The Completed Self: An Immunological View of the Human-Microbiome Superorganism and Risk of Chronic Diseases


Abstract
:1. Introduction
1.1. Signs, Biomarkers, and Health Outcomes
1.2. Development of Self Identity from an Immunological Perspective
1.3. Prenatal Symbiosis
1.4. Birth Delivery as the Transition
2. Results
2.1. The Completed Self Concept
2.2. Sources of Microbiota for Infant Self-Completion
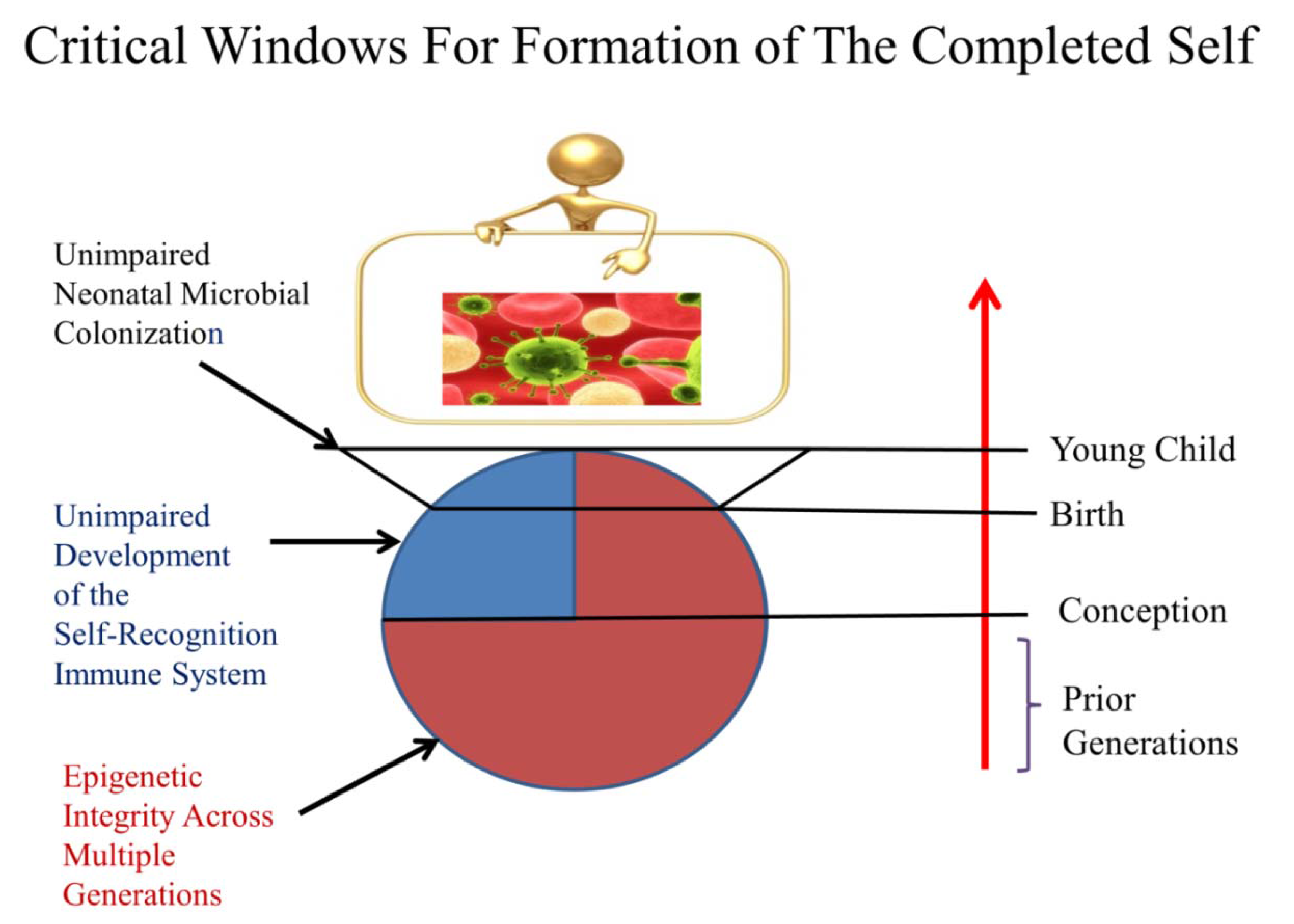
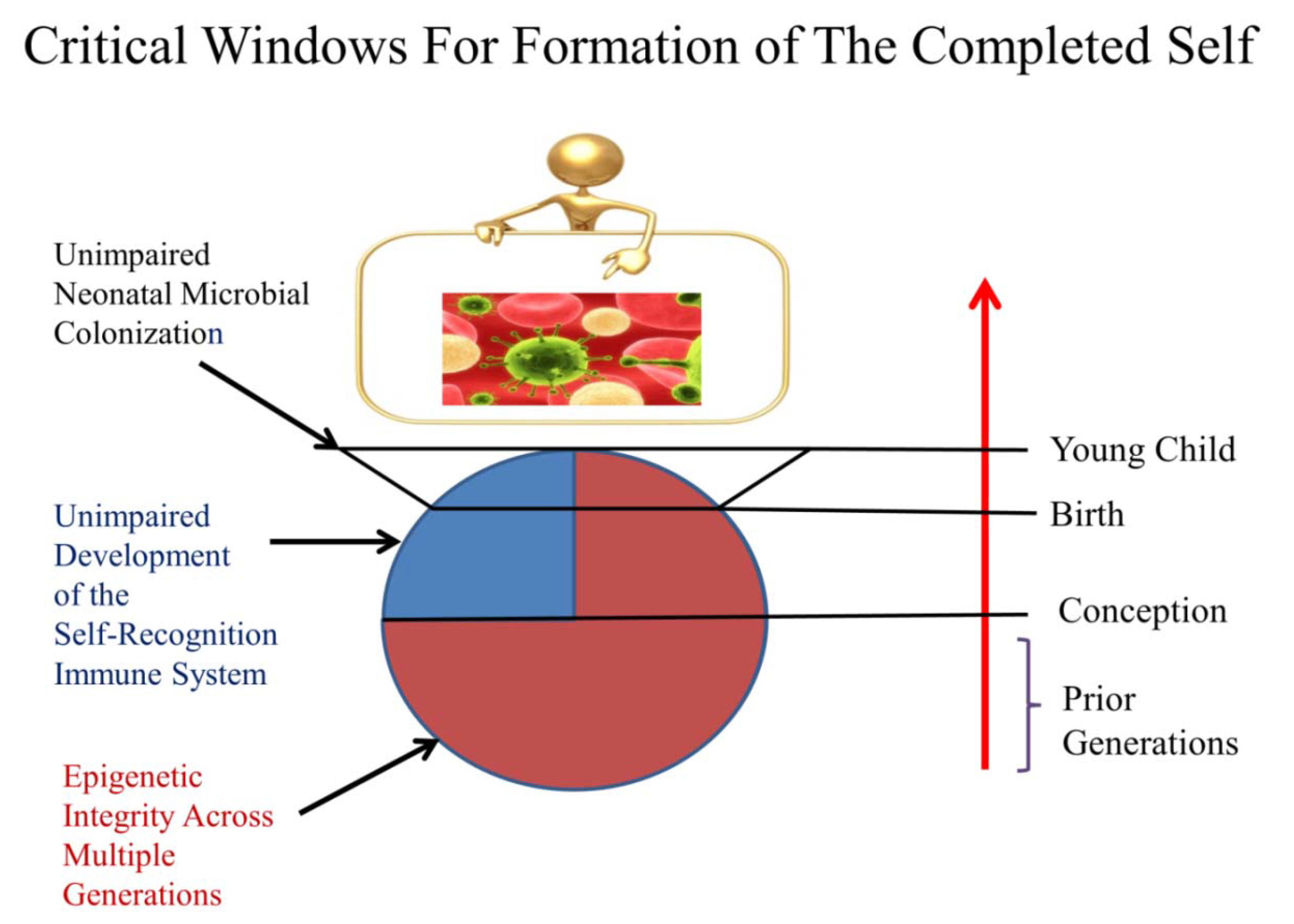
2.3. Critical Timing for Self-Completion
2.4. Health vs. Disease: the Epidemic of Chronic Diseases and Conditions

{kind=link}
{kind=link}
| Entryway Disease or Condition | Population studied | Period of study | Category of costs | Per annum amount per patient | Reference |
|---|---|---|---|---|---|
| Asthma | School age children in a 1996 Medical Expenditure Panel Survey | 1996 | Total economic impact, medical and lost parental wages | $791 | [146] |
| Asthma (difficult to control) | Children 6-12 years of age with difficult to control asthma from several US sites | 2001–2004 | Total asthma costs: medications, physician visits, hospital visits; lost work/school days | $7,846 | [147] |
| Autism spectrum disorders | US: Medicaid database from 42 states | 2003 | Total health care expenditures in Medicaid per child per annum | $22,079 | [148] |
| Childhood chronic kidney disease | Children from Canada (British Columbia and Yukon) | 2009 | Annual pharmaceutical cost | $1,800 | [149] |
| Pediatric Crohn’s disease | Pediatric patients in Canterbury, New Zealand | 2010-2011 | Total (direct and indirect) cost per person per annum | $14,375 NZD | [150] |
| Pediatric food allergies | Children 0–18 years of age as one categories in the study | 2007 | Mean direct medical cost per child per annum | $3,635 | [151] |
| Kawasaki disease | Across US | 1997–1999 | Median hospitalization cost | $6,169 | [152] |
| Pediatric arthritis | 2nd Children’s Hospital at Berlin-Buch cohort | 1998–2000 | Mean cost per patient per annum | 3,500 Euros | [153] |
| Type 1 diabetes | Texas | 2004–2005 | Median direct costs per person per annum | $4,730 | [154] |
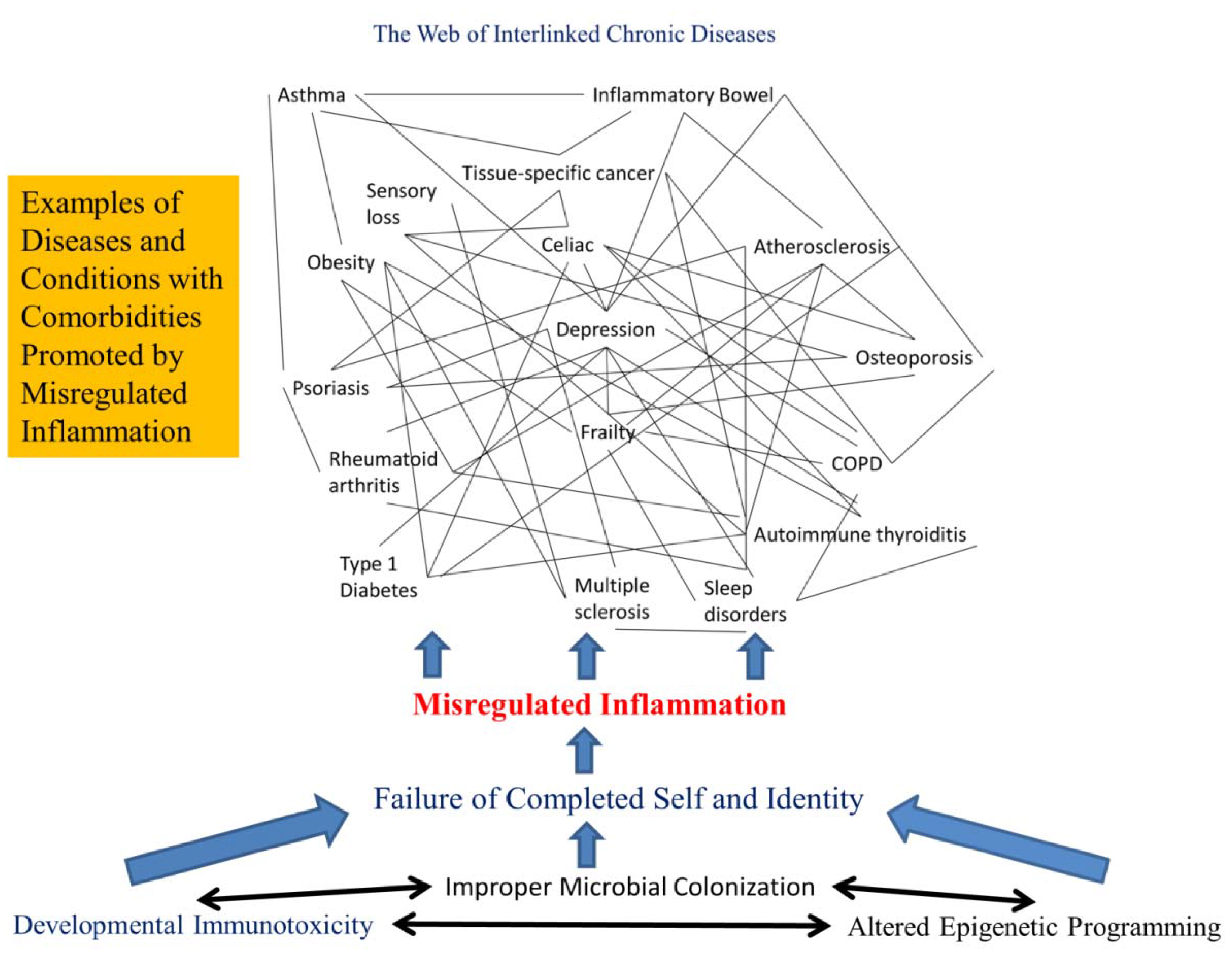
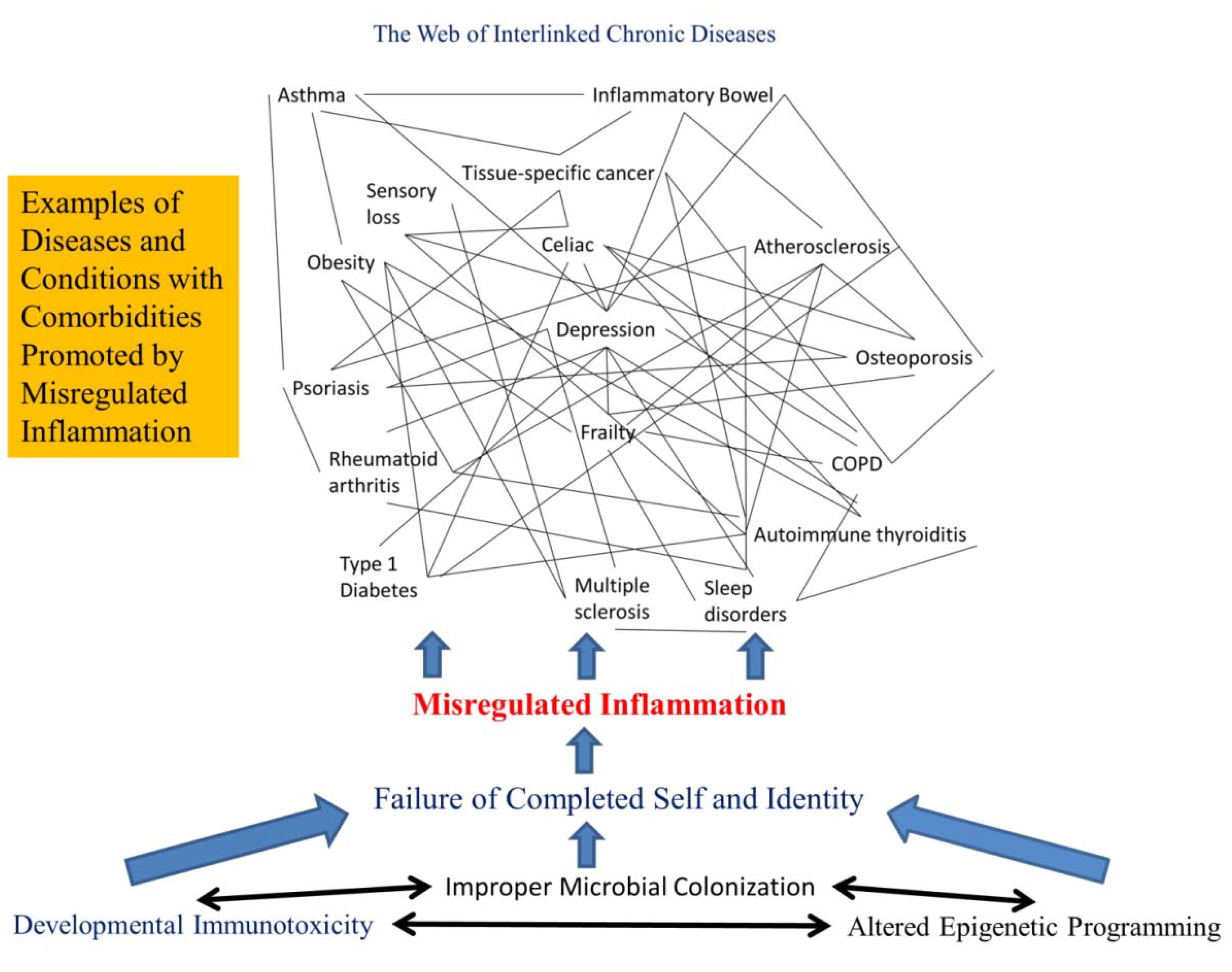
2.5. Chronic Diseases Are Connected as Interlinked Patterns
2.6. Chronic Disease Patterns Begin Early in Life
2.7. Misregulated Inflammation Is the Root of Most Chronic Diseases
| Disease/condition | Suggested biomarkers | Reference(s) |
|---|---|---|
| Alzheimer’s disease | Microglia and astrocyte proinflammatory cytokines IL-1, IL-6, TNF-alpha with inflammasome activation; Low M-CSF production by macrophages containing amyloid-B deposits; Altered TLR regulation | [155,156,157] |
| Asthma and respiratory allergies | Altered signaling and/or levels of Toll-like receptors 2, 4, and 7; Increased IgE and/or IL-4 levels; Eosinophilic infiltration of tissues; Production of specific microRNAs | [158,159,160] |
| Atherosclerosis | TLR 4, MyD88, and the inflammasome activation; Elevation of IL-1 and IL-18, C-reactive protein; Lipoprotein-associated phospholipase A2 | [161,162] |
| Autism | Increased proinflammatory cytokines and abnormal innate immune responses | [163] |
| Chronic obstructive pulmonary disease | Improper neutrophillic inflammation; Excessive ROS production; Excessive Th1 and Th17 activity; Impeded immune repair | [164,165] |
| Depression | Increased proinflammatory cytokines IL-1, IL-6, TNF-alpha; Elevated cortisol | [166,167] |
| Food allergies | Decline in TGF-beta producing T cells in the gut | [168,169] |
| Grave’s disease | Dendritic cell polarization; impaired CD4+CD25+ regulatory T cell capacity | [170] |
| Hashimoto’s thyroiditis | Decreased CD4+CD152+ T cells | [171] |
| Myalgic encephalomyelitis (Chronic Fatigue Syndrome) | Elevated proinflammatory cytokines; Elevated serum neopterin and PMN-elastase; blunted T cell memory | [172−174] |
| Multiple sclerosis | CD46 regulation; Activated microglial production of proinflammatory cytokines; Dysregulated Treg/Th17axis; Neuropilin-1 regulation | [156,175,176,177] |
| Osteoporosis | Urinary ratio of native (alpha) to isomerized (beta) CTX; Fragments of interalpha-trypsin-inhibitor heavy chain H4 precursor (ITIH4) | [178,179] |
| Parkinson’s disease | Change in olfaction; Depression | [180] |
| Psoriasis | IL-1F6 as well as upregulation of TNF-alpha, IL-17A, and IL-23 | [181] |
| Rheumatoid arthritis | Macrophage overproduction of IL-1, IL-6 and TNF-alpha; CD4+ Th1- and Th17-driven inflammation; inflammasome activation; Autoantibodies to Fc portion of host immunoglobulins | [182,183,184] |
| Sarcoidosis | A cytokine cascade of IL-1, IL-2, IL-6, IL-8, IL-12, IL-18, IFN-gamma, and TNF-alpha promotes the recruitment, activation, and proliferation of mononuclear cells and a Th1-driven granulomatous response; Overabundance of activated CD4+ T cells; Altered dendritic cell maturation | [164,185] |
