In this section, we present the results of the experimentation carried out in order to assess the reliability and usefulness of EnGNet. We first compared EnGNet with three standard information theory approaches commonly used in the literature to infer large GNs (based on NMI, Spearman and Kendall measures). Moreover, we compared our proposal with the ensemble strategy of these methods (i.e., only the first step of EnGNet). The aim of these experiments was to test the performance of EnGNet against other classical methods from the literature to infer large co-expression networks, and also to test the relevance of the prune step in the final results obtained. Thus, we not only tested the reliability of the inferred networks, but also the ability of EnGNet to reduce the size of final networks and their topological features.
In the second experiment, we also tested the performance of EnGNet against different algorithms from the literature for generating small gene networks. In particular, we present a study on 20 yeast genes that encode the Cell Cycle G1 phase.
Finally, with the aim of proving the effectiveness of our proposal in a biomedical study, we applied EnGNet to a human dataset regarding post traumatic stress disorder (PTSD).
3.1. Comparative Analysis Of EnGNet For Large Gene Networks
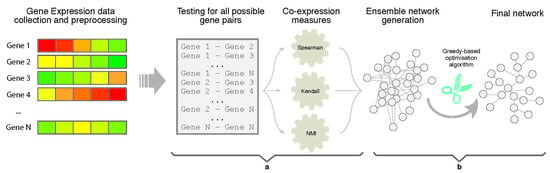
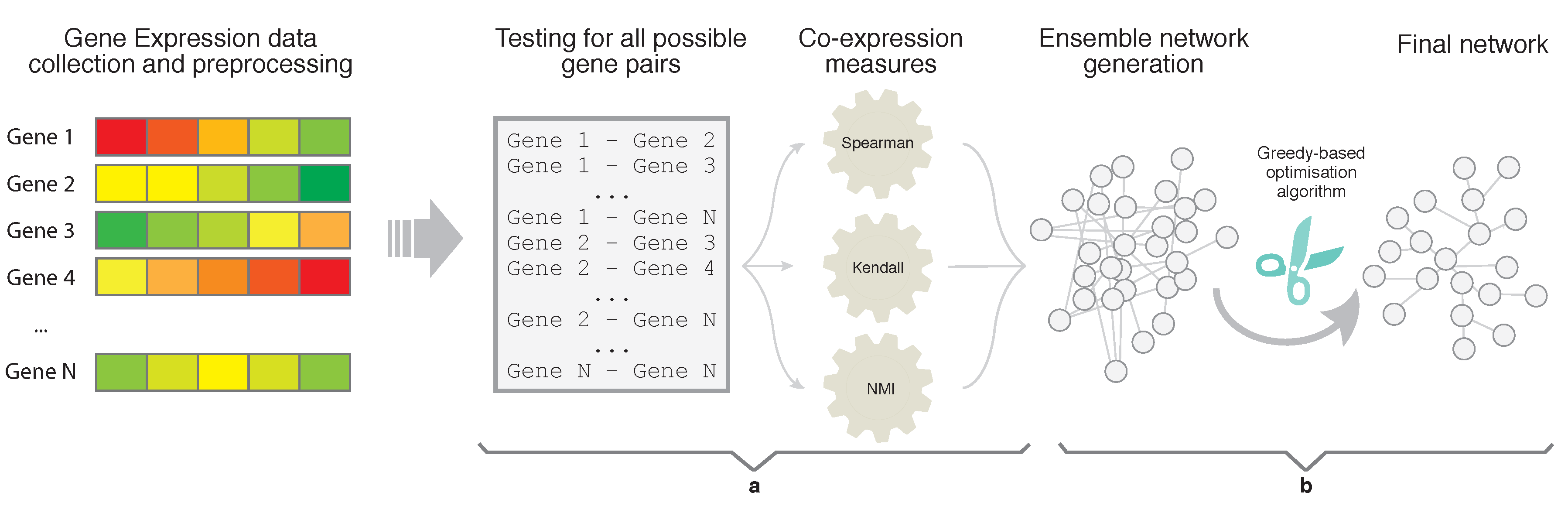
In the experiments, we used five approaches to generate networks from each dataset. In particular, we used EnGNet, the first phase of EnGNet, i.e., only the ensemble strategy without the pruning phase, and three information theory based methods. These last three methods are based on the NMI, Kendall and Spearman measures, in a similar way as the experiments presented in [
7,
15]. These approaches have been widely used in the biomedical literature for studying with gene co-expression networks (e.g., Xu et al. [
48], Johnson et al. [
49] and Liu et al. [
50]).
