1. Introduction
In recent years we have developed a new simulation method for calculating the absolute entropy,
S and the absolute Helmholtz free energy,
F called the hypothetical scanning Monte Carlo (HSMC) (or HSMD), where molecular dynamics (MD) is used [
1,
2,
3,
4,
5,
6,
7]. HSMC(D) has been applied to systems of increasing complexity, where the most recent ones have been mobile loops in proteins [
8,
9,
10]. Typically, a mobile loop has an “open” conformation in the unbound protein, which is changed to a “closed” conformation upon ligand binding,
i.e., the loop becomes a “lid” which covers the bound ligand. In these studies the main focus has been on entropy and free energy differences, Δ
Floop between these two loop conformations in the bound and free proteins, rather than on the absolute values themselves. (As discussed later, Δ
Floop not only sheds light on the mechanism of ligand binding, but in some cases it is an (ignored) component of the absolute free energy of binding.) In the present work we develop HSMD further, extending its applicability to more complex models, in particular to more complex models of loops, as explained below.
Before describing HSMC(D) and its planned enhancements in detail, it should be emphasized that calculation of
S and
F still constitutes a central problem in computer simulation, in spite of the significant progress achieved in the last 50 years [
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23]. This problem is in particular severe in structural biology due to the flexibility and strong long-range interactions characterizing bio-macromolecules such as proteins. More specifically, the potential energy surface of a protein,
E(
x) is rugged (
x is the 3
N-dimensional vector of the Cartesian coordinates of the molecule’s
N atoms),
i.e., this surface is “decorated” by a tremendous number of localized wells and “wider” ones, defined over regions, Ω
m (called microstates) each consisting of many localized wells. A microstate Ω
m, (e.g., the α-helical region of a peptide) which typically constitutes only a tiny part of the entire conformational space, Ω, can be represented by a sample (trajectory) generated by a local MD [
24,
25] simulation. A molecule will visit a localized well only for a very short time [several femtoseconds (fs)] while staying much longer within a microstate [
26,
27] which is thus of a greater physical significance (for further discussions about microstates and their problematic definition in simulations, see [
8,
9]).
Typically one is interested in calculating the free energy of the most stable microstates rather than calculating the total free energy (of Ω). Thus, flexible protein segments (e.g., sidechains and surface loops), cyclic peptides and ligands bound to proteins can populate significantly several Ωm in thermodynamic equilibrium, which should be identified and their populations, pm = exp[–Fm/kBT] calculated (kB is Boltzmann constant and T is the absolute temperature). In particular, identifying the global free energy minimum of a protein is the daunting task of protein folding.
As stated above, we are primarily interested in entropy and free energy differences, which are commonly calculated by thermodynamic integration (TI) techniques where the integration can be carried out over physical quantities such as the energy, temperature, and the specific heat, as well as over non-thermodynamic parameters [free energy perturbation (FEP), and histogram analysis methods are also included in this category] [
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23]. While TI is a robust and highly versatile approach, it has some limitations; for example, if one seeks to calculate Δ
Fm,n = ∫dF between two microstates
m and
n with significant structural variance the integration from
m to
n becomes difficult and for large molecules unfeasible. This drawback of TI can be overcome to a large extent by methods that provide the
absolute Fm (and
Sm)
from a given sample; thus, one is required to carry out (only) two
separate local MD simulations of microstates
m and
n, calculating directly the absolute
Fm and
Fn hence their difference Δ
Fmn =
Fm –
Fn, where the complex TI process is avoided. HSMC(D) (and other techniques [
28,
29,
30,
31] such as the quasi-harmonic approximation [
32,
33,
34]) enable one to calculate the absolute
F (and
S) (for a more extensive discussion on TI and other techniques for calculating differences, Δ
F (and Δ
S) see [
17]).
With HSMC(D) each conformation
i of a given MD or MC sample is reconstructed step-by-step (from nothing) using transition probabilities (TPs); the product of these TPs leads to an approximation for the correct Boltzmann probability
PiB from which various free energy functionals can be defined. While the TPs of HSMC(D) are stochastic in nature (calculated by MD or MC simulations), all the system interactions are taken into account; in this respect HSMC(D) can be viewed as exact [
3], where the only approximation involved is due to insufficient MC (MD) sampling for calculating the TPs. HSMC(D) has unique features: it provides rigorous lower and upper bounds for
F, which enable one to determine the accuracy from HSMD results alone without the need to know the correct answer. Furthermore,
F can be obtained from a very small sample and in principle even from any single conformation (e.g., see results for argon in [
3]).