| Sjogren syndrome | Enhanced production of type 1 interferons by dendritic and other cells; Increased oxidative stress; Autoantibodies against the RNA-binding proteins SSA/SSB/RNP | [186,187] |
| Sleep disorders | IL-6 levels in relation to other inflammation-regulating cytokines | [188] |
| Systemic sclerosis | CD70 overexpression; Endothelial adhesion molecules; Soluble TNF-alpha receptor; Altered pulmonary surfactant production | [189,190,191] |
| Type 1 diabetes | Macrophage overproduction of inflammatory cytokines and CD4+ Th1 subpopulation driven inflammation | [192] |
2.8. Human Avoidance of Misregulated Inflammation and Chronic Diseases
2.9. Three Key Factors That Affect Our Capacity for Self-Completion
2.9.1. Altered Epigenetic Programming (AEP) Across Generations
2.9.2. Developmental Immunotoxicity (DIT)
2.9.2.1. DIT and Misregulated Inflammation
2.9.2.2. DIT and Infectious Agents Can Combined to Initiate Chronic Diseases
2.9.3. Altered Neonatal Microbial Colonization
3. Discussion
3.1. DIT, Missing Microbes, and Future Health
3.2. Reducing the Risk of Chronic Diseases and Conditions: Prevention and Proactive Strategies
4. Conclusions
Acknowledgments
References
- Lamkanfi, M.; Walle, L.V.; Kanneganti, T.D. Deregulated inflammasome signaling in disease. Immunol. Rev. 2011, 243, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2012. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.E. Persistent systemic inflammation in chronic critical illness. Respir. Care 2012, 57, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; DeWitt, J.C.; Luebke, R.W. Reducing the Prevalence of Immune-Based Chronic Disease. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 419–440. [Google Scholar]
- Davidson, R.M.; Seneff, S. The initial common pathway of inflammation, disease, and death. Entropy 2012, 14, 1399–1442. [Google Scholar] [CrossRef]
- Oller, J.W.J. The Antithesis of Entropy: Biosemiotic Communication from Genetics to Human Language with Special Emphasis on the Immune Systems. Entropy 2010, 12, 631–705. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Ottman, N.; Smidt, H.; de Vos, W.M.; Belzer, C. The function of our microbiota: who is out there and what do they do? Front. Cell Infect. Microbiol. 2012, 2, 104. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F. Host responses to the human microbiome. Nutr. Rev. 2012, 70, S14–17. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R. Metabolomics of a superorganism. J. Nutr. 2007, 137, 259S–266S. [Google Scholar] [PubMed]
- Grice, E.A.; Segre, J.A. The Human Microbiome: Our Second Genome. Annu. Rev. Genomics Hum. Genet. 2012, 13, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Eberl, G. Development and evolution of RORgammat+ cells in a microbe's world. Immunol. Rev. 2012, 245, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.A.; Aitken, J.D.; Vijay-Kumar, M.; Gewirtz, A.T. Toll-like receptor-gut microbiota interactions: perturb at your own risk! Annu. Rev. Physiol. 2012, 74, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Pang, I.K.; Iwasaki, A. Control of antiviral immunity by pattern recognition and the microbiome. Immunol. Rev. 2012, 245, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, R.D.; Wegmann, T.G. Placental immunoregulation. Crit. Rev. Immunol. 1986, 6, 245–285. [Google Scholar] [PubMed]
- Gluckman, P.D.; Pinal, C.S. Maternal-placental-fetal interactions in the endocrine regulation of fetal growth: role of somatotrophic axes. Endocrine 2002, 19, 81–89. [Google Scholar] [CrossRef]
- Myatt, L. Placental adaptive responses and fetal programming. J. Physiol. 2006, 572, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Sandovici, I.; Hoelle, K.; Angiolini, E.; Constancia, M. Placental adaptations to the maternal-fetal environment: implications for fetal growth and developmental programming. Reprod. Biomed. Online 2012, 25, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Nauta, A.J.; Ben, A.K.; Knippels, L.M.; Knol, J.; Garssen, J. Early life: gut microbiota and immune development in infancy. Benef. Microbes 2010, 1, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; Dietert, J.M. Strategies for Protecting Your Child's Immune System; World Scienctific Publishing Company: Singapore, 2010. [Google Scholar]
- Sykes, L.; MacIntyre, D.A.; Yap, X.J.; Teoh, T.G.; Bennett, P.R. The Th1:th2 dichotomy of pregnancy and preterm labour. Mediators Inflamm. 2012, 2012, 967629. [Google Scholar] [CrossRef] [PubMed]
- Challis, J.R.; Lockwood, C.J.; Myatt, L.; Norman, J.E.; Strauss, J.F.; Petraglia, F. Inflammation and pregnancy. Reprod. Sci. 2009, 16, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, T.G.; Lin, H.; Guilbert, L.; Mosmann, T.R. Bidirectional cytokine interactions in the maternal-fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol. Today 1993, 14, 353–356. [Google Scholar] [CrossRef]