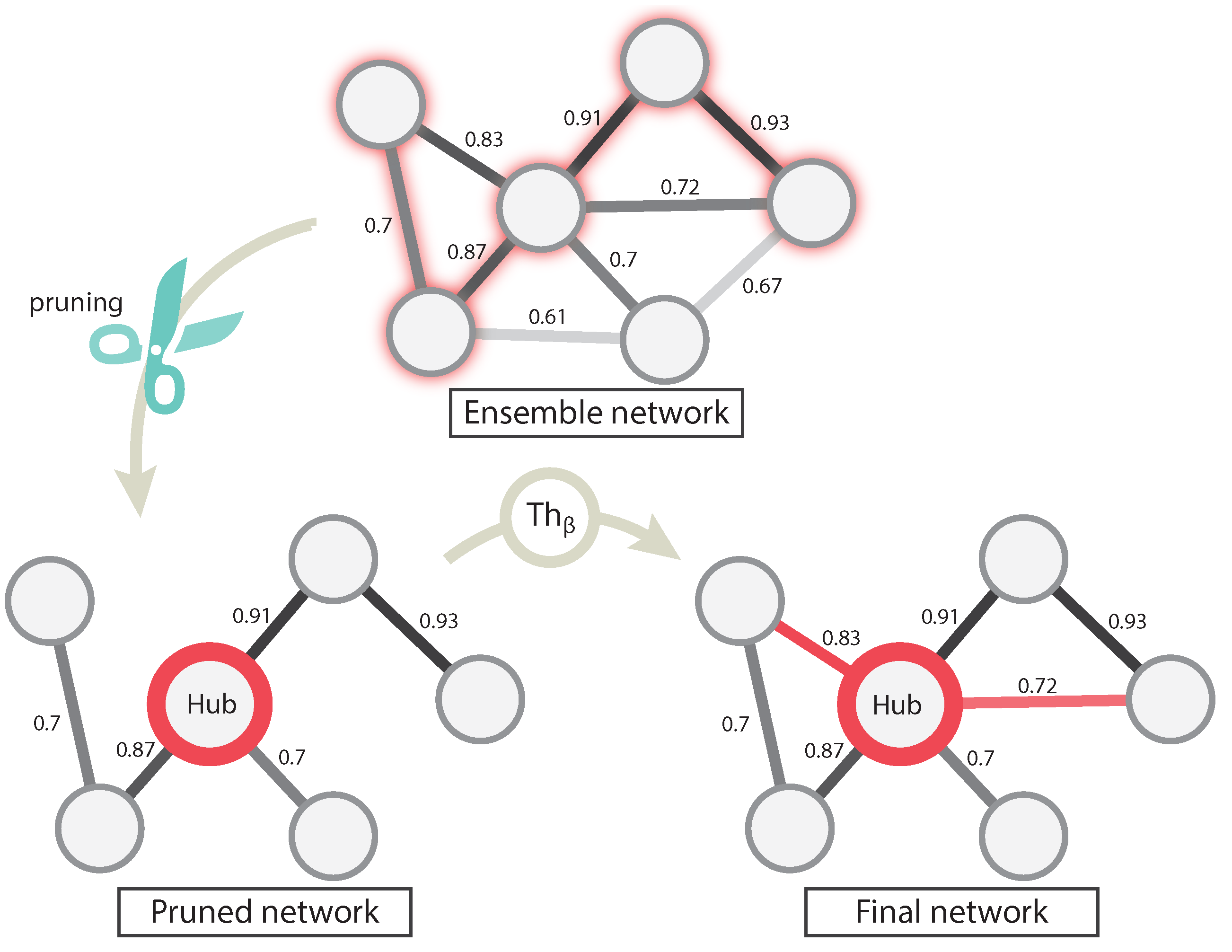
For each information theory method used, we needed to set a validity threshold, and in the case of EnGNet, we needed four thresholds (see
Section 2.1). For this experiment, we selected three different thresholds for all methods: 0.7, 0.8, 0.9. For a fair comparison, EnGNet and the ensemble approach also used the same thresholds for
and
. These thresholds represent a complete full spectrum from a mid correlation (0.7) to a very strong one (0.9). Thus, 60 networks were generated and analyzed (5 methods × 3 thresholds × 4 datasets).
3.1.1. Networks Performance Against GeneMANIA
As mentioned above, we first tested the biological significance of the obtained networks in a direct comparison with GeneMANIA database. The results obtained, in terms of nodes, edges, precision and recall, are presented in
Table 2,
Table 3,
Table 4 and
Table 5, respectively.
Table 2 shows how EnGNet achieved the second best results of the experiment (only behind Kendall’s) in terms of average precision. However, it is important to notice that EnGNet is the method that presents the most stable precision and size values for the different thresholds, obtaining the sparser networks for all methods considered (almost half the average size compared to Kendall’s). This result confirms the overall stability of EnGNet.
The experiment carried out on the Human SNP dataset shows that EnGNet obtains the best results in terms of average precision (see last row of
Table 4). We can also notice that the NMI approach infers smaller networks than EnGNet. However, the precision is so low that these networks do not appear to be biologically significant.
For the experiments with “Pre” and “Post” PTSD datasets (
Table 3 and
Table 5, respectively), the results present the same pattern: EnGNet obtains the best results in term of precision and size of the networks.
