Analysis of Time Reversible Born-Oppenheimer Molecular Dynamics †

Abstract
: We analyze the time reversible Born-Oppenheimer molecular dynamics (TRBOMD) scheme, which preserves the time reversibility of the Born-Oppenheimer molecular dynamics even with non-convergent self-consistent field iteration. In the linear response regime, we derive the stability condition, as well as the accuracy of TRBOMD for computing physical properties, such as the phonon frequency obtained from the molecular dynamics simulation. We connect and compare TRBOMD with Car-Parrinello molecular dynamics in terms of accuracy and stability. We further discuss the accuracy of TRBOMD beyond the linear response regime for non-equilibrium dynamics of nuclei. Our results are demonstrated through numerical experiments using a simplified one-dimensional model for Kohn-Sham density functional theory.1. Introduction
Ab initio molecular dynamics (AIMD) [1–6] has been greatly developed in the past few decades, so that nowadays, it is able to quantitatively predict the equilibrium and non-equilibrium properties for a vast range of systems. AIMD has become widely used in chemistry, biology, materials science, etc. A coherent and comprehensive presentation of AIMD with both the basic theory and advanced methods can be found in [7]. Most AIMD methods treat the nuclei as classical particles following Newtonian dynamics (known as the time-dependent Born-Oppenheimer approximation), and the interactive force among nuclei is provided directly from electronic structure theory, such as the Kohn-Sham density functional theory [8,9] (KSDFT), without the need of using empirical atomic potentials. KSDFT consists of a set of nonlinear equations that are solved at each molecular dynamics time step self-consistently via the self-consistent field (SCF) iteration. In Born-Oppenheimer molecular dynamics (BOMD), KSDFT is solved until full self-consistency for each atomic configuration per time step. Since many iterations are usually needed to reach full self-consistency and each iteration takes a considerable amount of time, until recently, this procedure was still found to be prohibitively expensive for producing meaningful dynamical information. On the other hand, if the self-consistent iterations are truncated before convergence is reached, it is often the case that the energy of the system is no longer conservative, even for an NVE system. The error in SCF iteration acts as a sink or source, gradually draining or adding energy to the atomic system within a short period of molecular dynamics simulation [10]. This is one of the main challenges for accelerating Born-Oppenheimer molecular dynamics.
AIMD was made practical by the ground-breaking work of Car-Parrinello molecular dynamics (CPMD) [11]. CPMD introduces an extended Lagrangian, including the degrees of freedom of both nuclei and electrons without the necessity of a convergent SCF iteration. The dynamics of electronic orbitals can be loosely viewed as a special way for performing the SCF iteration at each molecular dynamics (MD) step. Thanks to the Hamiltonian structure, numerical simulation for CPMD is stable, and the energy is conservative over a much longer time period compared to that for BOMD with non-convergent SCF iteration. When the system has a spectral gap, the accuracy of CPMD is controlled by a single parameter, the fictitious electron mass, μ. The result of CPMD approaches that of BOMD as μ goes to zero [12,13]. However, it has also been shown that CPMD does not work as well for systems with a vanishing gap, for example, for metallic systems [12].
To reduce the cost of BOMD, in particular, the number of SCF iterations needed per MD time step, a new type of AIMD method, the time reversible Born-Oppenheimer molecular dynamics (TRBOMD) method has been recently proposed by Niklasson, Tymczak and Challacombe in [14]. The method has been further developed in [15–18]. The idea of TRBOMD can be summarized as follows: TRBOMD assumes that the SCF iteration is a deterministic procedure, with the outcome determined only by the initial guess of the variable to be determined self-consistently. For instance, this variable can be the electron density, and the SCF iteration procedure can be simple mixing with a fixed number of iteration steps without reaching full self-consistency. Then, a fictitious dynamics governed by a second order ordinary differential equation (ODE) is introduced on this initial guess variable. The resulting coupled dynamics is then time-reversible and supposed to be more stable, since it has been found that time-reversible numerical schemes are more stable for long time simulation [19,20]. Besides TRBOMD, alternative ideas based on time-reversible predictor-corrector methods [21] and Langevin dynamics [22,23] can also relax the requirement on the accuracy of the force for AIMD simulation. For these methods, we refer the readers to a recent review paper [24] for more information.
Although TRBOMD has been found to be effective and significantly reduces the number of SCF iterations needed in practice, to the extent of our knowledge, there has been so far no detailed analysis of TRBOMD, other than the numerical stability condition of the Verlet or generalized Verlet scheme for time discretization [17]. Accuracy, stability, as well as the applicability range of TRBOMD remain unclear. In particular, it is not known how the choice of SCF iteration scheme affects TRBOMD. These are crucial issues for guiding the practical use of TRBOMD. The full TRBOMD method for general systems is highly nonlinear and is difficult to analyze. In this work, we first focus on the linear response regime, i.e., we assume that each atom oscillates around their equilibrium position and the electron density stays around the “true” electron density. Under such assumptions, we analyze the accuracy and stability of TRBOMD. We then extend the results to the regime where the atom position is not near equilibrium using the averaging principle.
The rest of the paper is organized as follows. We illustrate the idea of TRBOMD and its analysis in the linear response regime using a simple model in Section 2 and introduce TRBOMD for AIMD in Section 3. We analyze TRBOMD in the linear response regime and compare TRBOMD with CPMD in Section 4. The numerical results for TRBOMD in the linear response regime are given in Section 5. We present the analysis of TRBOMD beyond the linear response regime, such as the non-equilibrium dynamics in Section 6, and conclude with a few remarks in Section 7.
2. An Illustrative Model
To start, let us illustrate the main idea for a simple model problem, which provides the essence of TRBOMD in a much simplified setting. Consider the following nonlinear ODE:
In the linear response regime, we assume the linear approximation of force for x around x*:
From Equation (14), we conclude that if the time reversible numerical scheme (4) is used for simulating the ODE Equation (1) and if we neglect the error due to the Verlet scheme, the error introduced in computing the frequency, Ω, is proportional to ω−2. This seems to indicate that very large ω (i.e., very small time step Δt) might be needed to obtain accurate results. Fortunately, the ω−2 term in Equation (14) has the prefactor, f′(x*)−1. Equation (6) shows that f′(x*) ≈ −Ω2, which is small compared to ω2. If gs(x*, f(x*)) is small, then ≈ 1, and the accuracy of Ω̃ is determined by or gs(x*, f(x*)), which indicates the sensitivity of the computed force with respect to the initial guess, or the accuracy of the iterative procedure for computing the force. If a “good” iterative procedure is used, gs(x*, f(x*)) will be small. Therefore, the presence of the term, , allows one to obtain relatively accurate approximation to the frequency, Ω, without using a large ω. The same behavior can be observed when using TRBOMD to approximate BOMD (vide post).
Finally, we remark that even though Equation (1) is a much simplified system, it will be seen below that for BOMD with M atoms and N interacting electrons, the analysis in the linear response regime follows the same line, and the result for the frequency is similar to Equation (14).