- Liu, F.; Guo, J.; Tian, T.; Wang, H.; Dong, F.; Huang, H.; Dong, M. Placental trophoblasts shifted Th1/Th2 balance toward Th2 and inhibited Th17 immunity at fetomaternal interface. APMIS 2011, 119, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Doria, A.; Iaccarino, L.; Arienti, S.; Ghirardello, A.; Zampieri, S.; Rampudda, M.E.; Cutolo, M.; Tincani, A.; Todesco, S. Th2 immune deviation induced by pregnancy: the two faces of autoimmune rheumatic diseases. Reprod. Toxicol. 2006, 22, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.P.; Fan, D.X.; Zhang, T.; Guo, P.F.; Li, D.J. The costimulatory signal upregulation is associated with Th1 bias at the maternal-fetal interface in human miscarriage. Am. J. Reprod. Immunol. 2011, 66, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Giannubilo, S.R.; Landi, B.; Pozzi, V.; Sartini, D.; Cecati, M.; Stortoni, P.; Corradetti, A.; Saccucci, F.; Tranquilli, A.L.; Emanuelli, M. The involvement of inflammatory cytokines in the pathogenesis of recurrent miscarriage. Cytokine 2012, 58, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Patni, S.; Flynn, P.; Wynen, L.P.; Seager, A.L.; Morgan, G.; White, J.O.; Thornton, C.A. An introduction to Toll-like receptors and their possible role in the initiation of labour. BJOG 2007, 114, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Oomomian, Y.; Stephen, G.; Shynlova, O.; Tower, C.L.; Garrod, A.; Lye, S.J.; Jones, R.L. Macrophages infiltrate the human and rat decidua during term and preterm labor: evidence that decidual inflammation precedes labor. Biol. Reprod. 2012, 86, 39. [Google Scholar] [CrossRef] [PubMed]
- Shynlova, O.; Lee, Y.H.; Srikhajon, K.; Lye, S. Physiologic Uterine Inflammation and Labor Onset: Integration of Endocrine and Mechanical Signals. Reprod. Sci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; Etzel, R.A.; Chen, D.; Halonen, M.; Holladay, S.D.; Jarabek, A.M.; Landreth, K.; Peden, D.B.; Pinkerton, K.; Smialowicz, R.J.; et al. Workshop to identify critical windows of exposure for children's health: immune and respiratory systems work group summary. Environ. Health Perspect. 2000, 108, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Selevan, S.G.; Kimmel, C.A.; Mendola, P. Identifying critical windows of exposure for children's health. Environ. Health Perspect. 2000, 108, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Makris, S.L.; Thompson, C.M.; Euling, S.Y.; Selevan, S.G.; Sonawane, B. A lifestage-specific approach to hazard and dose-response characterization for children's health risk assessment. Birth Defects Res. B Dev. Reprod. Toxicol. 2008, 83, 530–546. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D. The human superorganism- of microbes and men. Med. Hypotheses 2010, 74, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Wang, X.; Li, L. Human gut microbiome: the second genome of human body. Protein Cell 2010, 1, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, T.B.; Detsky, A.S. Time to Recognize Our Fellow Travellers. J. Gen. Intern. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Eberl, G. A new vision of immunity: homeostasis of the superorganism. Mucosal. Immunol. 2010, 3, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Colloada, M.C.; Cernada, M.; Bauerl, C.; Vento, M.; Perez-Martinez, G. Microbial ecology and host-microbiota interactions during early life stages. Gut Microbes 2012, 3, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Stencel-Gabriel, K.; Gabriel, I.; Wiczkowski, A.; Paul, M.; Olejek, A. Prenatal priming of cord blood T lymphocytes by microbiota in the maternal vagina. Am. J. Reprod. Immunol. 2009, 61, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Fouhy, F.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C.; Cotter, P.D. Composition of the early intestinal microbiota: Knowledge, knowledge gaps and the use of high-throughput sequencing to address these gaps. Gut Microbes 2012, 3, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, C.; Burton, J.P.; Reid, G. Breast, milk and microbes: a complex relationship that does not end with lactation. Womens Health (Lond. Engl.) 2012, 8, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rubio, R.; Collado, M.C.; Laitinen, K.; Salminen, S.; Isolauri, E.; Mira, A. The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am. J. Clin. Nutr. 2012, 96, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Nagata, R.; Nagano, H.; Ogishima, D.; Nakamura, Y.; Hiruma, M.; Sugita, T. Transmission of the major skin microbiota, Malassezia, from mother to neonate. Pediatr. Int. 2012, 54, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Capone, K.A.; Dowd, S.E.; Stamatas, G.N.; Nikolovski, J. Diversity of the human skin microbiome early in life. J. Invest. Dermatol. 2011, 131, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Brulc, J.M.; Iovieno, A.; Bates, B.; Garoutte, A.; Miller, D.; Revanna, K.V.; Gao, X.; Antonopoulos, D.A.; Slepak, V.Z.; et al. Diversity of bacteria at healthy human conjunctiva. Invest. Ophthalmol. Vis. Sci. 2011, 52, 5408–5413. [Google Scholar] [CrossRef] [PubMed]