The HSMC(D) methodology has been developed systematically, as applied to systems of increasing complexity. The initial (HSMC) calculations of liquid argon, TIP3P water [
3], self-avoiding walks [
5] and polyglycine molecules [
6] have verified the validity of the theoretical predictions stated above by comparisons with accurate results obtained by other well established techniques; a subsequent application of HSMD to peptides has led to a 100 times reduction in computer time [
7].
As mentioned earlier, HSMD has also been applied to mobile loops calculating Δ
Fm,n (and Δ
Sm,n) between the open and closed microstates. First, we treated the 7-residue mobile loop, 304–310 (Gly-His-Gly-Ala-Gly-Gly-Ser) of the enzyme porcine pancreatic α-amylase [
8,
9], where the system is modeled by the AMBER96 force field [
35] alone and by AMBER96 with the GB/SA implicit solvent of Still and coworkers [
36]. In this work only protein atoms close to the loop (the template) within a sphere of radius,
Rtmpl were considered, where they were kept fixed in their x-ray crystallography positions, while only the loop’s atoms were moved by MD. Later the implicit solvent was replaced by explicit solvent,
i.e., the α-amylase loop was capped with 70 TIP3P [
37] water molecules (within a sphere of radius
Rwater = 14 Å), which (together with the loop’s atoms) were moved in the MD simulation. Because the application of HSMD to water has not been optimized yet, the contribution of water to the free energy was calculated by a TI procedure incorporated within the framework of HSMD. This HSMD-TI process leads to a relatively small statistical error because only the water-loop interactions are involved, where their full values are gradually decreased to zero, for a fixed loop structure.
The number of water molecules used (70) is relatively small relying on a previous paper [
38] where the minimal number of water molecules,
required to cap a loop was studied.
was determined from a stability criterion based on the loop’s root mean square deviation (RMSD) from the Protein Data Bank (pdb) structure during 4 ns MD simulations. Surprisingly,
(which depends on the loop’s sequence and environment) was found to be relatively small on average 12 waters per residue, in accord with a study of Steinbach and Brooks [
39] For the loop of amylase, which consists of small residues,
= 70 seemed to be adequate. Clearly, these conclusions should further be tested and corroborated by free energy calculations.
A detailed such study was carried out in a following paper [
10] where HSMD-TI was applied to the loop 287–290 with the bulky residues, Ile, Phe, Arg, and Phe of acetylcholinesterase (AChE) from
Torpedo california; reaction of AChE with the inhibitor diisopropylphosphorofluoridate (DFP) leads to a displacement of the loop’s backbone roughly by 4 Å [
40,
41,
42,
43,
44,
45], and experimental evidence suggests that the free energy penalty for the loop displacement is on the order of 4 kcal/mol (
i.e., Δ
Floop =
Ffree –
Fbound ~ −4 kcal/mol) [
40,
42,
46]. Therefore AChE has been an ideal system for checking the performance of HSMD-TI, and in particular for examining the minimal number of water molecules needed to cap the loop. Thus we carried out two sets of calculations for systems of increasing size defined by
Rtmpl = 12 and
Rwater = 13 Å, and by
Rtmpl = 13 and
Rwater = 14 Å, for an increasing number of water molecules,
Nwater = 80, 100, 120, 180 and 80, 100, 120, …, 220, respectively. We have found that to recover the experimental free energy difference the water density should be close to that of bulk water and our results for the first set, Δ
Floop = −3.1 ± 2.5 and −3.6 ± 4 kcal/mol for a sphere containing 160 and 180 waters are equal within error bars to the experimental value [
10].
Because the crystal structures of the free (unbound) [
47] and bound [
40] proteins are similar, in [
10], (but also in our previous loop studies [
8,
9] and studies by others [
48,
49]) the template was “cut” from the crystal structure of the unbound protein, where the loop conformation in the bound protein was attached to the free template by superimposing the two crystal structures. Obviously, it would be more realistic to carry out the calculations with respect to two templates, which are cut separately from the free and bound x-ray structures. However, this would require defining an additional TI procedure where the water-template interactions are also gradually eliminated. This TI is expected to be computationally demanding since, unlike the original TI where (relatively small) loop-water interactions are considered, the number of template-water interacting atoms is relatively large increasing with the increase of the system size.