3. Time Reversible Born-Oppenheimer Molecular Dynamics
Consider a system with M atoms and N electrons. The position of the atoms at time t is denoted by R(t) = (R1(t), …, RM(t))T. In BOMD, the motion of atoms follows Newton’s law:
Starting from an inaccurate input electron density, ρin, one first computes the output electron density by solving the lowest N eigenfunctions of the problem:
The map, ρSCF, is usually highly nonlinear, which makes it difficult to correct the error in the force. The TRBOMD scheme avoids the direct correction for the inaccurate ρSCF, but allows the initial guess to dynamically evolve together with the motion of the atoms. We denote by ρ(x, t) the initial guess for the SCF iteration at time t. When ρ(·, t) is used as an argument, we also write ρSCF(x; R(t), ρ(t)) ≔ ρSCF(x; R(t), ρ(·, t)). The Hellmann-Feynman formula (19) is used to compute the force at the electron density, ρSCF(x; R(t), ρ(t)), even though ρ*(x; R(t)) is not available. Thus, the equation of motion in TRBOMD reads:
Let us mention that TRBOMD is closely related to CPMD. In CPMD, the equation of motion is given by:
Note that the frequency of the evolution equation for {ψi} in CPMD is adjusted by the fictitious mass parameter, μ. Comparing with TRBOMD, the parameter, μ, plays a similar role as ω−2, which controls the frequency of the fictitious dynamics of the initial density guess in SCF iteration. This connection will be made more explicit in the sequel.
We remark that the papers, [16,17], took a further step in viewing TRBOMD by an extended Lagrangian approach in a vanishing mass limit. This was also interpreted differently in [24] by starting from a Lagrangian and, then, using inaccurate forces in the equation of motions. However, unless a very specific and restrictive form of the error due to non-convergent SCF iterations is assumed, the equation of motion in TRBOMD does not have an associated Lagrangian in general. The connection to Lagrangian dynamics remains formal, and hence, we will not further explore it here.
4. Analysis of TRBOMD in the Linear Response Regime
In this section, we consider Equation (25) in the linear response regime, in which each atom, I, oscillates around its equilibrium position, . The displacement of the atomic configuration, R, from the equilibrium position is denoted by R̃(t) ≔ R(t) − R*, and the deviation of the electron density from the converged density is denoted by ρ̃(x, t) ≔ ρ(x, t) − ρ*(x; R(t)). Both R̃(t) and ρ̃(x, t) are small quantities in the linear response regime and contain the same information as R(t) and ρ(x, t). Using R̃(t) and ρ̃(x, t) as the new variables and noting the chain rule due to the R-dependence in ρ*(x; R(t)), the equation of motion in TRBOMD becomes:
In the linear response regime, the operator, , carries all the information of the SCF iteration scheme. Let us now derive the explicit form of for the k-step simple mixing scheme with mixing parameter (step length) α (0 < α ≤ 1). If k = 1, the simple mixing scheme reads:
From Equation (30b), we have:
The operator, (x, y), in Equation (39) is directly related to the stability of the dynamics. Equation (42b) also suggests that in the linear response regime, the spectrum of (x, y) must be on the real line, which requires that the matrix, , be diagonalizable with real eigenvalues. This has been shown for the simple mixing scheme. However, we remark that the condition that all eigenvalues of (x, y) are real may not hold for general preconditioners or for more complicated SCF iterations (for instance, Anderson mixing). This is one important restriction of the linear response analysis. Of course, this may not be a restriction for practical TRBOMD simulation for real systems. We will leave further understanding of this to future works.
Let us now assume that all eigenvalues of are real. The lower bound of the spectrum of , denoted by λmin(), should satisfy:
Let us compare TRBOMD with CPMD. It is well known that CPMD accurately approximates the results of BOMD, provided that the electronic and ionic degrees of freedom remain adiabatically separated, as well as the electrons stay close to the Born-Oppenheimer surface [12,13]. More specifically, the fictitious electron mass should be chosen, so that the lowest electronic frequency is well above ionic frequencies:
Note that condition (51) implies that CPMD no longer works if the system has a small gap or is even metallic. The usual work-around for this is to add a heat bath for the electronic degrees of freedom in CPMD [33], so that it maintains a fictitious temperature for the electronic degree of freedom. Nonetheless, the adiabaticity is lost for metallic systems, and CPMD is no longer accurate over long time simulation. In contrast, as we have discussed previously, TRBOMD may work for both insulating and metallic systems without any modification, provided that the SCF iteration is accurate and no resonance occurs. This is an important advantage of TRBOMD, which we will illustrate using numerical examples in the next section.
When the system has a gap, we can take μ sufficiently small to satisfy the adiabatic separation condition (51). Compare Equation (52) with Equation (50); we see that μ in CPMD plays a similar role as ω−2 in TRBOMD. The accuracy (in the linear regime) for CPMD and TRBOMD is the first order in μ and ω−2, respectively. At the same time, as taking a small μ or large ω increases the stiffness of the equation, the computational cost is proportional to μ−1 and ω2, respectively.
Let us remark that the above analysis is done in the linear response regime. As shown in [12,13], the accuracy of CPMD, in general, is only (μ1/2) instead of (μ) for the linear regime. Due to the close connection between these two parameters, we do not expect (ω−2) accuracy for TRBOMD in general, either. Actually, as will be discussed in Section 6, if the deviation of atom positions from equilibrium is not so small that we cannot linearize the nuclei motion, the error of TRBOMD in general will be (ω−1).
5. Numerical Results in the Linear Response Regime
In this section, we present numerical results for TRBOMD in the linear response regime using a one-dimensional (1D) model for KSDFT without the exchange correlation functional. The model problem can be tuned to exhibit both metallic and insulating features. Such a model was used before in mathematical analysis of ionization conjecture [36].
The total energy functional in our 1D density functional theory (DFT) model is given by:
Instead of using a bare Coulomb interaction, which diverges in 1D, we adopt a Yukawa kernel:
The parameters used in the 1D DFT model are chosen as follows. Atomic units are used throughout the discussion unless otherwise mentioned. The Yukawa parameter, κ = 0.01, is small enough so that the range of the electrostatic interaction is sufficiently long, and ∊0 is set to 10.00. The nuclear charge, ZI, is set to one for all atoms. Since spin is neglected, ZI = 1 implies that each atom contributes to one occupied state. The Hamiltonian operator is represented in a planewave basis set. All the examples presented in this section consists of 32 atoms. Initially, the atoms are at their equilibrium positions, and the distance between each atom and its nearest neighbor is set to 10 au. Starting from the equilibrium position, each ion is given a finite velocity, so that the velocity on the centroid of mass is zero. In the numerical experiments below, the system contains only one single phonon, which is obtained by assigning an initial velocity, v0 ∝ (1, −1, 1, −1, · · ·), to the atoms. We denote by ΩRef the corresponding phonon frequency. We choose v0, so that , where kB is the Boltzmann constant and Tion is 10 K, to make sure that the system is in the linear response regime. In the atomic unit, the mass of the electron is one, and the mass of each nuclei is set to 42, 000. By adjusting the parameters, {σI}, the 1D DFT model model can be tuned to resemble an insulating (with σI = 2.0) or a metallic system (with σI = 6.0) throughout the MD simulation. Figure 1 shows the spectrum of the insulating and the metallic system after running 1, 000 BOMD steps with converged SCF iteration.