Finally,
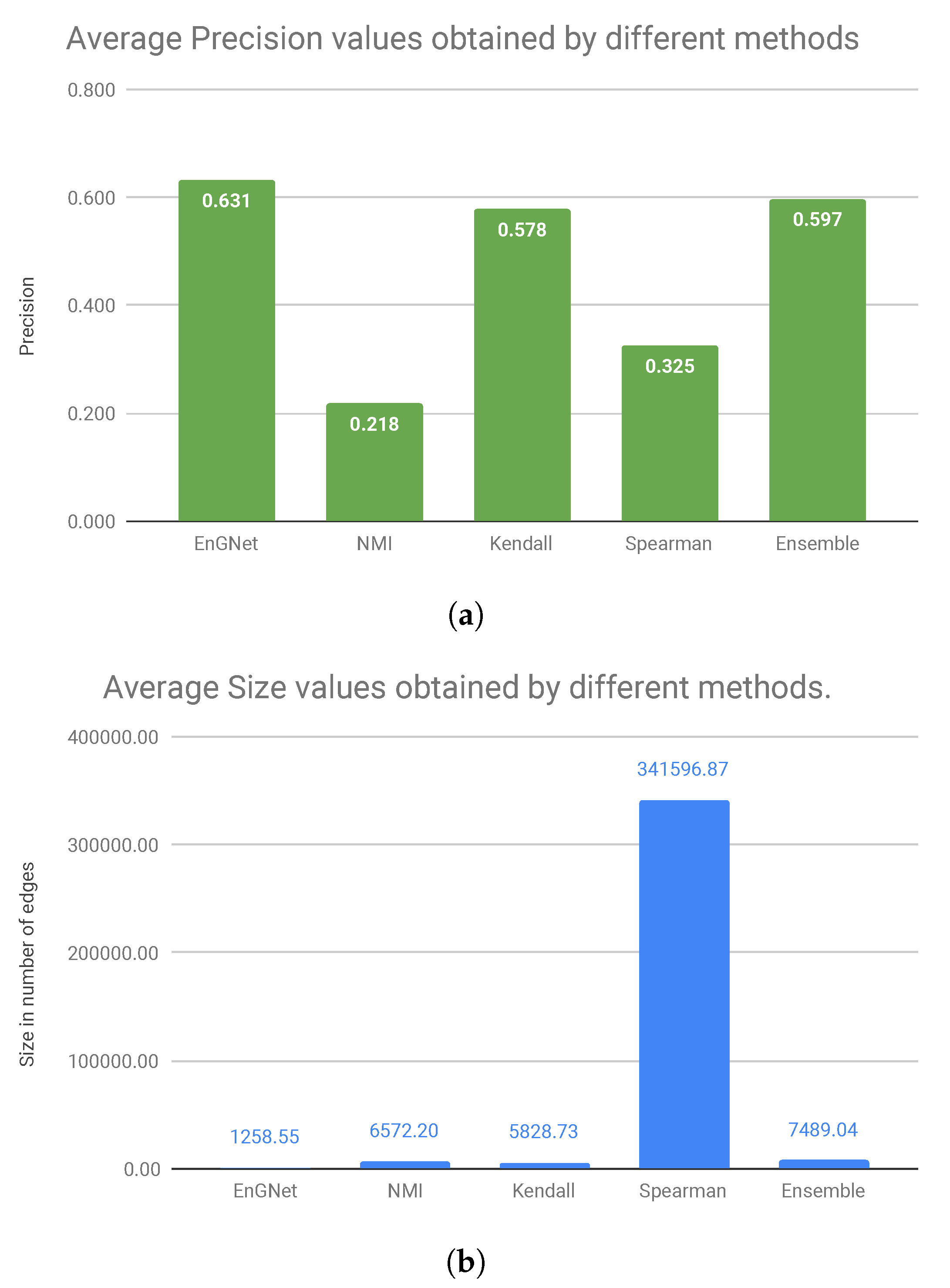
Figure 4 shows the average values of precision and size of the networks for all experiments presented above. Considering the precision results presented in
Figure 4a, we can observe that our algorithm is the one that obtains the best values, followed by the Ensemble approximation and Kendall’s. Regarding the size of the networks, it can be verified in
Figure 4bthat EnGNet obtains the smallest networks (approximately 271 times smaller than Spearman’s network or six times smaller than Ensemble’s network, which is the second approximation in precision values) with the highest precision values.
In summary, we can conclude that EnGNet is successful in reducing the size of the networks while keeping competitive results in terms of precision and recall (against other methods studied). In fact, networks generated by EnGNet are significantly sparser than those obtained by other methods (see
Figure 4). As stated above, this is a significant result, since sparseness is a desirable feature in GNs reconstruction from a large dataset. In fact, the smaller is the networks, the easier is their analysis [
51]. Additionally, although networks are sparser in terms of the number of edges, precision and recall values do not suffer a relevant loss. This observation is confirmed from the results presented, since EnGNet obtains average precision values above
in all the cases studied (presented in the tables).
Finally,
Figure 4 shows that EnGNet obtains the best average precision value, whilst the size of the network is significantly reduced (especially against the Spearman’s approach). This result indicates that EnGNet networks do not lose biological significance upon pruning. As a conclusion, we can affirm that EnGNet is a competitive and reliable method for the generation of large gene networks.
3.1.2. Topological Features Analysis
In addition to network sparseness, the topological properties of gene networks should be considered in order to estimate the performance of EnGNet upon network reconstruction [
7,
9,
16]. As discussed in
Section 1, biological networks tend to be sparse and to follow a scale-free topology. Therefore, it is desirable for the reconstruction methods to provide networks that present such topological features.