AChE is expected to be a suitable system for studying the effect of two different templates because the experimental Δ
Floop is known. Therefore, the loop of AChE is treated here again, where the simulations are carried out with respect to two different templates which are cut from the free and bound crystal structures; this requires applying the (relatively) elaborate HSMD-TI procedure mentioned above. While the basic theory is similar to that described in [
10], we derive it here in a new and shorter way for better clarity.
The foregoing (mainly technical) discussion did not touch on the relation of Δ
Floop to ligand binding. One question of interest in this area is whether the conformational change adopted by a mobile loop upon ligand binding is induced by the ligand (induced fit [
50,
51]) or alternatively, the free loop (in the apo protein) interconverts among different microstates, one of which is selected upon binding (selected fit [
52]). While some recent studies favor the latter mechanism, there is no general consensus, and the adopted mechanism is probably system dependent. The present study is not aimed at solving this problem, which would require to apply the longest simulation possible to the entire AChE protein soaked in explicit water, as done, for example, in [
53] for studying loop 6 of TIM. Notice, however, that in the case of induced fit Δ
Floop (in the apo protein) should be added to the total absolute free energy of binding of the protein-ligand complex. Thus, if the template remains unchanged (due to binding), calculation of Δ
Floop carried out in [
10] is expected to be adequate, whereas for a changed template the present calculation should be used. Indeed, in our recent study of the avidin-biotin complex Δ
Floop is taken into account (for the first time) in the calculation of the absolute free energy of binding [
54].
However, even if the behavior of the loop of AChE is of a selected fit type (i.e., the loop conformations in the apo protein also cover the structural region of the bound loop) the crystal structures and other experimental data suggest that the loop spends appreciable amount of time in its unbound microstate. Therefore, calculating ΔFloop, which measures the difference in stability between the two (possibly) meta-stable microstates is of interest. For this purpose, generating the longest possible MD trajectories might be a disadvantage because the loop is expected to escape from the original microstates during the simulations. Therefore, ΔFloop is calculated from relatively short trajectories (0.5 ns) where the loop remains in its original microstates (bound and free).
In this context it should be pointed out that determining the exact limits of a microstate in conformational space is practically impossible and therefore it is commonly defined by an MC or MD sample initiated from a microstate’s structure. Thus, the microstate’s size typically increases with simulation time,
t and estimation of
E,
S, and
F depend on
t as well; in previous publications we have developed procedures for checking that the system remains in its microstate during a simulation. Thus, in the case of two microstates one should verify that the differences, Δ
Eloop(
t), Δ
Sloop(
t) and Δ
Floop(
t) are stable during a long enough time,
t; for more details see [
8,
9].
Finally, while the differences Δ
Eloop, Δ
Sloop and Δ
Floop between two loop microstates might be small, the fluctuations in
E and
S increase as
N1/2, where
N is the number of atoms simulated. Therefore, simulating the entire protein might be prohibitive and to keep the fluctuations manageable the coordinates of the template in our calculations are held fixed. Notice, however, that this is not an inherent limitation as in our present application of HSMD-TI to the avidin-biotin complex [
54] part of the template is also moved in the MD simulation since the size of the fluctuations remains under control.
2. Theory and Methodology
2.1. The Loop and the Protein’s Template
As in [
10] we study the 4-residue loop, Ile, Phe, Arg, Phe, (287-290) of AChE in two microstates related to the free and bound loop structures. The starting point is the available crystal structures of AChE from the protein data bank (PDB), where the unbound (free) structure 2ace [
47] is based on residues 4 to 535 and 2dfp, the structure of AChE with DFP consists of residues 2 to 535 (the bound structure) [
40]. To be able to compare these structures 2dfp was trimmed so that both structures consist of the 531 residues, 4–535 and crystal water molecules were retained.
Moreover changing the bound structure also consisted in recovering the catalytic residue Serine (200) of this serine hydrolase (2ace) by remodeling the mono-isoprophyl-phosphoryl residue (MIS) (200) of the aged phosphorylated enzyme (2dfp). Remodeling consisted in removing the d-isopropyl-phosphoryl chain without changing the heavy atoms of the modified Serine residue and adding the corresponding hydrogen atoms.
