- Purps, J.; Siegert, S.; Willuweit, S.; Nagy, M.; Alves, C.; Salazar, R.; Angustia, S.M.T.; Santos, L.H.; Anslinger, K.; Bayer, B.; et al. A global analysis of Y-chromosomal haplotype diversity for 23 STR loci. Forensic Sci. Int. Genet. 2014, 12, 12–23. [Google Scholar] [CrossRef]
- Kimura, M. The number pf alleles that can be maintained in a finite population. Genetics 1964, 49, 725–738. [Google Scholar]
- Hjort, N.; Holmes, C.; Müller, P.; Walker, S. Bayesian Nonparametrics; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Ghosal, S.; Van der Vaart, A. Fundamentals of Nonparametric Bayesian Inference; Cambridge University Press: Cambridge, UK, 2017. [Google Scholar]
- Pitman, J.; Yor, M. The two-parameter Poisson-Dirichlet distribution derived from a stable subordinator. Ann. Probab. 1997, 25, 855–900. [Google Scholar] [CrossRef]
- Feng, S. The Poisson-Dirichlet Distribution and Related Topics: Models and Asymptotic Behaviors; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Buntine, W.; Hutter, M. A Bayesian view of the Poisson-Dirichlet process. arXiv 2012, arXiv:1007.0296. [Google Scholar]
- Pitman, J. Combinatorial Stochastic Processes; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Zabell, S.L. The Continuum of Inductive Methods Revisited; Cambridge Studies in Probability, Induction and Decision Theory; Cambridge University Press: Cambridge, UK, 2005; pp. 243–274. [Google Scholar]
- Pitman, J. The Two-Parameter Generalization of Ewens’ Random Partition Structure; Technical Report 345; Department of Statistics U.C.: Berkeley, CA, USA, 1992.
- Pitman, J. Exchangeable and partially exchangeable random partitions. Probab. Theory Relat. Fields 1995, 102, 145–158. [Google Scholar] [CrossRef]