From these results, we can observe that EnGNet obtains the most stable results over the experiments carried with respect to the majority of the topological features studied (see “Average” rows in the tables). To clarify these results, we also calculated the average values for all datasets and thresholds presented. These results are reported in
Table 10. In the table, it is possible to observe that, for all topological features studied, EnGNet is the algorithm achieving the best results, except for the network diameter. For the network’s diameter, only the Spearman’s method obtains better results. This is a logical result since Spearman’s method generates the biggest networks (271 times bigger than EnGNet). It is remarkable, from a topological point of view, that our method reaches a diameter in a similar range with a significantly smaller size than Spearman’s network.
In summary, EnGNet obtains the best results on all topological features, for all the networks, indicating that EnGNet networks follow a biological pattern (scale-free topology). Furthermore, EnGNet-generated networks improve the results obtained by information theory methods and ensemble networks. Bearing this in mind and the results presented in the comparison with the network contained in GeneMANIA, we can affirm that EnGNet is a suitable tool for large co-expression GNs reconstruction in biomedical research.
3.2. Comparative Analysis Of EnGNet For Small Networks
The ability of our approach to infer small gene networks was also tested. To do so, we performed a similar experiment to the one presented by Gallo et al. [
52]. In this experiment, precision was used as quality measure to rate the reliability of the input GNs. The main objective of the experiment is to compare the precision values of different gene networks algorithms from the literature on the same dataset.
To obtain the input networks, we used different methods from the literature, which are described in the works by:
Soinov et al. [
53], a C4.5-based method;
Bulashevska et al. [
54], a Bayesian-based method;
Ponzoni et al. [
55], a combinatorial optimization algorithm (GRNCOP);
Gallo et al. [
52], an upgraded version of the previous algorithm named GRNCOP2; and
Gomez-Vela et al. [
15], a fuzzy method to infer gene co-expression networks named FyNe.
These methods were applied to the same dataset from the Yeast Cell Cycle—more specifically, to a subset of 20 well-described genes. These genes code for key proteins in cell-cycle regulation, as presented by Martinez-Ballesteros et al. [
56].
As in the experiment performed by Gallo et al. [
52], the quality of the networks was assessed regarding the precision values obtained against the data stored in YeastNet [
57]. YeastNet is a repository that comprises a probabilistic functional GN generated from verified protein-coding open reading frames (ORFs) of the yeast genome. This repository combines protein–protein interactions, protein–DNA interactions, co-expression, phylogenetic conservation and literature information, in total covering more than 102,803 linkages among 5483 yeast proteins (95% of the validated proteome).
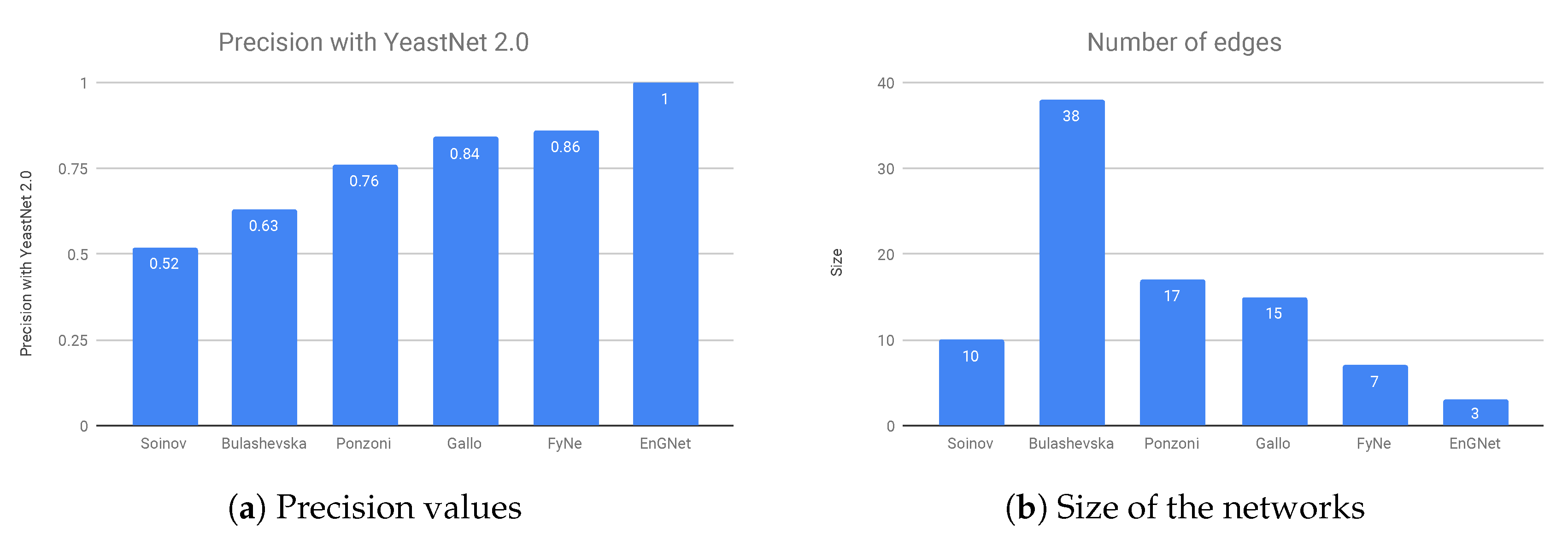
The results of the experiment are presented in
Figure 5a,b, where it can be verified that EnGNet yields the best results amongst all studied methods, and again with the smaller network. Note that the inference of small gene networks usually provides higher precision results than in the case of large ones, as detailed in Hecker et al. [
16]. The results show that not only is EnGNet suitable for large gene networks studies, but also obtains competitive results for studies with small datasets.