A close analysis shows that these conformations differ overall by (all heavy atoms) root mean square deviation (RMSD) of 1.22 Å, while the loop conformations differ by RMSD = 1.38, 2.91, and 2.71 Å for the backbone, sidechains, and all loop heavy atoms, respectively. The RMSD of the templates, i.e., all atoms excluding the loop’s atoms, is 1.10 Å. It should be emphasized that the coordinates of the x-ray bound structure (2dfp) appear with significantly larger B-factors than those of the free structure (2ace).
Using the program TINKER [
55], and the AMBER force field [
35], where Lys, Arg, and His are positively charged and Glu and Asp have a negative charge. Hydrogens were added to the free and bound structures (including the crystal water) and the potential energy was minimized, with all heavy atoms restrained to their crystal positions by harmonic forces (with a force constant of 1 kcal/Å
2). Afterwards the loop and (TIP3P) water atoms were allowed to relax in the presence of a fixed template. These minimizations eliminate bad atomic overlaps and strains in the original structures, while keeping the atoms reasonably close to the PDB coordinates.
The relatively weak force constant (1 kcal/Å
2) (as compared to 100 kcal/Å
2 used in [
10] for 2ace) was chosen to allow more extensive changes in the bound structure due to its relatively large B-factors and the chemical change imposed on it by removing the phosphate group; correspondingly, larger structural changes were also allowed to occur in the free structure. To assess the change in the structure during minimization we list the various RMSD values (Å) in pairs, for the beginning and the end of the process: RMSD for all atoms, (0.98, 0.99), backbone atoms, (0.43, 0.41), sidechains, (1.20, 1.21), whole template, (0.85, 0.86), template backbone, (0.35, 032), template sidechains, (1.05, 1.06), whole loop (2.71, 2.87), loop backbone (1.39, 1.45), loop sidechains (2.92, 3.10). Thus, the strongest changes occurred for the loops.
Taking into account the whole protein (of 8,284 atoms) would be computationally prohibitive; therefore (as in [
8,
9,
10]), the template size is reduced to the
Ntmpl atoms closest to the loop, where the rest of the protein atoms are ignored. More specifically, the center of mass of the backbone atoms of the free loop (in the 2ace structure) is calculated as a (3D) reference point denoted
xcmb and a distance (
Rtmpl) is chosen. If the distance of any atom of a residue from
xcmb is less than
Rtmpl, the entire residue is included in the template; otherwise, the residue is eliminated. We use
Rtmpl = 12 Å which was found in [
10] to lead to a large enough template consisting of
Ntmpl = 944 atoms; however, for the present (somewhat different) structure
Ntmpl = 961, and exactly all these atoms were included in the template which was “cut” from the crystal structure of the bound protein (2dfp). We added to each template 33 crystal water molecules selected from those which are closest to the loop (30 in [
10]).
After defining the templates, the water molecules and the orientations of the polar hydrogens of the free and bound loops (on different templates) were subjected to an optimization procedure. This procedure (where all the loop’s heavy atoms are kept fixed), was carried out in several rounds, each consisting of a 0.1 ns MD run (1 fs time step) at high temperature (1,250 K) followed by energy minimization; the process is stopped after a fixed number of unsuccessful rounds (typically 100), namely rounds which do not reduce the energy further. Although the heavy atoms are fixed, considerable gain in potential energy is achieved. Next, keeping the template atoms fixed, the energy of each system was minimized where the loop and water molecules were allowed to move freely. Each of the final “free” and “bound” structures constitute starting points for a further analysis. The RMSD values between the final (optimized) loop structure and its crystal structure are relatively small. For the free loop they are 0.18, 0.45, and 0.42 Å, and for the bound loop, 0.27, 0.47, and 0.44 Å for the backbone, sidechains, and all loop atoms, respectively.
2.2. Addition of Water
While our considerations thus far are based on the crystal structures, we seek to simulate the loop in solution. Therefore, it is not clear whether the positions of the crystal waters are relevant for the solution environment. In particular, water molecules that are caged within the crystal structure are expected to stay there during the simulation, and thus can be considered as part of the template. Therefore, the number and arrangement of these waters should be globally optimized, practically by energy minimization, which, however, is a non-trivial problem (see below).