In the linear response regime, we measure the error of the phonon frequency calculated from TRBOMD. This can be done in two ways. The first is given by Equation (50), namely, all quantities in the big parentheses in Equation (50) can be directly obtained by using the finite difference method at the equilibrium position, R*. The second is to explore the fact that in the linear response regime, there is a linear relation between the force and the atomic position, as in Equation (32), i.e., Hooke’s law:
In order to compare the performance among BOMD, TRBOMD and CPMD, we define the following relative errors:
5.1. Numerical Comparison between BOMD and TRBOMD
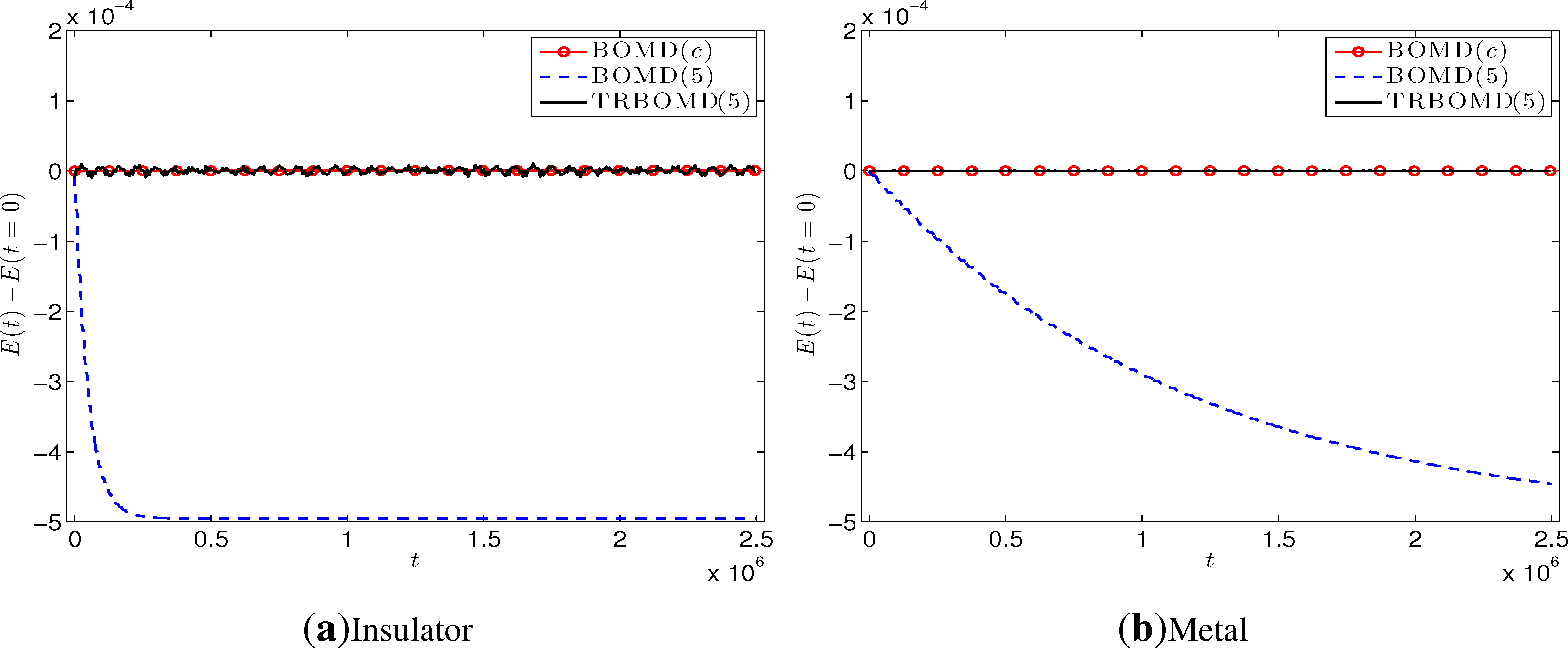
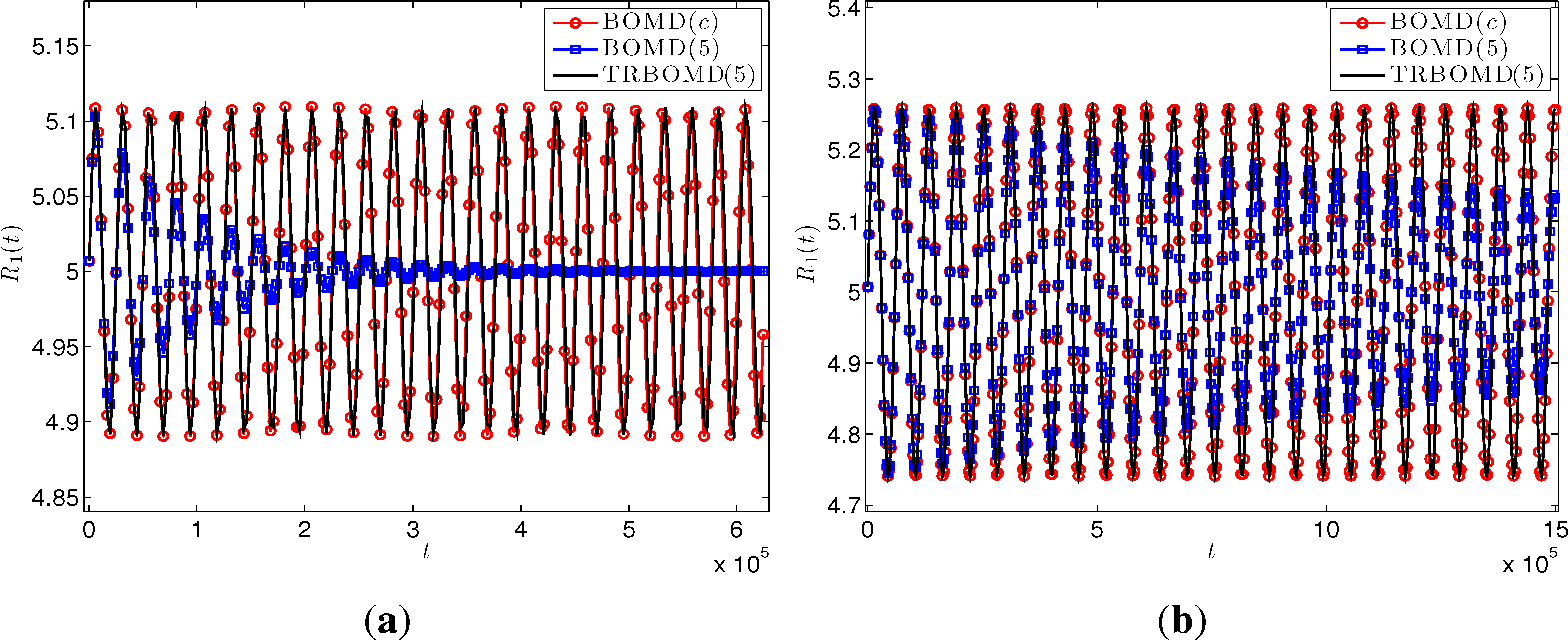
The first run is to validate the performance of TRBOMD. We set the time step Δt = 250, the artificial frequency , the final time T = 2.50E+06 and employ the simple mixing with step length α= 0.3 and the Kerker preconditioner in SCF cycles. Figure 2 plots the energy drift for BOMD with the converged SCF iteration (denoted by BOMD(c)) where the tolerance is 1.00E-08; BOMD with five SCF iterations per time step (denoted by BOMD(5)) and TRBOMD with five SCF iterations per time step (denoted by TRBOMD(5)). We see clearly there that BOMD(5) produces large drift for both insulator and metal, but TRBOMD(5) does not. Actually, from Table 1, the relative error in the average total energy over time between TRBOMD(5) and BOMD(c) is under 1.30E-05, but BOMD(c) needs about an average of 45 SCF iterations per time step to reach the tolerance 1.00E-08. Figure 3 plots corresponding trajectory of the left-most atom during about the first 25 periods and shows that the trajectory from TRBOMD (five) almost coincides with that from BOMD (c), which is also confirmed by the data of and in Table 1. However, for BOMD(5), the atom will cease oscillation after a while. A similar phenomena occurs for other atoms. In Table 1, we present more results for TRBOMD(n) with n = 3, 5, 7. We observe there that TRBOMD(n) gives more accurate results with larger n, and has a similar behavior as n increases to , which is in accord with our previous linear response analysis in Section 4.