- Raskind, C.H.; Sabo, B.E.; Callan, D.A.; Farrel, P.A.; Dembry, L.M.; Gallagher, P.G. Conjunctival colonization of infants hospitalized in a neonatal intensive care unit: a longitudinal analysis. Infect. Control Hosp. Epidemiol. 2004, 25, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Rampersaud, R.; Randis, T.M.; Ratner, A.J. Microbiota of the upper and lower genital tract. Semin. Fetal Neonatal Med. 2012, 17, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Biasucci, G.; Rubini, M.; Riboni, S.; Morelli, L.; Bessi, E.; Retetangos, C. Mode of delivery affects the bacterial community in the newborn gut. Early Hum. Dev. 2010, 86, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Malamitsi-Puchner, A.; Protonotariou, E.; Boutsikou, T.; Makrakis, E.; Sarandakou, A.; Creatsas, G. The influence of the mode of delivery on circulating cytokine concentrations in the perinatal period. Early Hum. Dev. 2005, 81, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Mulder, I.E.; Schmidt, B.; Lewis, M.; Delday, M.; Stokes, C.R.; Bailey, M.; Aminov, R.I.; Gill, B.P.; Pluske, J.R.; Mayer, C.D.; et al. Restricting microbial exposure in early life negates the immune benefits associated with gut colonization in environments of high microbial diversity. PLoS One 2011, 6, e28279. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S.U.N. Meeting brings attention but little action on chronic diseases. Science 2011, 333, 1561. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.E.; Cafiero, E.T.; Jané-Llopis, E.; Abrahams-Gessel, S.; Bloom, L.R.; Fathima, S.; Feigl, A.B.; Gaziano, T.; Mowafi, M.; Pandya, A.; et al. The Global Economic Burden of Noncommunicable Diseases; World Economic Forum: Geneva, Switzerland, 2011. [Google Scholar]
- Theodore, K. Chronic non-communicable diseases and the economy. West Indian Med. J. 2011, 60, 392–396. [Google Scholar] [PubMed]
- Thorpe, K.E.; Philyaw, M. The medicalization of chronic disease and costs. Annu. Rev. Public Health 2012, 33, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Stafford, N. Switzerland needs more GPs if it is to tackle growing burden of chronic disease. BMJ 2011, 343, d7057. [Google Scholar] [CrossRef] [PubMed]
- Dans, A.; Ng, N.; Varghese, C.; Tai, E.S.; Firestone, R.; Bonita, R. The rise of chronic non-communicable diseases in southeast Asia: time for action. Lancet 2011, 377, 680–689. [Google Scholar] [CrossRef]
- Parry, J. Chronic illness and disability are a hidden economic burden in Australia. Brit. Med. J. 2012, 344, e491. [Google Scholar] [CrossRef] [PubMed]
- Mayosi, B.M.; Flisher, A.J.; Lalloo, U.G.; Sitas, F.; Tollman, S.M.; Bradshaw, D. The burden of non-communicable diseases in South Africa. Lancet 2009, 374, 934–947. [Google Scholar] [CrossRef]
- Schmidt, M.I.; Duncan, B.B.; Azeved e Silva, G.; Menezes, A.M.; Monteiro, C.A.; Barreto, S.M.; Chor, D.; Menezes, P.R. Chronic non-communicable diseases in Brazil: burden and current challenges. Lancet 2011, 377, 1949–1961. [Google Scholar] [CrossRef]
- Stuckler, D.; Basu, S.; McKee, M. Commentary: UN high level meeting on non-communicable diseases: an opportunity for whom? Brit. Med. J. 2011, 343, d5336. [Google Scholar] [CrossRef] [PubMed]
- Mayes, R.; Oliver, T.R. Chronic disease and the shifting focus of public health: is prevention still a political lightweight? J. Health Polit. Policy Law 2012, 37, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, D. Why we don't spend enough on public health. N. Engl. J. Med. 2010, 362, 1657–1658. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, N.; Olden, K. Getting serious about the prevention of chronic diseases. Prev. Chronic Dis. 2011, 8, A90. [Google Scholar] [PubMed]
- Dietert, R.R. Role of developmental immunotoxicity and immune dysfunction in chronic disease and cancer. Reprod. Toxicol. 2011, 31, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; Luebke, R.W. Immunotoxicity, Immune Dysfunction, and Chronic Disease; Humana Press: New York, NY, USA, 2012. [Google Scholar]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Main, K.M. Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Dalgaard, M.D.; Weinhold, N.; Edsgard, D.; Silver, J.D.; Pers, T.H.; Nielsen, J.E.; Jorgensen, N.; Juul, A.; Gerds, T.A.; Giwercman, A.; Giwercman, Y.L.; et al. A genome-wide association study of men with symptoms of testicular dysgenesis syndrome and its network biology interpretation. J. Med. Genet. 2012, 49, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Balkau, B.; Eschwege, E. The metabolic syndrome. Lancet 2005, 366, 192. [Google Scholar] [CrossRef]
- Dietert, R.R.; Zelikoff, J.T. Pediatric Immune Dysfunction and Health Risks Following Early-Life Immune Insult. Curr. Pediat. Rev. 2009, 5, 35–51. [Google Scholar] [CrossRef]