As in [
10], our strategy is to add more water molecules to the already existing crystallographic waters. Thus, we define a sphere centered at
xcmb with a radius,
Rwater (
Rwater =
Rtmpl + 1 Å) where waters are added at random to the hemisphere oriented towards the exterior of the template. To hold these waters around the loop they are restrained with a flat-welled half-harmonic potential (with a force constant of 10 kcal mol
-1Å
-2) based on their distance from
xcmb. That is, if the distance of a water oxygen from
xcmb is greater than
Rwater a harmonic restoring force is applied, otherwise the restraining force is zero.
Even though we have found in [
10] that for the above
Rtmpl and
Rwater,
Nwater should be 140–180, it is of interest to check this conclusion again for the present case of two different templates; therefore, we carry out calculations for
Nwater = 40, 60, 80, 100, 120, 140, and 160.
Each of the two fully hydrated systems is then treated by a sequence of MD/minimization procedure similar to that outlined above, applied only to the hydrogen atoms of the loop and
all water molecules restrained within the full sphere of radius
Rwater. Again, at the end of this optimization the energy is minimized where the loop’s atoms and the water molecules are free to move. The change in the loops’ conformations from their crystal structures measured by the RMSD are relatively small; for the free loop we obtain 0.31, 1.03, and 0.94 Å for backbone, side chains and all loop atoms, respectively, where the corresponding results for the bound loop are 0.37, 1.06, and 0.98 Å (see the discussion related to
Table 1).
For each of the optimized “free” and “bound” structures an MD run at 300 K is performed, where only the loop and water atoms are moved while the template atoms are kept fixed. Thus, 1,000 loop/water configurations are collected by retaining a configuration every 0.5 ps along the 0.5 ns MD trajectory. An equilibration run of 0.5 ns is carried out prior to the production run. The total potential energy
Etotal is the sum of partial energies related to the loop and water (the template-template energy is constant and thus is ignored):
where
Eloop-loop is the intra loop energy,
Eloop-tmpl is the energy due to loop-template interactions; these energies define the total loop energy
Eloop, and the interactions related to water are defined in a similar way, where their total is defined as
Ewater. The reconstruction of the loop structure is carried out in internal coordinates; therefore, the loop conformations simulated by MD are transferred from Cartesians to the dihedral angles φ
i, ψ
i, and ω
i (i = 1,
Nres = 4), the bond angles θ
i,
l (
i = 1,
Nres,
l = 1,3), the side chain angles χ, and the corresponding bond angles. For convenience, all these angles (ordered along the backbone) are denoted by α
k,
k = 1,37 =
K. We have argued in [
7,
8] that to a good approximation bond stretching can be ignored.
2.3. Statistical Mechanics of a Loop in Internal Coordinates
The theory of HSMD-TI has been developed in previous publications; therefore, the present description is shorter, providing mainly equations that are related directly to the calculations.
The reconstruction of the loop structure (of
Nres = 4 residues) is carried out in internal coordinates; therefore, the loop conformations simulated by MD are transferred from Cartesians to the dihedral angles φ
i, ψ
i, and ω
i (
i = 1,
Nres), the bond angles θ
i,
l (
i = 1,
Nres,
l = 1, 3), the side chain angles χ, and the corresponding bond angles. For convenience, all these angles (ordered along the backbone) are denoted by α
k,
k = 1,
K; We have argued in [
7,
8] that to a good approximation bond stretching can be ignored, thus the bond lengths are considered to be constant.
The partition function of the loop/water /template system is:
where
E(
xloop,
xN) =
Etotal is defined in Equation (1),
xloop is the Cartesian coordinates of the loop in microstate
m and
is the 9
N Cartesian coordinates of the water molecules;
Etotal also depends on the coordinates of the “frozen” template which are omitted for simplicity. For the same reason the letter
m will be omitted in most of the equations and
Nwater will be replaced in the theoretical section by
N (
N =
Nwater). After changing the variables of integration from
xloop to internal coordinates, the integral becomes a function of the
K dihedral and bond angles, α
k,
k = 1,
K and a Jacobian, cos(θ
i,
l) that depends (only) on each of the bond angles θ
i,
l [
27,
28,
32]:
where
and
. In Equation (3) we have omitted a factor, which depends on the bond lengths and is
assumed to be the same (
i.e., constant) for different microstates of the same loop and therefore does not affect entropy
differences (e.g., see discussion in [
10]). The Jacobian is also omitted for simplicity because we have shown [
8] that it cancels out (within the error bars) in entropy and free energy differences. The Boltzmann probability density corresponding to
Z [Equation (3)] is:
and the exact entropy,
S and exact free energy,
F (defined up to an additive constant) are:
and:
It should be noted that the fluctuation of the
exactF is zero [
56,
57], because by substituting
[Equation (4)] inside the curly brackets of Equation (6) one obtains,
,
i.e., the expression in the curly brackets is constant and equal to
F for any set
within
m. This means that the free energy can be obtained from
any single conformation if its Boltzmann probability density is known. [Notice, however, that calculation of
for a single conformation depends on the entire microstate as is also evident from the HSMC(D) procedure discussed later]. Still, the fluctuation of an approximate free energy (
i.e., which is based on an approximate probability density) is finite and is expected to decrease as the approximation improves [
3,
4,
5,
56,
57]. Because HSMC(D) provides an approximation for
, one can,
in principle, estimate the free energy of the system from
any single structure [
3,
4,
5]. This is the reason why in practice reliable HSMC(D) results for
F (but not necessarily for
S and
E) can be obtained from a relatively small sample.