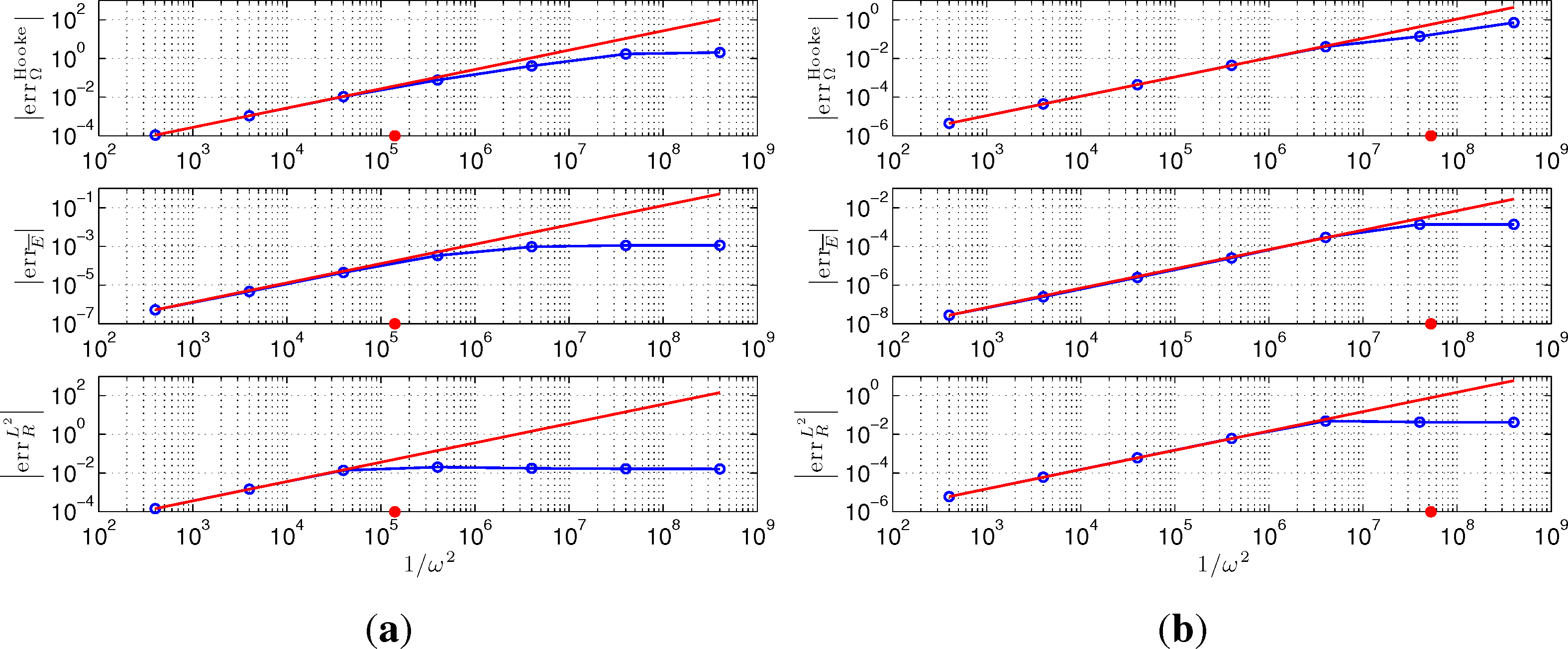
According to Equation (50), we have that is proportional to 1/ω2 for large ω. We verify this behavior using TRBOMD(3) as an example. In this example, a smaller time step, Δt = 20, is set to allow bigger artificial frequency ω. The final time is T = 6.00E+05, and the simple mixing with α= 0.3 and the Kerker preconditioner is applied in SCF iterations. For TRBOMD (three) under these settings, we have λmin() ≃ 8.81E-03 for the insulator and λmin() ≃ 5.92E-01 for the metal, and thus, the critical values of (ΩRef)2/λmin() in Equation (49) are about 7.12E-06 and 1.90E-08, respectively. We choose ω2 = 2.50E-03, 2.50E-04, 2.50E-05, 2.50E-06, 2.50E-07, 2.50E-08, 2.50E-09, and plot in Figure 4 the absolute values of , errĒ, for TRBOMD (three) as a function of 1/ω2 in logarithmic scales. When 1/ω2 ≪ λmin()/(ΩRef)2, Figure 4 shows clearly that all of ,|errĒ|, depend linearly on 1/ω2. The error, , has a similar behavior to and is skipped here for saving space.