- Dietert, R.R.; Zelikoff, J.T. Identifying patterns of immune-related disease: use in disease prevention and management. World J. Pediatr. 2010, 6, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; deWitt, J.C.; Germolec, D.R.; Zelikoff, J.T. Breaking patterns of environmentally influenced disease for health risk reduction: immune perspectives. Environ. Health Perspect. 2010, 118, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P. Late insights into early origins of disease. Basic Clin. Pharmacol. Toxicol. 2008, 102, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Halfon, N.; Verhoef, P.A.; Kuo, A.A. Childhood antecedents to adult cardiovascular disease. Pediatr. Rev. 2012, 33, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The fetal and infant origins of adult disease. Brit. Med. J. 1990, 301, 1111. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The intrauterine environment and adult cardiovascular disease. Ciba Found. Symp. 1991, 156, 3–10. [Google Scholar] [PubMed]
- Mu, M.; Wang, S.F.; Sheng, J.; Zhao, Y.; Li, H.Z.; Hu, C.L.; Tao, F.B. Birth weight and subsequent blood pressure: a meta-analysis. Arch. Cardiovasc. Dis. 2012, 105, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Regnault, T.R. In utero origins of adult insulin resistance and vascular dysfunction. Semin. Reprod. Med. 2011, 29, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Portha, B.; Chavey, A.; Movassat, J. Early-life origins of type 2 diabetes: fetal programming of the beta-cell mass. Exp. Diabetes Res. 2011, 2011, 105076. [Google Scholar] [CrossRef] [PubMed]
- Leduc, L.; Levy, E.; Bouity-Voubou, M.; Delvin, E. Fetal programming of atherosclerosis: possible role of the mitochondria. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 149, 127–130. [Google Scholar] [CrossRef] [PubMed]
- White, S.L.; Perkovic, V.; Cass, A.; Chang, C.L.; Poulter, N.R.; Spector, T.; Haysom, L.; Craig, J.C.; Salmi, I.A.; Chadban, S.J.; et al. Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Am. J. Kidney Dis. 2009, 54, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Kelishadi, R. Inflammation-induced atherosclerosis as a target for prevention of cardiovascular diseases from early life. Open Cardiovasc. Med. J. 2010, 4, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Bethell, C.D.; Kogan, M.D.; Strickland, B.B.; Schor, E.L.; Robertson, J.; Newacheck, P.W. A national and state profile of leading health problems and health care quality for US children: key insurance disparities and across-state variations. Acad. Pediatr. 2011, 11, S22–S33. [Google Scholar] [CrossRef] [PubMed]
- Rappange, D.R.; Brouwer, W.B.; Hoogenveen, R.T.; van Baal, P.H. Healthcare costs and obesity prevention: drug costs and other sector-specific consequences. Pharmacoeconomics 2009, 27, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Asche, C.V.; Singer, M.E.; Jhaveri, M.; Chung, H.; Miller, A. All-cause health care utilization and costs associated with newly diagnosed multiple sclerosis in the United States. J. Manag. Care Pharm. 2010, 16, 703–712. [Google Scholar] [PubMed]
- Rius, B.; Lopez-Vicario, C.; Gonzalez-Periz, A.; Moran-Salvador, E.; Garcia-Alonso, V.; Claria, J.; Titos, E. Resolution of inflammation in obesity-induced liver disease. Front. Immunol. 2012, 3, 257. [Google Scholar] [CrossRef] [PubMed]
- Despres, J.P. Abdominal Obesity and Cardiovascular Disease: Is Inflammation the Missing Link? Can. J. Cardiol. 2012. [Google Scholar] [CrossRef] [PubMed]
- De Heredia, F.P.; Gomez-Martinez, S.; Marcos, A. Obesity, inflammation and the immune system. Proc. Nutr. Soc. 2012, 71, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Angata, T.; Fujinawa, R.; Kurimoto, A.; Nakajima, K.; Kato, M.; Takamatsu, S.; Korekane, H.; Gao, C.X.; Ohtsubo, K.; Kitazume, S.; et al. Integrated approach toward the discovery of glyco-biomarkers of inflammation-related diseases. Ann. N. Y. Acad. Sci. 2012, 1253, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Sung, B.; Aggarwal, B.B. Age-associated chronic diseases require age-old medicine: role of chronic inflammation. Prev. Med. 2012, 54, S29–S37. [Google Scholar] [CrossRef] [PubMed]
- Aller, M.A.; Arias, N.; Fuentes-Julian, S.; Blazquez-Martinez, A.; Argudo, S.; Miguel, M.P.; Arias, J.L.; Arias, J. Coupling inflammation with evo-devo. Med. Hypotheses 2012, 78, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Renz, H.; Autenrieth, I.B.; Brandtzaeg, P.; Cookson, W.O.; Holgate, S.; von Mutius, E.; Valenta, R.; Haller, D. Gene-environment interaction in chronic disease: a European Science Foundation Forward Look. J. Allergy Clin. Immunol. 2011, 128, S27–S49. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson's disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef] [PubMed]
- Manabe, I. Chronic inflammation links cardiovascular, metabolic and renal diseases. Circ. J. 2011, 75, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.B.; Storkus, W.J. Chronic inflammation and immunologic-based constraints in malignant disease. Immunotherapy 2011, 3, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Shanmugam, M.K.; Ramachandran, L.; Kumar, A.P.; Tergaonkar, V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012, 32, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Butler, J. From risk factors to structural heart disease: the role of inflammation. Heart Fail. Clin. 2012, 8, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Atherosclerosis as an Inflammatory Disease. Curr. Pharm. Des. 2012. [Google Scholar] [CrossRef]