2.4. Exact Future Scanning Procedure
It should first be pointed out that MC and MD are exact dynamical methods that enable one to sample system configuration i correctly with its Boltzmann probability, PiB, while the value of PiB is not provided (due to the dynamical character of these methods). [To simplify the discussion we use the discrete probability, PiB rather than the probability density defined in Equation (4).] Thus, properties such as the energy that are “written” on i can easily be calculated, while a direct calculation of the absolute S is difficult because lnPiB is unknown (it depend not only on i but on the entire ensemble through the partition function Z, which cannot be obtained from a finite sample).
Unlike the dynamical MC and MD, the exact future scanning method [that constitutes the basis of HSMC(D)] is a growth procedure which enables one (at least in principle) generating any system configuration (from nothing) by determining the positions of the atoms step-by-step with the help of transition probabilities (TPs); the product of these TPs leads to the value of PiB hence to S and F. Practically, a loop/water/template configuration would be generated by initially building a loop structure (in the presence of moving water) followed by the construction of a configuration of the surrounding water molecules (in the presence of a fixed loop conformation). In this way a sample of statistically independent system configurations can be obtained.
For simplicity this construction is described for a loop without side chains, consisting of M Gly residues, i.e., ordered along the chain are the 3M heavy atoms denoted k’ = 1…3M and the 6M dihedral and bond angles denoted αk, 1 ≤ αk ≤ 6M = K with values within microstate m; the loop is surrounded by Nwater water molecules moving within the volume defined by a sphere of radius, Rwater, the template, and the loop. We seek to generate a configuration of the entire system by first generating a loop conformation and then a configuration of the water molecules.
With the scanning procedure the position of the heavy atoms (
xloop) is determined step-by step. Thus, the position of the first atom
k’ = 1 is defined by the simultaneous determination of the first pair of dihedral and bond angles α
1, and α
2. The maximum range Δα
1Δα
2 which will keep the loop within
m is defined, and each of Δα
1 and Δα
2 is divided into
nb small bins (of sizes Δα
1/
nb and Δα
2/
nb) denoted
j1 and
j2,
j1 = 1…
nb,
j2 = 1…
nb, respectively. A long MD simulation of the whole system (loop + water) is carried out within microstate
m, where a conformation is retained every
l fs leading to a huge sample of size
n; then, the number of conformations n
j1j2 that visit simultaneously the (double) bin
j1j2 is calculated from which the corresponding TP is obtained,
pj1j2 =
nj1j2/
n (or ρ
j1j2 = [
nj1j2/
n]/[Δα
1Δα
2/
nb2]). A double bin is then selected by a random number according to the probabilities
pj1j2 which defines the position of atom
k’ = 1 (and its hydrogen or oxygen). The position of this atom is not changed in the next steps of the build-up process,
i.e., it becomes part of the “past”. The position of the second atom (
k’ = 2) is determined in the same manner from a long MD simulation of the future part of the system (
i.e., atoms
k’ = 2…3
M and water) where α
3 and α
4 are considered, bins Δα
3/
nb and Δα
4/
nb are defined, probabilities are calculated and a “lottery” (like above) determines the values of α
3 and α
4 which define the position of atom
k’ = 2; the process continues until the positions of all the loop’s atoms (and their hydrogens or oxygens) have been determined. A configuration of the
Nwater molecules is then determined in a similar way step-by-step in the presence of the fixed loop structure previously constructed (for details see [
3]). Obviously, the smaller are the bins the higher is the accuracy of the construction process, provided that the statistics is adequate,
i.e., that the (future) MD simulations are long enough; this stochastic scanning method becomes exact as the bin size → 0 (
nb → ∞) and
n→ ∞. Notice that in applications of the (deterministic) scanning method to lattice models, only part of the future has been considered, (
i.e., only
f steps ahead), where this part has been scanned completely; therefore, the corresponding TPs are approximate but deterministic (rather than stochastic), and accurate results were obtained by using an additional importance sampling procedure [
58].