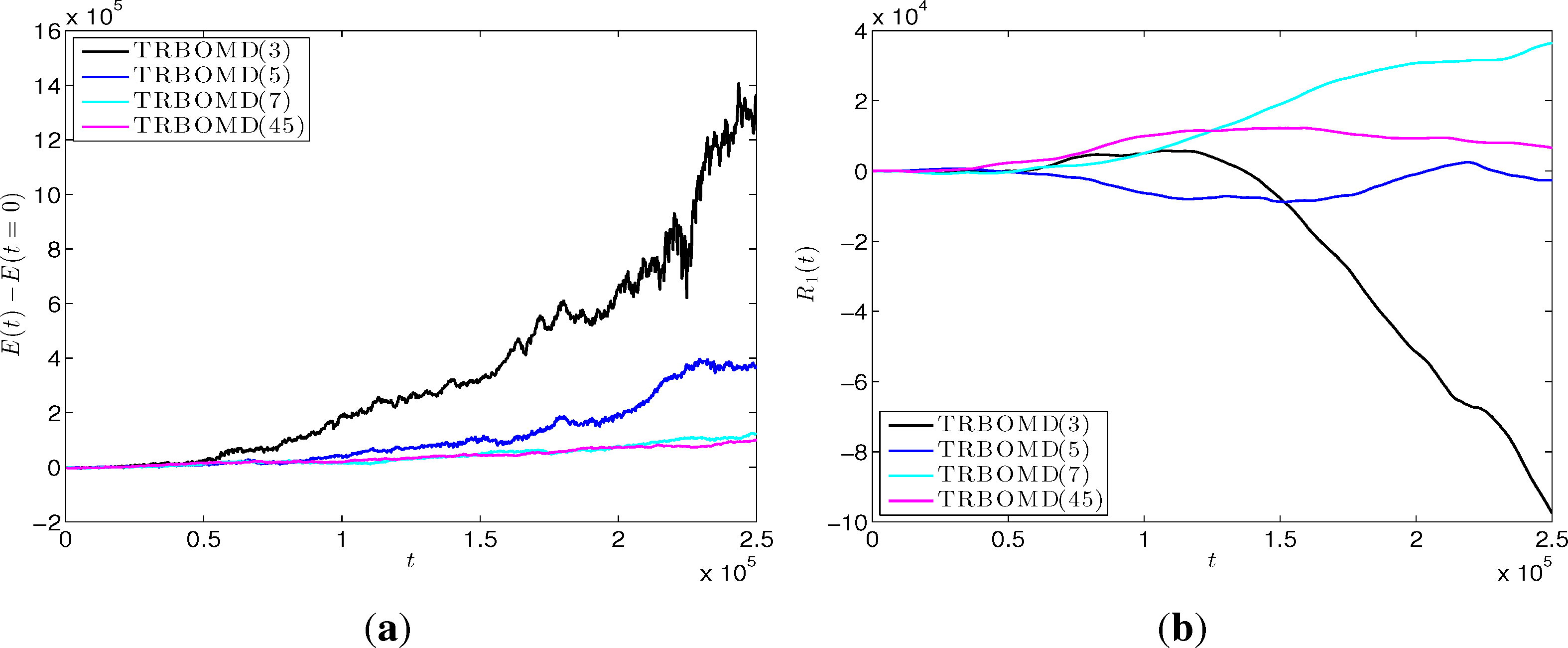
The last example illustrates the possible unstable behavior of TRBOMD when the stability condition λmin() > 0 in Equation (48) is violated. Here, we take the insulator as an example and set the time step Δt = 250, the final time to 2.50E+05 and the artificial frequency . The simple mixing with α= 0.3 is now applied in SCF iterations. Under these setting, we have λmin() < 0, e.g., λmin() = −2.42E+03 for TRBOMD (three). Figure 5a plots the energy drift for TRBOMD (n) with n = 3, 5, 7, 45. We see clearly there that TRBOMD is unstable even using 45 SCF iterations per time step (recall that BOMD (c) in the first run needs about average 45 SCF iterations per time step). Figure 5b plots the corresponding trajectory of the left-most atom and shows that the atom is driven wildly by the non-convergent SCF iteration.
5.2. Numerical Comparison between TRBOMD and CPMD
We now present some numerical examples for CPMD illustrating the difference between CPMD and TRBOMD. As we have discussed, TRBOMD is applicable to both metallic and insulting systems, while CPMD becomes inaccurate when the gap vanishes. To make this statement more concrete, we apply CPMD to the same atom chain system. We implement CPMD using a standard velocity Verlet scheme combined with RATTLEfor the orthonormality constraints [37–39].
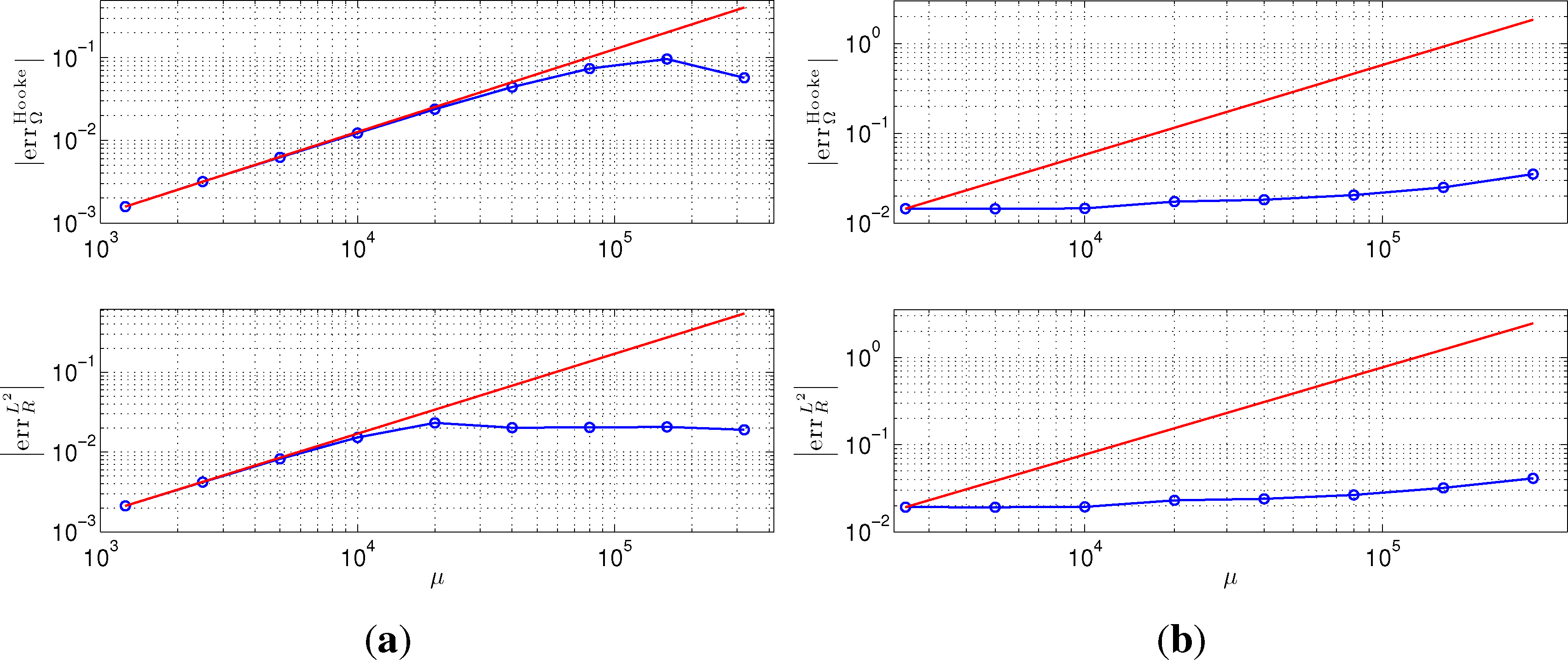
We present in Figure 6 the error of CPMD simulation for different choices of fictitious electron mass μ. We study the relative error of the phonon frequency, , the relative error of the position of the left-most atom measured in L2 norm, i.e., . We observe in Figure 6a linear convergence of CPMD to the BOMD result as the parameter, μ, decreases. This is consistent with our analysis. Recall that in CPMD, μ plays a similar role as ω−2 in TRBOMD. For the metallic example, the behavior is quite different; actually, Figure 6b shows a systematic error as μ decreases. For metallic system, as the spectral gap vanishes, the adiabatic separation between ionic and electronic degrees of freedom cannot be achieved no matter how small μ is. The adiabatic separation for TRBOMD, on the other hand, relies on the choice of an effective ρSCF, and hence, TRBOMD also works for a metallic system, as Figure 4 indicates.
The different behavior of CPMD for insulating and metallic systems is further illustrated by Figure 7, which shows the trajectory of the position of the left-most atom during the simulation. The phase error is apparent from the two subfigures. While the phase error decreases so that the trajectory approaches that of BOMD for the insulator in Figure 7a, the result in Figure 7b shows a systematic error for a metallic system.