- Straub, R.H. Evolutionary medicine and chronic inflammatory state-known and new concepts in pathophysiology. J .Mol. Med. (Berl.) 2012, 90, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Liu, J.; Geletka, L.; Delaney, C.; Delproposto, J.; Desai, A.; Oatmen, K.; Martinez-Santibanez, G.; Julius, A.; Garg, S.; et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J. Immunol. 2011, 187, 6208–6216. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Sun, L.; Klein, B.E.; Lee, K.E.; Muntner, P.; Nieto, F.J.; Tsai, M.Y.; Cruickshanks, K.J.; Schubert, C.R.; Brazy, P.C.; et al. Markers of inflammation predict the long-term risk of developing chronic kidney disease: a population-based cohort study. Kidney Int. 2011, 80, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Carrero, J.J.; Stenvinkel, P. Inflammation as a risk factor and target for therapy in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H. Resolvins as new fascinating drug candidates for inflammatory diseases. Arch. Pharm. Res. 2012, 35, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Gressens, P.; Mallard, C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 2012, 71, 444–4457. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, F.; Shafique, K.; Mirza, S.S.; Ayoob, Z.; Vart, P.; Rao, S. Neutrophil lymphocyte ratio as a measure of systemic inflammation in prevalent chronic diseases in Asian population. Int. Arch. Med. 2012, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Netzer, N.; Goodenbour, J.M.; David, A.; Dittmar, K.A.; Jones, R.B.; Schneider, J.R.; Boone, D.; Eves, E.M.; Rosner, M.R.; Gibbs, J.S.; et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 2009, 462, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Mechtcheriakova, D.; Svoboda, M.; Meshcheryakova, A.; Jensen-Jarolim, E. Activation-induced cytidine deaminase (AID) linking immunity, chronic inflammation, and cancer. Cancer Immunol. Immunother. 2012, 61, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Marusawa, H.; Ushijima, T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 2012, 143, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Rau, T.T.; Rogler, A.; Frischauf, M.; Jung, A.; Konturek, P.C.; Dimmler, A.; Faller, G.; Sehnert, B.; El-Rifai, W.; Hartmann, A.; et al. Methylation-Dependent Activation of CDX1 through NF-kappaB: A Link from Inflammation to Intestinal Metaplasia in the Human Stomach. Am. J. Pathol. 2012, 181, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Goel, A. Genetic and epigenetic signatures in human hepatocellular carcinoma: a systematic review. Curr. Genomics 2011, 12, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.; Larsen, G.; Manikkam, M.; Guerrero-Bosagna, C.; Savenkova, M.I.; Skinner, M.K. Environmentally induced epigenetic transgenerational inheritance of ovarian disease. PLoS One 2012, 7, e36129. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.T.; Ram, S.; Kuo, A.; Gilbert, J.L.; Yan, W.; Pellegrini, M.; Nickerson, D.W.; Chatila, T.A.; Gomperts, B.N. Identification of an interleukin 13-induced epigenetic signature in allergic airway inflammation. Am. J. Transl. Res. 2012, 4, 219–228. [Google Scholar] [PubMed]
- Yao, H.; Rahman, I. Role of histone deacetylase 2 in epigenetics and cellular senescence: implications in lung inflammaging and COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C.; Reedquist, K.A.; Gay, S.; Tak, P.P. Inflammatory memories: is epigenetics the missing link to persistent stromal cell activation in rheumatoid arthritis? Autoimmun. Rev. 2011, 10, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R. Developmental immunotoxicology: focus on health risks. Chem. Res. Toxicol. 2009, 22, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Luebke, R.W.; Chen, D.H.; Dietert, R.; Yang, Y.; King, M.; Luster, M.I. The comparative immunotoxicity of five selected compounds following developmental or adult exposure. J. Toxicol. Environ. Health B Crit. Rev. 2006, 9, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Tonk, E.C.; Verhoef, A.; Gremmer, E.R.; van Loveren, H.; Piersma, A.H. Relative sensitivity of developmental and immune parameters in juvenile versus adult male rats after exposure to di(2-ethylhexyl) phthalate. Toxicol. Appl. Pharmacol. 2012, 260, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Tonk, E.C.; Verhoef, A.; de la Fonteyne, L.J.; Waalkens-Berendsen, I.D.; Wolterbeek, A.P.; van Loveren, H.; Piersma, A.H. Developmental immunotoxicity in male rats after juvenile exposure to di-n-octyltin dichloride (DOTC). Reprod. Toxicol. 2011, 32, 341–348. [Google Scholar] [CrossRef] [PubMed]
- US Environmental Protection Agency. Integrated Risk Information System (IRIS): Trichloroethylene (CASRN 79-01-6); US Environmental Protection Agency: Washington, DC, 2011. Available online: http://www.epa.gov/iris/subst/0199.htm accessed on 24 October 2012.