3.3. Application to the Study of Human Post Traumatic Stress Disorder
The second objective of this study was to prove the usefulness of EnGNet in actual life sciences research. To do so, EnGNet was applied to a human PTSD dataset obtained by Breen et al. [
42], so as to shed some light over the genes involved in this pathology.
In this case-control study, expression data were obtained from US marines peripheral blood leukocytes both before and after deployment to conflict zones (that called “Pre” and “Post”). As stated above, 94 marines (47 cases and 47 controls) were analyzed. According to the original article by Breen et al. [
42], controls refer to selected marines who did not show signs of PTSD. These are used as a reference for cases, which are marines who show a broad spectrum of signs that classify them as under PTSD after battlefield deployment. PTSD was scored through a diagnostic interview and annotated in the Clinician Administered PTSD Scale (CAPS) [
58]. In the experimental design, cases are analogous to controls prior to battlefield deployment, i.e., none are under PTSD symptomatology. On the other hand, after battlefield deployment cases significantly differ from controls in terms of the CAPS score (see the original article by Breen et al. [
42] for further details).
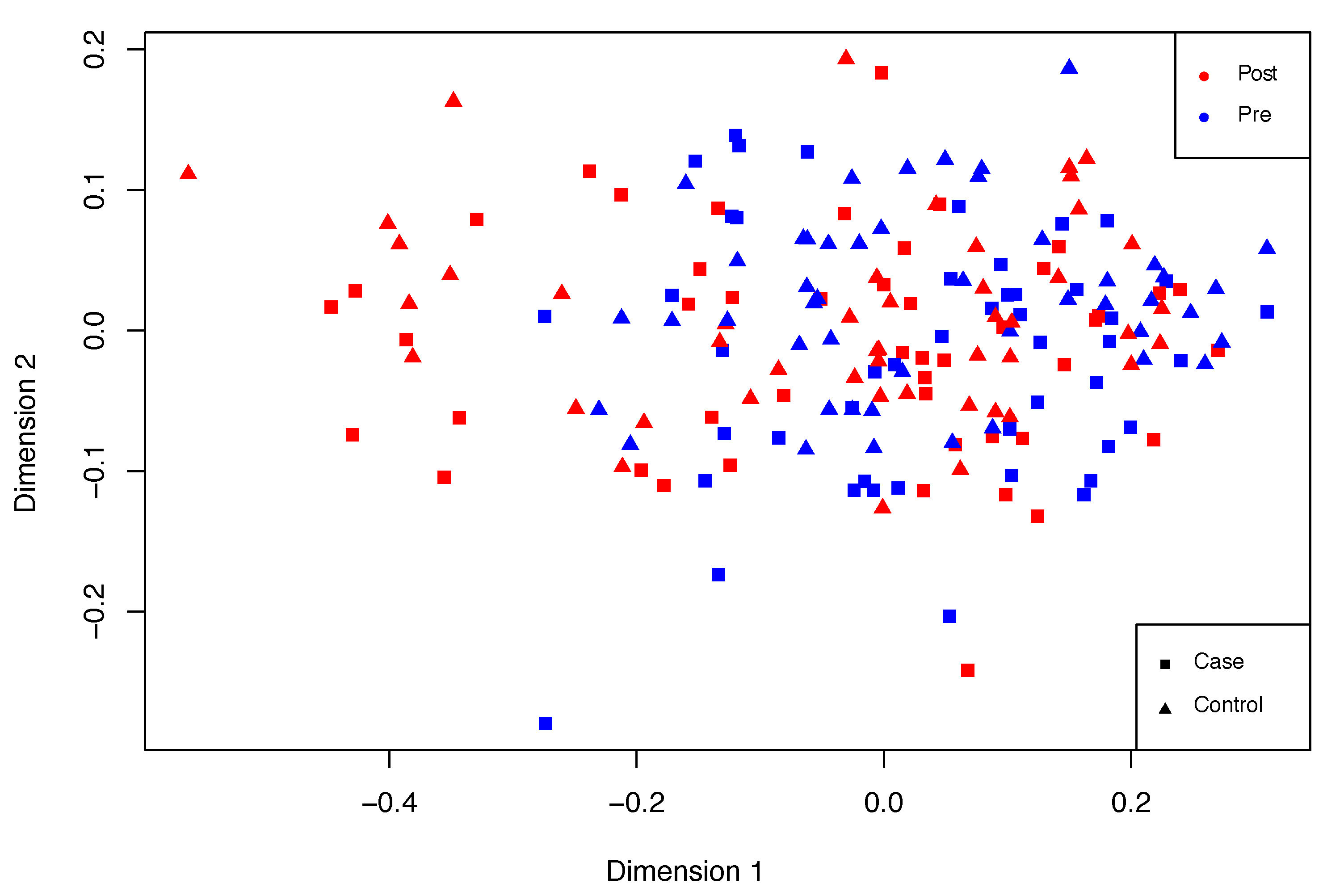
Overall, PTSD signs may be observed in the second group when compared to the first one. An exploratory multidimensional scaling (MDS) plot or Principal Coordinates Analysis (PCoA) was performed in order to roughly examine these differences. MDS assisted the examination of sample similarity. On this occasion, the classical MDS method was applied, assuming Euclidean distances. An illustrative distribution of this dataset is shown in
Figure 6, in which differences can be observed between post- and pre-deployment marines. However, these differences are fuzzy and there is a spectrum of sample states between pre- and post-deployment situations.