6. Beyond the Linear Response Regime: Non-Equilibrium Dynamics
The discussion so far has been limited to the linear response regime so that we can make linear approximations for the degrees of freedom of both nuclei and electrons. In this case, as the system becomes linear, explicit error analysis has been given. For practical applications, we will be also interested in non-equilibrium nuclei dynamics, so that the deviation of atom positions is no longer small. In this section, we will investigate the non-equilibrium case using the averaging principle (see e.g., [40,41] for a general introduction on the averaging principle).
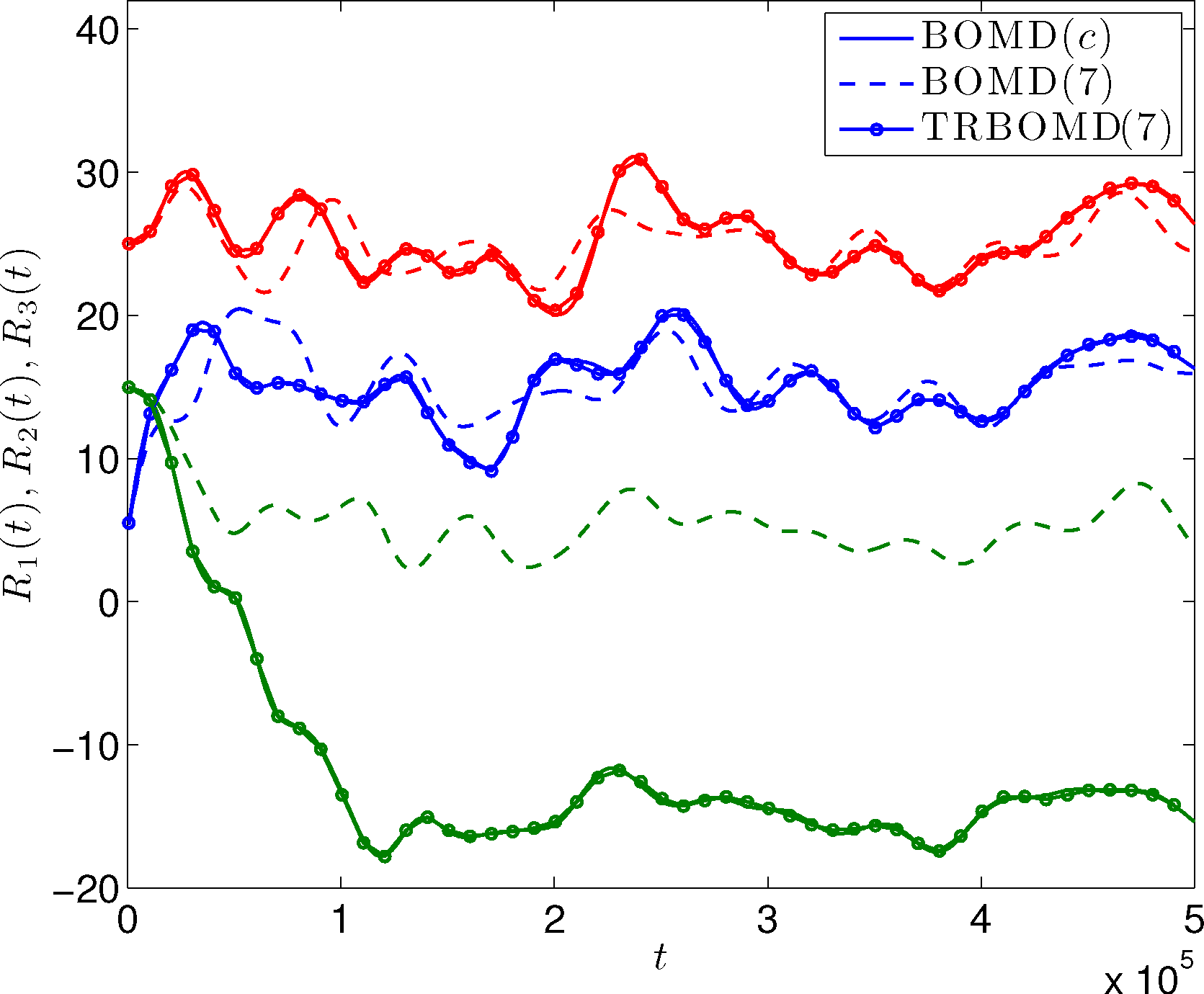
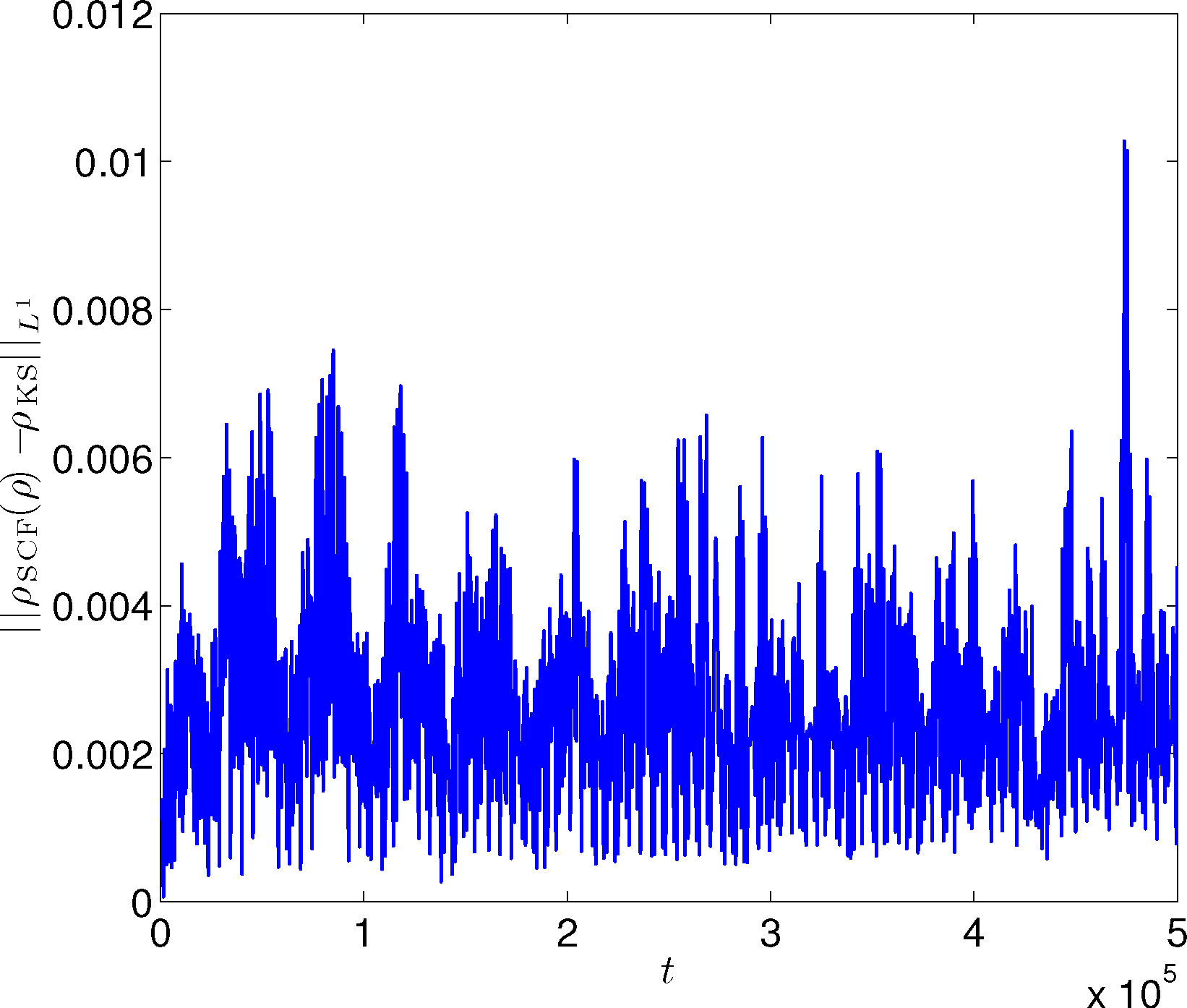
Let us first show numerically a non-equilibrium situation for the atom chain example discussed before. Initially, the 32 atoms stay at their equilibrium position. We set the initial velocity so that the left-most atom has a large velocity towards the right and other atoms have equal velocity towards the left. The mean velocity is equal to zero; so, the center of mass does not move. Figure 8 shows the trajectory of the positions of the first three atoms from the left. We observe that the results from TRBOMD agree very well with the BOMD results with convergent SCF iterations. Let us note that in the simulation, the left-most atom crosses over the second left-most atom. This happens since, in our model, we have taken a 1D analog of Coulomb interaction, the nuclei background charges are smeared out and, hence, the interaction is “soft” without hard-core repulsion. In Figure 9, we plot the difference between ρSCF and the converged electron density of the SCF iteration (denoted by ρKS) along the TRBOMD simulation. We see that the electron density used in TRBOMD stays close to the ground state electron density corresponding to the atom configuration.