- Grandjean, P.; Poulsen, L.K.; Heilmann, C.; Steuerwald, U.; Weihe, P. Allergy and sensitization during childhood associated with prenatal and lactational exposure to marine pollutants. Environ. Health Perspect. 2010, 118, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Hoppin, J.A.; Umbach, D.M.; London, S.J.; Henneberger, P.K.; Kullman, G.J.; Alavanja, M.C.; Sandler, D.P. Pesticides and atopic and nonatopic asthma among farm women in the Agricultural Health Study. Am. J. Respir. Crit. Care Med. 2008, 177, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; Zelikoff, J.T. Early-life environment, developmental immunotoxicology, and the risk of pediatric allergic disease including asthma. Birth Defects Res. B Dev. Reprod. Toxicol. 2008, 83, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; Piepenbrink, M.S. Perinatal immunotoxicity: why adult exposure assessment fails to predict risk. Environ. Health Perspect. 2006, 114, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Bunn, T.L.; Parsons, P.J.; Kao, E.; Dietert, R.R. Gender-based profiles of developmental immunotoxicity to lead in the rat: assessment in juveniles and adults. J. Toxicol. Environ. Health Part A 2001, 64, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Leifer, C.A.; Dietert, R.R. Early life environment and developmental immunotoxicity in inflammatory dysfunction and disease. Toxicol. Environ. Chem. 2011, 93, 1463–1485. [Google Scholar] [CrossRef] [PubMed]
- Kasten-Jolly, J.; Heo, Y.; Lawrence, D.A. Impact of developmental lead exposure on splenic factors. Toxicol. Appl. Pharmacol. 2010, 247, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Hogaboam, J.P.; Moore, A.J.; Lawrence, B.P. The aryl hydrocarbon receptor affects distinct tissue compartments during ontogeny of the immune system. Toxicol. Sci. 2008, 102, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R. Misregulated Inflammation as an Outcome of Early-Life Exposure to Endocrine Disrupting Chemicals. Rev. Environ. Health 2012, 27, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R. Distinguishing Environmental Causes of Immune Dysfunction from Pediatric Triggers of Disease. The Open Pediatr. Med. J. 2009, 3, 38–44. [Google Scholar] [CrossRef]
- Deleidi, M.; Isacson, O. Viral and inflammatory triggers of neurodegenerative diseases. Sci. Transl. Med. 2012, 4, 121ps3. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, M.F.; Walker, W.A. Neonatal microbial flora and disease outcome. Nestle Nutr. Workshop Ser. Pediatr. Program 2008, 61, 211–224. [Google Scholar] [PubMed]
- Gravitz, L. Microbiome: The critters within. Nature 2012, 485, S12–S13. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.L.; Shi, H.N.; Walker, W.A. The role of microbes in developmental immunologic programming. Pediatr. Res. 2011, 69, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Renz, H.; Brandtzaeg, P.; Hornef, M. The impact of perinatal immune development on mucosal homeostasis and chronic inflammation. Nat. Rev. Immunol. 2011, 12, 9–23. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Kamanova, J.; Cinova, J.; Olivares, M.; Drasarova, H.; Tuckova, L.; Sanz, Y. Modulation of phenotypic and functional maturation of dendritic cells by intestinal bacteria and gliadin: relevance for celiac disease. J. Leukoc. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, H.; Li, N.; Bonnelykke, K.; Chawes, B.L.; Skov, T.; Paludan-Muller, G.; Stokholm, J.; Smith, B.; Krogfelt, K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011, 128, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.O.; Newson, R.B.; Smith, G.D.; Henderson, A.J. Prenatal paracetamol exposure and asthma: further evidence against confounding. Int. J. Epidemiol. 2010, 39, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Jedrychowski, W.; Galas, A.; Whyatt, R.; Perera, F. The prenatal use of antibiotics and the development of allergic disease in one year old infants. A preliminary study. Int. J. Occup. Med. Environ. Health 2006, 19, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Siracusa, M.C.; Abt, M.C.; Kim, B.S.; Kobuley, D.; Kubo, M.; Kambayashi, T.; Larosa, D.F.; Renner, E.D.; Orange, J.S.; et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat. Med. 2012, 18, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Probst-Hensch, N.; Tanner, M.; Kessler, C.; Burri, C.; Kunzli, N. Prevention-a cost-effective way to fight the non-communicable disease epidemic: an academic perspective of the United Nations High-level NCD Meeting. Swiss Med. Wkly. 2011, 141, w13266. [Google Scholar] [CrossRef] [PubMed]
- Sears, M.E.; Genuis, S.J. Environmental determinants of chronic disease and medical approaches: recognition, avoidance, supportive therapy, and detoxification. J. Environ. Public Health 2012, 2012, 356798. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Zhong, Y.; Wheeler, L. Direct and indirect costs of asthma in school-age children. Prev. Chronic Dis. 2005, 2, A11. [Google Scholar] [PubMed]
- Szefler, S.J.; Zeiger, R.S.; Haselkorn, T.; Mink, D.R.; Kamath, T.V.; Fish, J.E.; Chipps, B.E. Economic burden of impairment in children with severe or difficult-to-treat asthma. Ann. Allergy Asthma Immunol. 2011, 107, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leslie, D.L. Health care expenditures for children with autism spectrum disorders in Medicaid. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 1165–1171. [Google Scholar] [PubMed]
- Dionne, J.M.; Lou, K.; Er, L.; Collin, K.; White, C.T. Pharmaceutical cost distribution in childhood chronic kidney disease. Pediatr. Nephrol. 2012, 27, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Lion, M.; Gearry, R.B.; Day, A.S.; Eglinton, T. The cost of paediatric and perianal Crohn's disease in Canterbury, New Zealand. N. Z. Med. J. 2012, 125, 11–20. [Google Scholar] [PubMed]
- Patel, D.A.; Holdford, D.A.; Edwards, E.; Carroll, N.V. Estimating the economic burden of food-induced allergic reactions and anaphylaxis in the United States. J. Allergy Clin. Immunol. 2011, 128, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Belay, E.D.; Holman, R.C.; Maddox, R.A.; Foster, D.A.; Schonberger, L.B. Kawasaki syndrome hospitalizations and associated costs in the United States. Public Health Rep. 2003, 118, 464–469. [Google Scholar] [CrossRef]
- Minden, K.; Niewerth, M.; Listing, J.; Biedermann, T.; Schontube, M.; Zink, A. Burden and cost of illness in patients with juvenile idiopathic arthritis. Ann. Rheum. Dis. 2004, 63, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Ying, A.K.; Lairson, D.R.; Giardino, A.P.; Bondy, M.L.; Zaheer, I.; Haymond, M.W.; Heptulla, R.A. Predictors of direct costs of diabetes care in pediatric patients with type 1 diabetes. Pediatr. Diabetes 2011, 12, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; McGeer, P.L. Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J. Alzheimers Dis. 2008, 13, 359–69. [Google Scholar] [PubMed]
- Bynoe, M.S.; Viret, C. Multiple Sclerosis, Alzheimer’s Disease, and Inflammation: A Hypothetical View. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 215–252. [Google Scholar]
- Fiala, M.; Liu, P.T.; Espinosa-Jeffrey, A.; Rosenthal, M.J.; Bernard, G.; Ringman, J.M.; Sayre, J.; Zhang, L.; Zaghi, J.; Dejbakhsh, S.; et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer's disease patients are improved by bisdemethoxycurcumin. Proc. Natl. Acad. Sci. USA 2007, 104, 12849–12854. [Google Scholar] [CrossRef] [PubMed]
- Roponen, M.; Yerkovich, S.T.; Hollams, E.; Sly, P.D.; Holt, P.G.; Upham, J.W. Toll-like receptor 7 function is reduced in adolescents with asthma. Eur. Respir. J. 2010, 35, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, D.M.; Williams, M.A. Asthma and Respiratory Allergic Disease. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 51–101. [Google Scholar]