First, a differential gene expression analysis was carried out to verify the mentioned differences using the
[
59] R package, a tool for the estimation of differentially-expressed genes (DEGs). The information on gene up- or down-regulation was of especial interest in the analysis of the biological processes underlying PTSD development. Hence, data provided by
were latter imported into Cytoscape for network interpretation purposes.
EnGNet was used to reconstruct two different networks corresponding to pre-deployment and post-deployment samples, respectively. To this aim, the EnGNet
thresholds were set to the values that yield the best results in the experimentation presented in
Section 2.3, namely
= 0.7,
= 0.8 and
= 0.9. As far as the
threshold is concerned, a new analysis was carried out to determine the optimal threshold for each sample. The results of this study are presented in
Table 11 and show the values of the precision and recall measure obtained by different networks against GeneMANIA.
Therefore, considering the results presented in the table, candidate networks for this study correspond to = 0.8 in the pre-deployment case and = 0.9 in the post-deployment situation.
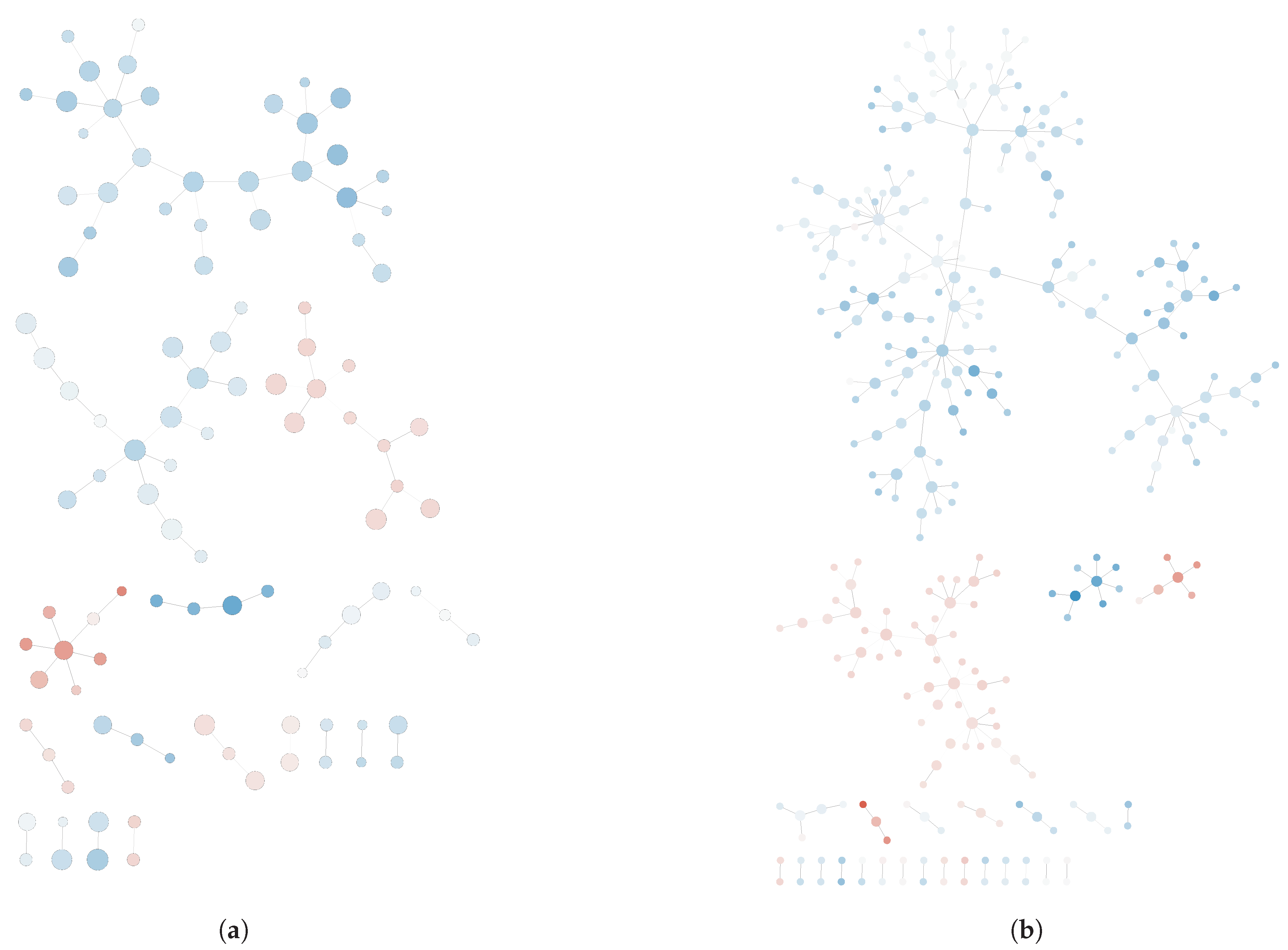
Once the networks were generated, a significant increase in the number of genes was found in the post-deployment network compared to its pre-deployment counterpart, which is indicative of gene up-regulation in lymphocytes upon PTSD development. Pre- and post-deployment networks are shown in
Appendix A (see
Figure A1). Remarkably, the reconstructed networks for pre-deployment and post-deployment samples were significantly different, which is indicative for the discrimination power of the GN reconstruction approach over other unsupervised techniques such as PCoA.