To understand the performance of TRBOMD, recall that the equations of motion are given by:
Let us consider the limit, ω → ∞. In this case, we may freeze the R degree of freedom in the equation of motion for ρ, as ρ changes on a much faster time scale. To capture the two time scale behavior, we introduce a heuristic two-scale asymptotic expansion with faster time variable given by τ = ωt (with some abuse of notation):
To proceed, we consider the scenario that ρ (t, τ) is close to the ground state electron density corresponding to the current atom configuration, ρ*(R(t)). We have seen from numerical examples (Figure 9) that this is indeed the case for a good choice of SCF iteration, while we do not have a proof of this in the general case. Hence, we linearize the map: ρSFC.
Under the stability condition (48), it is easy to see that for ρ(t, τ) satisfying Equation (75), the limit of the time average:
Remark. If we do not make the linear approximation for the electronic degree of freedom, as the map, ρSCF, is quite nonlinear and complicated, the analysis of the long time (in τ) behavior of Equation (73) is not as straightforward. In particular, it is not clear to us whether the limit:
As a result of this discussion, in practice, when we apply TRBOMD to a particular system, we need to be cautious whether the electronic degree of freedom remains around the converged Kohn-Sham electron density, which is not necessarily guaranteed (in contrast to CPMD for systems with gaps).
7. Conclusions
The recently developed time reversible Born-Oppenheimer molecular dynamics (TRBOMD) scheme provides a promising way for reducing the number of self-consistent field (SCF) iterations in molecular dynamics simulation. By introducing auxiliary dynamics to the initial guess of the SCF iteration, TRBOMD preserves the time-reversibility of the NVE dynamics, both at the continuous and at the discrete level, and exhibits improved long time stability over the Born-Oppenheimer molecular dynamics with the same accuracy. In this paper we analyze, for the first time, the accuracy and the stability of the TRBOMD scheme, and our analysis is verified through numerical experiments using a one-dimensional density functional theory (DFT) model without exchange correlation potential. The validity of the stability condition in TRBOMD is directly associated with the quality of the SCF iteration procedure. In particular, we demonstrate in the case in which the SCF iteration procedure is not very accurate, the stability condition can be violated, and TRBOMD becomes unstable. We also compare TRBOMD with the Car-Parrinello molecular dynamics (CPMD) scheme. CPMD relies on the adiabatic evolution of the occupied electron states, and therefore, CPMD works better for insulators than for metals. However, TRBOMD may be effective for both insulating and metallic systems. The present study is restricted to the NVE system and to simplified DFT models. Moreover, the analysis in the present work is mainly focused on the accuracy of trajectories and harmonic frequencies in the perturbation regime. However, in practice, the more important question is how the introduced artificial dynamics influence static properties, like distribution functions, and the most critical capability is to reproduce the correct distribution functions. The performance of TRBOMD for the NVT system and for realistic DFT systems with emphasis on the accuracy of static properties will be our future work.
Acknowledgments
This work was partially supported by the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory under the US Department of Energy contract number DE-AC02-05CH11231 and the Scientific Discovery through Advanced Computing (SciDAC) program funded by the US Department of Energy, Office of Science, Advanced Scientific Computing Research and Basic Energy Sciences (L.L.), the Alfred P. Sloan Foundation and the National Science Foundation (J.L.), the National Natural Science Foundation of China under the Grant Nos. 11101011 and 91330110 and the Specialized Research Fund for the Doctoral Program of Higher Education under the Grant No. 20110001120112 (S.S.). The authors would also like to thank the referees for many useful suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix
Here, we derive the perturbation analysis result in Equation (50). When deriving the perturbation analysis below, we use linear algebra notation and do not distinguish matrices from operators. We use the linear algebra notation, replace all the integrals by matrix-vector multiplication and drop all the dependencies of the electron degrees of freedom, x and y. For instance, ρ̃ should be understood as ∫(x,y)ρ̃(y) dy. We also denote simply by ; then, Equation (42) can be rewritten as:
References
- Marx, D.; Hutter, J. Ab initio molecular dynamics: Theory and implementation. Mod. Methods Algorithms Quantum Chem 2000, 1, 301–449. [Google Scholar]
- Kirchner, B.; di Dio, P.J.; Hutter, J. Real-world predictions from ab initio molecular dynamics simulations. Top. Curr. Chem 2012, 307, 109–153. [Google Scholar]
- Payne, M.C.; Teter, M.P.; Allen, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total energy calculation: Molecular dynamics and conjugate gradients. Rev. Mod. Phys 1992, 64, 1045–1097. [Google Scholar]
- Deumens, E.; Diz, A.; Longo, R.; Öhrn, Y. Time-dependent theoretical treatments of the dynamics of electrons and nuclei in molecular systems. Rev. Mod. Phys 1994, 66, 917–983. [Google Scholar]
- Tuckerman, M.E.; Ungar, P.J.; von Rosenvinge, T.; Klein, M.L. Ab initio molecular dynamics simulations. J. Phys. Chem 1996, 100, 12878–12887. [Google Scholar]
- Parrinello, M. From silicon to RNA: The coming of age of ab initio molecular dynamics. Solid State Commun 1997, 102, 107–120. [Google Scholar]
- Marx, D.; Hutter, J. Ab initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev 1964, 136, B864–B871. [Google Scholar]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev 1965, 140, A1133–A1138. [Google Scholar]