- Aldous, D.J. Exchangeability and Related Topics; Springer: New York, NY, USA, 1985. [Google Scholar]
- Ramos, D.; Gonzales-Rodriguez, J.; Zadora, G.; Aitken, C. Information-Theoretical Assessment of the Performance of Likelihood Ratio Computation Methods. J. Forensic Sci. 2013, 58, 1503–1517. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
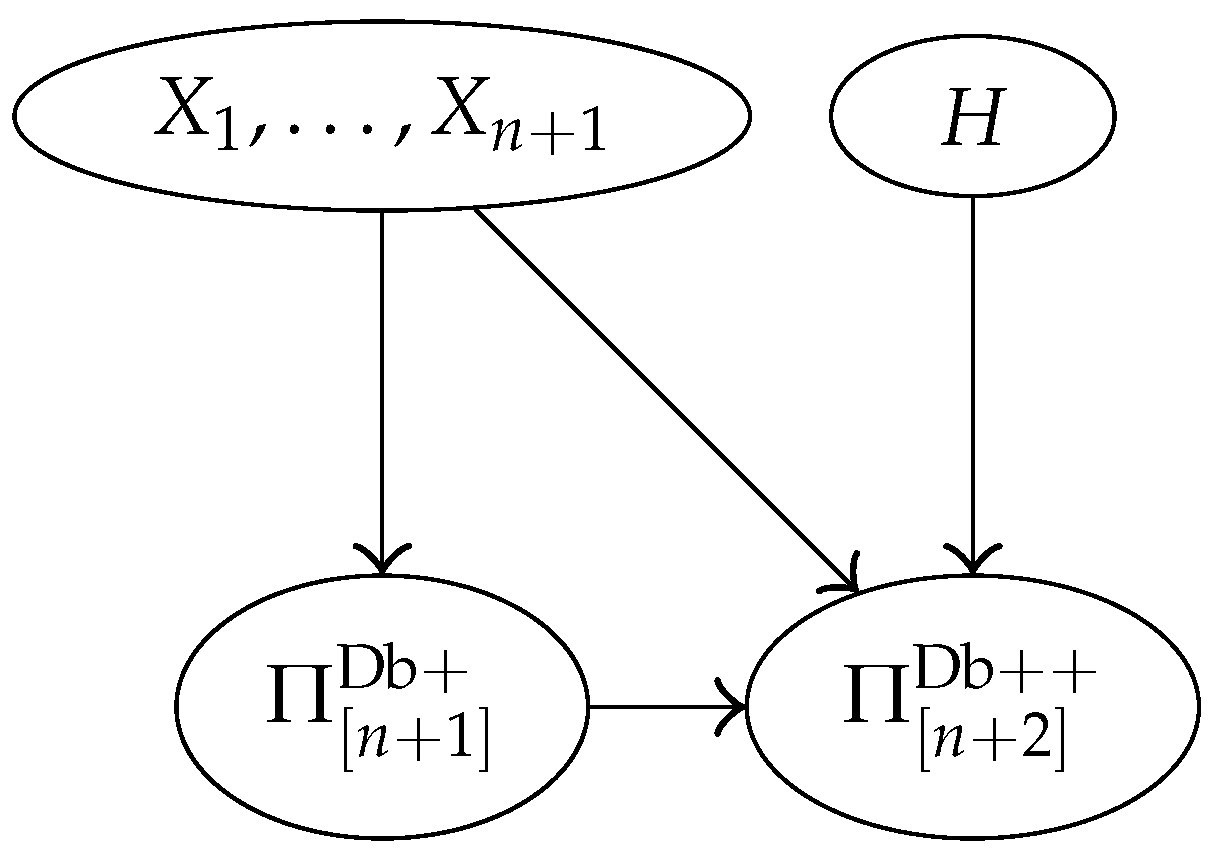{kind=link}
{kind=link}
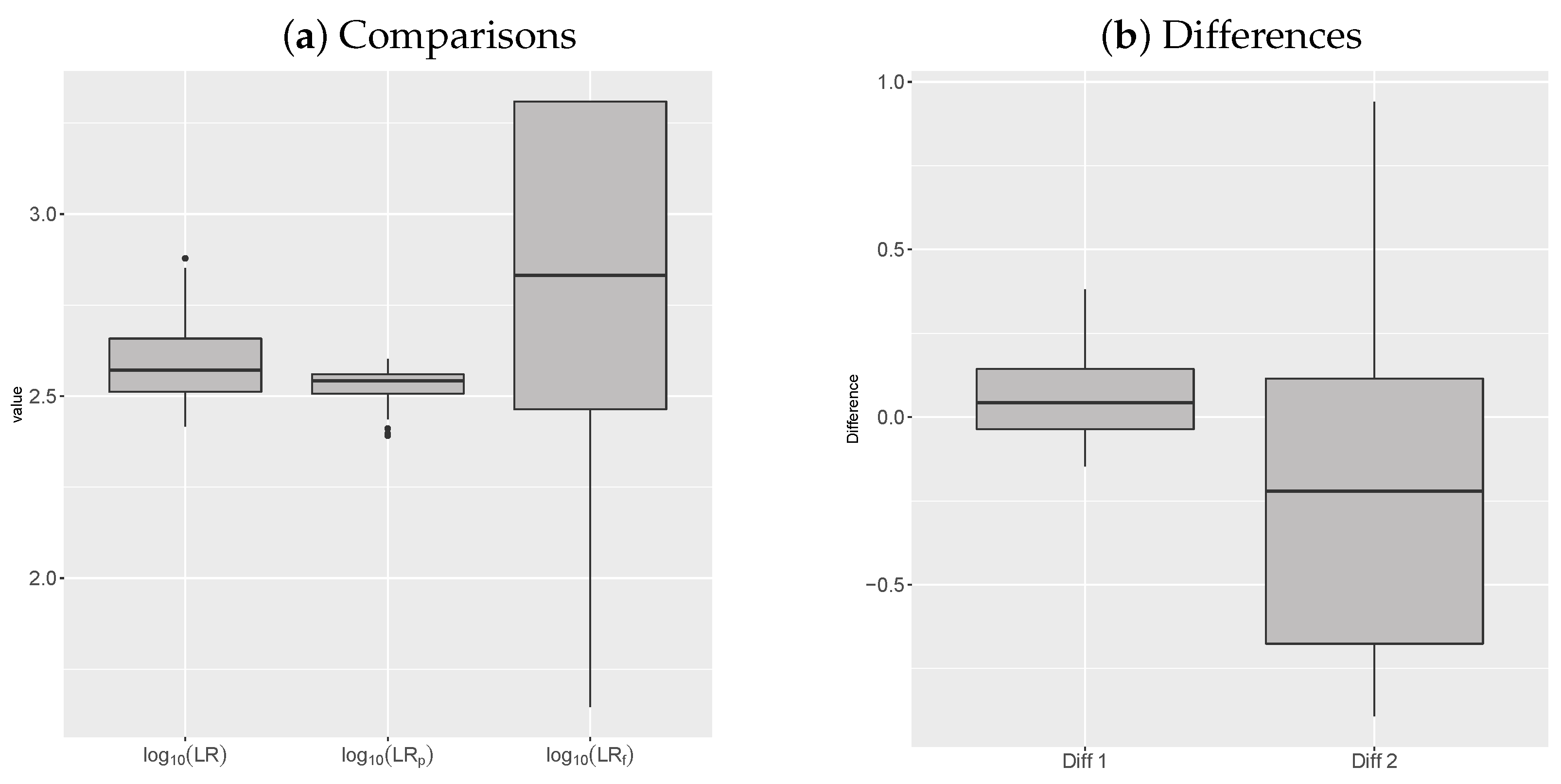{kind=link}
| Min | 1st Qu. | Median | Mean | 3rd Qu. | Max | sd | |
|---|---|---|---|---|---|---|---|
| 2.417 | 2.512 | 2.572 | 2.59 | 2.658 | 2.879 | 0.102 | |
| 2.392 | 2.507 | 2.542 | 2.529 | 2.56 | 2.602 | 0.045 | |
| 1.646 | 2.464 | 2.832 | 2.803 | 3.309 | 3.309 | 0.463 |
| Min | 1st Qu. | Median | Mean | 3rd Qu. | Max | sd | |
|---|---|---|---|---|---|---|---|
| Diff | −0.146 | −0.036 | 0.044 | 0.06 | 0.144 | 0.381 | 0.126 |
| Diff | −0.891 | −0.676 | −0.221 | −0.213 | 0.115 | 0.94 | 0.472 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cereda, G.; Gill, R.D. A Nonparametric Bayesian Approach to the Rare Type Match Problem. Entropy 2020, 22, 439. https://doi.org/10.3390/e22040439
Cereda G, Gill RD. A Nonparametric Bayesian Approach to the Rare Type Match Problem. Entropy. 2020; 22(4):439. https://doi.org/10.3390/e22040439
Chicago/Turabian StyleCereda, Giulia, and Richard D. Gill. 2020. "A Nonparametric Bayesian Approach to the Rare Type Match Problem" Entropy 22, no. 4: 439. https://doi.org/10.3390/e22040439
APA StyleCereda, G., & Gill, R. D. (2020). A Nonparametric Bayesian Approach to the Rare Type Match Problem. Entropy, 22(4), 439. https://doi.org/10.3390/e22040439