Pre- and post-deployment networks were merged in order to graphically observe the differences in gene expression upon PTSD development. Overall, 73.8% of the nodes in this merged network were found to be upregulated in the post-deployment situation compared to pre-deployment, which suggest the importance of gene activation upon PTSD development. Genes up/down-regulation in the merged network is shown in
Appendix A (see
Figure A2).
Enrichment analysis was performed by means of Cytoscape’s plugins ClueGO [
60] and CluePedia [
61], which shows over-represented GO-terms in a ensemble of genes. ClueGO + CluePedia analyses provided useful information about the biological processes in which the genes comprised at the pre-deployment and post-deployment networks were involved.
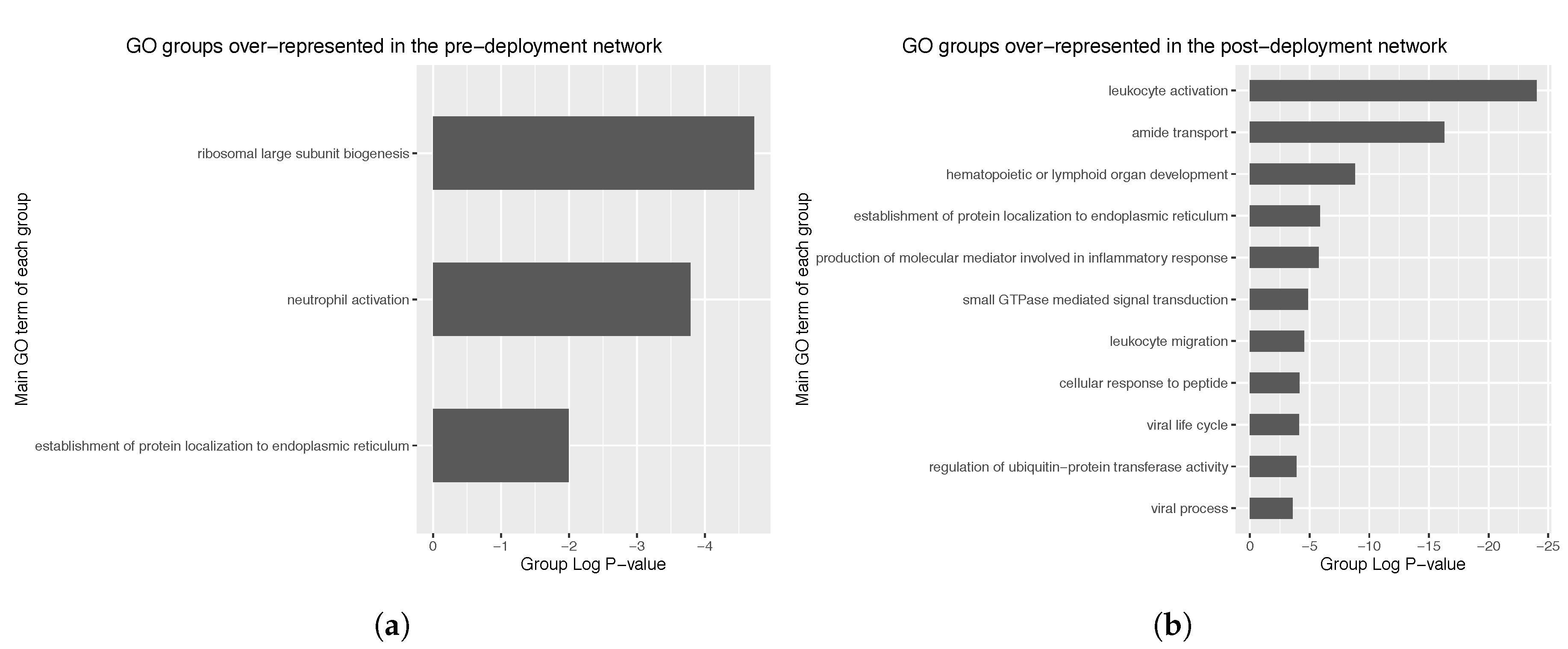
Regarding the pre-deployment network (105 nodes), three different GO groups were identified, corresponding to ribosomal biogenesis, neutrophil activation and establishment of protein localization to endoplasmic reticulum (
Figure 7a). Group
p-values observed were of the order of 10
−6.