- Ariel, D.; Upadhyay, D. The role and regulation of microRNAs in asthma. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Yu, X.; Gao, P.; Wang, Y.; Baek, S.H.; Chen, X.; Kim, H.L.; Subjeck, J.R.; Wang, X.Y. Pattern recognition scavenger receptor SRA/CD204 down-regulates Toll-like receptor 4 signaling-dependent CD8 T-cell activation. Blood 2009, 113, 5819–5828. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D. Atherosclerosis and Inflammatory Heart Disease. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 271–289. [Google Scholar]
- Careaga, M.; Van de Water, J.; Ashwood, P. Immune Dysfunction in Autism Spectrum Disorders. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 253–269. [Google Scholar]
- Beamer, C.A.; Seaver, B.P.; Shepherd, D.M. COPD and Other Inflammatory Diseases of the Lung: Focus on AhR Signaling. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 313–343. [Google Scholar]
- Langereis, J.D.; Schweizer, R.C.; Lammers, J. W.; Koenderman, L.; Ulfman, L.H. A unique protein profile of peripheral neutrophils from COPD patients does not reflect cytokine-induced protein profiles of neutrophils in vitro. BMC Pulm. Med. 2011, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Anders, S.; Kinney, D.K. Environment, the Immune System, and Depression: An Integrative Review and Discussion of the Infection-Defense Hypothesis. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 345–385. [Google Scholar]
- O'Brien, S.M.; Scott, L.V.; Dinan, T.G. Cytokines: abnormalities in major depression and implications for pharmacological treatment. Hum. Psychopharmacol. 2004, 19, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C. Food Allergies. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 127–149. [Google Scholar]
- Ganeshan, K.; Neilsen, C.V.; Hadsaitong, A.; Schleimer, R.P.; Luo, X.; Bryce, P.J. Impairing oral tolerance promotes allergy and anaphylaxis: a new murine food allergy model. J. Allergy Clin. Immunol. 2009, 123, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Wang, S.; Xiao, Y.; Xu, J.; Jiang, Q.; Jin, M.; Jiang, X.; Guo, H.; Ning, G.; Zhang, Y. Impairment of regulatory capacity of CD4+CD25+ regulatory T cells mediated by dendritic cell polarization and hyperthyroidism in Graves' disease. J. Immunol. 2011, 186, 4734–4743. [Google Scholar] [CrossRef] [PubMed]
- Kucharska, A.M.; Gorska, E.; Wasik, M.; Pyrzak, B. Decreased CD4+CD152+ T cell subset and its correlation with the level of antithyroid antibodies in children with chronic autoimmune thyroiditis. Eur. J. Med. Res. 2010, 15, 72–75. [Google Scholar] [PubMed]
- Bansal, A.S.; Bradley, A.S.; Bishop, K.N.; Kiani-Alikhan, S.; Ford, B. Chronic fatigue syndrome, the immune system and viral infection. Brain Behav. Immun. 2012, 26, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; Dietert, J.M. Possible role for early-life immune insult including developmental immunotoxicity in chronic fatigue syndrome (CFS) or myalgic encephalomyelitis (ME). Toxicology 2008, 247, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Twisk, F.N.; Kubera, M.; Ringel, K. Evidence for inflammation and activation of cell-mediated immunity in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): increased interleukin-1, tumor necrosis factor-alpha, PMN-elastase, lysozyme and neopterin. J. Affect. Disord. 2012, 136, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Astier, A.L. T-cell regulation by CD46 and its relevance in multiple sclerosis. Immunology 2008, 124, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Kishimoto, T. IL-6: regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; Mueller, C.; Chae, W.J.; Alabanza, L.M.; Bynoe, M.S. Neuropilin-1 attenuates autoreactivity in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Garnero, P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol. Diagn. Ther. 2008, 12, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Siegel, E.R.; Achenbach, S.J.; Khosla, S.; Suva, L.J. Serum biomarker profile associated with high bone turnover and BMD in postmenopausal women. J. Bone Miner. Res. 2008, 23, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Marek, K.; Ross, G.W.; Poewe, W. Defining at-risk populations for Parkinson's disease: lessons from ongoing studies. Mov. Disord. 2012, 27, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, H.; Dinh, H.; Dean, C., Jr.; Trueblood, E.S.; Bailey, K.; Shows, D.; Bhagavathula, N.; Aslam, M.N.; Varani, J.; Towne, J.E.; Sims, J.E. IL-1RL2 and its ligands contribute to the cytokine network in psoriasis. J. Immunol. 2010, 185, 4354–4362. [Google Scholar] [CrossRef] [PubMed]
- Pfau, J.C. Rhematoid Arthritis. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 171–192. [Google Scholar]
- Kolly, L.; Busso, N.; Palmer, G.; Talabot-Ayer, D.; Chobaz, V.; So, A. Expression and function of the NALP3 inflammasome in rheumatoid synovium. Immunology 2010, 129, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; McDermott, M.F.; Kanneganti, T.D. Inflammasomes and autoimmunity. Trends Mol. Med. 2011, 17, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Zaba, L.C.; Smith, G.P.; Sanchez, M.; Prystowsky, S.D. Dendritic cells in the pathogenesis of sarcoidosis. Am. J. Respir. Cell. Mol. Biol. 2010, 42, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Nordmark, G.; Eloranta, M.L.; Ronnblom, L. Primary Sjogren's Syndrome and the Type I Interferon System. Curr. Pharm. Biotechnol. 2012, 13, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Norheim, K.B.; Jonsson, G.; Harboe, E.; Hanasand, M.; Goransson, L.; Omdal, R. Oxidative stress, as measured by protein oxidation, is increased in primary Sjogren's syndrome. Free Radic. Res. 2012, 46, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.S.; Motivala, S.; Olmstead, R.; Irwin, M.R. Sleep depth and fatigue: role of cellular inflammatory activation. Brain Behav. Immun. 2011, 25, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.V.; Jimenez, S.A. Biomarkers in systemic sclerosis. Biomark. Med. 2010, 4, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhao, M.; Lu, Q. Demethylation of promoter regulatory elements contributes to CD70 overexpression in CD4+ T cells from patients with subacute cutaneous lupus erythematosus. Clin. Exp. Dermatol. 2010, 35, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xiao, R.; Lian, X.; Kanekura, T.; Luo, Y.; Yin, Y.; Zhang, G.; Yang, Y.; Wang, Y.; Zhao, M.; et al. Demethylation of TNFSF7 contributes to CD70 overexpression in CD4+ T cells from patients with systemic sclerosis. Clin. Immunol. 2012, 143, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Cooke, A.; Zacone, P. Factors Involved in Onset of Type 1 Diabetes. In Immunotoxicity, Immune Dysfunction, and Chronic Disease; Dietert, R.R., Luebke, R.W., Eds.; Humana: New York, NY, USA, 2012; pp. 153–170. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dietert, R.; Dietert, J. The Completed Self: An Immunological View of the Human-Microbiome Superorganism and Risk of Chronic Diseases. Entropy 2012, 14, 2036-2065. https://doi.org/10.3390/e14112036
Dietert R, Dietert J. The Completed Self: An Immunological View of the Human-Microbiome Superorganism and Risk of Chronic Diseases. Entropy. 2012; 14(11):2036-2065. https://doi.org/10.3390/e14112036
Chicago/Turabian StyleDietert, Rodney, and Janice Dietert. 2012. "The Completed Self: An Immunological View of the Human-Microbiome Superorganism and Risk of Chronic Diseases" Entropy 14, no. 11: 2036-2065. https://doi.org/10.3390/e14112036
APA StyleDietert, R., & Dietert, J. (2012). The Completed Self: An Immunological View of the Human-Microbiome Superorganism and Risk of Chronic Diseases. Entropy, 14(11), 2036-2065. https://doi.org/10.3390/e14112036