- Remler, D.K.; Madden, P.A. Molecular dynamics without effective potentials via the Car-Parrinello approach. Mol. Phys 1990, 70, 921–966. [Google Scholar]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett 1985, 55, 2471–2474. [Google Scholar]
- Pastore, G.; Smargiassi, E.; Buda, F. Theory of ab initio molecular-dynamics calculations. Phys. Rev. A 1991, 44, 6334–6347. [Google Scholar]
- Bornemann, F.A.; Schütte, C. A mathematical investigation of the Car-Parrinello method. Numer. Math 1998, 78, 359–376. [Google Scholar]
- Niklasson, A.M.N.; Tymczak, C.J.; Challacombe, M. Time-reversible Born-Oppenheimer molecular dynamics. Phys. Rev. Lett 2006, 97, 123001:1–123001:4. [Google Scholar]
- Niklasson, A.M.N.; Tymczak, C.J.; Challacombe, M. Time-reversible ab initio molecular dynamics. J. Chem. Phys 2007, 126, 144103:1–144103:9. [Google Scholar]
- Niklasson, A.M.N. Extended Born-Oppenheimer molecular dynamics. Phys. Rev. Lett 2008, 100, 123004:1–123004:4. [Google Scholar]
- Niklasson, A.M.N.; Steneteg, P.; Odell, A.; Bock, N.; Challacombe, M.; Tymczak, C.J.; Holmström, E.; Zheng, G.; Weber, V. Extended Lagrangian Born-Oppenheimer molecular dynamics with dissipation. J. Chem. Phys 2009, 130, 214109. [Google Scholar] [CrossRef]
- Niklasson, A.M.N.; Cawkwell, M.J. Fast method for quantum mechanical molecular dynamics. Phys. Rev. B 2012, 86, 174308:1–174308:12. [Google Scholar]
- Hairer, E.; Lubich, C.; Wanner, G. Geometric Numerical Integration, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- McLachlan, R.I.; Perlmutter, M. Energy drift in reversible time integration. J. Phys. A: Math. Gen 2004, 37, L593–L598. [Google Scholar]
- Kolafa, J. Time-reversible always stable predictor-corrector method for molecular dynamics of polarizable molecules. J. Comput. Chem 2004, 25, 335–342. [Google Scholar]
- Kühne, T.D.; Krack, M.; Mohamed, F.R.; Parrinello, M. Efficient and accurate Car-Parrinello-like approach to Born-Oppenheimer molecular dynamics. Phys. Rev. Lett 2007, 98, 066401:1–066401:4. [Google Scholar]
- Dai, J.; Yuan, J. Large-scale efficient Langevin dynamics, and why it works. EPL 2009, 88, 20001. [Google Scholar] [CrossRef]
- Hutter, J. Car-Parrinello molecular dynamics. WIREs Comput. Mol. Sci 2012, 2, 604–612. [Google Scholar]
- Anderson, D.G. Iterative procedures for nonlinear integral equations. J. Assoc. Comput. Mach 1965, 12, 547–560. [Google Scholar]
- Pulay, P. Convergence acceleration of iterative sequences: The case of SCF iteration. Chem. Phys. Lett 1980, 73, 393–398. [Google Scholar]
- Johnson, D.D. Modified Broyden’s method for accelerating convergence in self-consistent calculations. Phys. Rev. B 1988, 38, 12807–12813. [Google Scholar]
- Kerker, G.P. Efficient iteration scheme for self-consistent pseudopotential calculations. Phys. Rev. B 1981, 23, 3082–3084. [Google Scholar]
- Lin, L.; Yang, C. Elliptic preconditioner for accelerating self consistent field iteration in Kohn-Sham density functional theory. SIAM J. Sci. Comput 2013, 35, S277–S298. [Google Scholar]
- McLachlan, R.I.; Atela, P. The accuracy of symplectic integrators. Nonlinearity 1992, 5, 541–562. [Google Scholar]
- Adler, S.L. Quantum theory of the dielectric constant in real solids. Phys. Rev 1962, 126, 413–420. [Google Scholar]
- Wiser, N. Dielectric constant with local field effects included. Phys. Rev 1963, 129, 62–69. [Google Scholar]
- Blöchl, P.E.; Parrinello, M. Adiabaticity in first-principles molecular dynamics. Phys. Rev. B 1992, 45, 9413–9416. [Google Scholar]
- Tangney, P.; Scandolo, S. How well do Car-Parrinello simulations reproduce the Born-Oppenheimer surface? Theory and examples. J. Chem. Phys 2002, 116, 14–24. [Google Scholar]
- Tangney, P. On the theory underlying the Car-Parrinello method and the role of the fictitious mass parameter. J. Chem. Phys 2006, 124, 044111:1–044111:14. [Google Scholar]
- Solovej, J.P. Proof of the ionization conjecture in a reduced Hartree-Fock model. Invent. Math 1991, 104, 291–311. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- Ciccotti, G.; Ferrario, M.; Ryckaert, J.P. Molecular dynamics of rigid systems in cartesian coordinates: A general formulation. Mol. Phys 1982, 47, 1253–1264. [Google Scholar]
- Andersen, H.C. Rattle: A “velocity” version of the Shake algorithm for molecular dynmiacs calculations. J. Comput. Phys 1983, 52, 24–34. [Google Scholar]
- E, W. Principles of Multiscale Modeling; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Pavliotis, G.; Stuart, A. Multiscale Methods: Averaging and Homogenization; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Insulator: ΩRef = 2.51E-04, ERef = 8.66E-01 | |||||
| n | errĒ | ||||
| 3 | −6.53E-03 | −1.63E-02 | −7.63E-05 | 2.26E-02 | 4.25E-02 |
| 5 | −1.08E-03 | −2.38E-03 | −1.30E-05 | 1.27E-02 | 2.92E-02 |
| 7 | −2.76E-04 | −5.41E-04 | −3.32E-06 | 3.02E-03 | 7.22E-03 |
| Metal: ΩRef = 1.06E-04, E¯Ref = 5.28E-01 | |||||
| 3 | −2.65E-04 | −6.92E-04 | −4.36E-06 | 3.86E-03 | 8.95E-03 |
| 5 | −3.65E-05 | −7.31E-05 | −4.44E-07 | 4.14E-04 | 9.60E-04 |
| 7 | −5.24E-06 | 2.93E-06 | −1.10E-07 | 1.63E-05 | 3.78E-05 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lin, L.; Lu, J.; Shao, S. Analysis of Time Reversible Born-Oppenheimer Molecular Dynamics. Entropy 2014, 16, 110-137. https://doi.org/10.3390/e16010110
Lin L, Lu J, Shao S. Analysis of Time Reversible Born-Oppenheimer Molecular Dynamics. Entropy. 2014; 16(1):110-137. https://doi.org/10.3390/e16010110
Chicago/Turabian StyleLin, Lin, Jianfeng Lu, and Sihong Shao. 2014. "Analysis of Time Reversible Born-Oppenheimer Molecular Dynamics" Entropy 16, no. 1: 110-137. https://doi.org/10.3390/e16010110
APA StyleLin, L., Lu, J., & Shao, S. (2014). Analysis of Time Reversible Born-Oppenheimer Molecular Dynamics. Entropy, 16(1), 110-137. https://doi.org/10.3390/e16010110