In the case of the post-deployment network (298 nodes), 10 GO groups were identified, mostly corresponding to leukocyte activation, amide transport and hematopoietic or lymphoid organ development (
Figure 7b). Observed group
P-values were of the order of 10
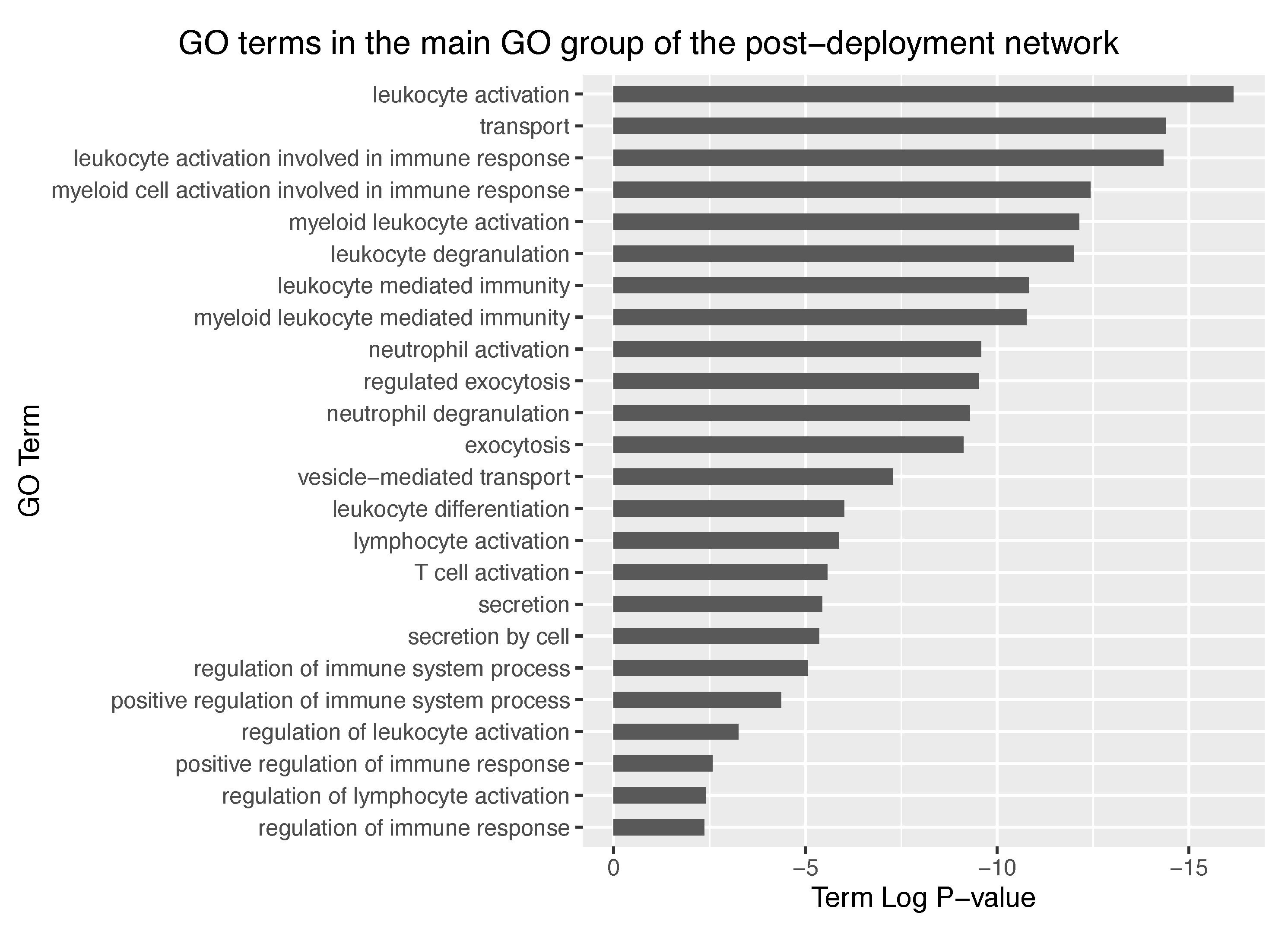
−25, thus representing a dramatic increase in significancy compared to the pre-deployment GO groups. Further exploration of the main GO group in the post-deployment revealed GO terms such as leukocyte activation involved in immune response, myeloid cell activation involved in immune response, myeloid leukocyte activation, and leukocyte degranulation. Main GO terms comprised in the main GO group of the post-deployment network are shown in
Figure 8.
Enrichment analyses thus revealed a dramatically different situation in the post-deployment network compared to pre-deployment one, in terms of the biological processes these represent. Whereas the pre-deployment network shows biological processes more related to an unexcited steady-state immune system, the post-deployment network displays several GO groups and GO terms which lie under the context of immunoenhancement. Reconstructed GNs thereby model two different situations in terms of the biological context. This also suggests the potential use of GNs for diagnostic purposes.
With regard to differential expression, a considerable gene up-regulation is observed, which correlates to immunoenhancement upon PTSD development. In general, the above mentioned GO terms are indicative of a nonspecific immune response, characteristic of innate immunity, suggesting the potential role of myeloid leukocytes in PTSD. Quite significant is also the GO group “hematopoietic or lymphoid organ development”, as the immune system is generated from multipotent hematopoietic stem cells, which branch in myeloid and lymphoid progenitors. This myeloid cell line comprises cells such as basophils, neutrophils, eosinophils and macrophages, which through immunosurveillance are responsible for the so-called unspecific or innate immunity. This is consistent with the results found by Breen et al. [
42], who predicted the intrinsic role of innate immunity upon PTSD. These findings were also highlighted in a previous study by Watson et al. [
62], who observed enhanced immunological features in PTSD-diagnosed Vietnam combat veterans in comparison with civilians.

 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}