This procedure can be described more formally as follows: at step
k’ (
k = 2
k’), the positions of
k’ − 1 atoms have already been determined (from the values of the corresponding
k − 2 angles α
1, …, α
k-2) and they are kept fixed (defining the “past”); α
k-1 and α
k (which will determine the position of atom
k’) are defined with the exact TP density
:
where
is a future partition function. The term “future” indicates that the integration defining
Zfuture is carried out over the positions of atoms
k’ =
k/2 + 1…
K/2 (which affect angles,
) and the 9
N coordinates
of the water molecules (which will be determined in future steps of the build-up process). Notice that this integration is carried out in a restrictive way where the corresponding conformations (of the loop) remain within microstate
m. Also, in this integration the atoms treated in the past (1…
k’ − 1) (which were determined by
) are held fixed in their coordinates. For simplicity the integrations below are written over the angles rather than the Cartesian coordinates (
xloop) of the loop atoms,
k’ =
k/2+1…
K/2. Thus:
where
E (Equation (1)) is the total potential energy of the loop/template/water system, which also imposes the loop closure condition. The product of the TPs (Equation (7)) leads to the (Boltzmann) probability density of the entire loop conformation,
. After the loop structure has been constructed a configuration of water molecules is generated step-by-step (in the presence of the constant loop structure where the product of the corresponding TPs leads to the probability density of the water configuration,
. The probability density
of the loop/water /template configuration is the product of
and
. One can define for
m “the loop entropy of mean force”,
:
where
Sloop is defined up to an additive constant. Extending the exact scanning procedure to side chains is straightforward, where again the position of a side chain atom is defined by two angles as described above.
However, implementation of the exact scanning procedure (as described prior to Equation (7)) for generating Boltzmann weighted configurations of a large loop/water/template system would be inefficient, due to the need to calculate (at each step) a large set of accurate TPs (
i.e., for an extremely large number of small bins); this would require extremely long (future) MD simulations. Also, with long simulations it is difficult to guarantee that the loop will remain in microstate
m (see discussion in [
8]). However, the exact scanning procedure provides the theoretical basis for HSMC(D). Thus, the exact scanning method is equivalent to any other exact simulation technique (in particular, to MC or MD) in the sense that large samples generated by such methods lead to the same averages and fluctuations (the sample does not carry a memory of the simulation method with which it has been generated). Therefore, one can assume that a given MC or MD sample has rather been generated by the exact scanning method, which enables one to reconstruct each conformation
i by calculating the TP densities that hypothetically were used to create it step-by-step. With HSMC(D) the efficiency problems discussed above for the exact (stochastic) scanning procedure are alleviated to a large extent since only a single TP is calculated at each step (rather than many TPs (
pj) required with the scanning method), as described below; also, because we are mainly interested in entropy differences, approximations (e.g., ignoring the Jacobians and bond stretching) can be applied without compromising the accuracy of the results.
2.5. Loop Reconstruction by HSMC(D)
The theory of HSMC(D) is described again for a loop consisting of
M Gly residues. Notice that while HSMD and the exact scanning method (described in the previous section) have some resemblance, they are different. With the scanning method a loop conformation is generated with
PiB whereas with HSMD an already generated structure (by MD) is reconstructed in order to obtain its probability. Thus, one starts by generating an MD sample of the loop in water in microstate
m; the configurations of this sample will be reconstructed by HSMD (application of HSMC is similar). First, the conformations are represented in terms of the dihedral and bond angles α
k, 1 ≤ α
k ≤ 6
M = K, and the variability range Δα
k is calculated:
where α
k(max) and α
k(min) are the maximum and minimum values of α
k found in the sample, respectively. Δα
k, α
k(max), and α
k(min) enable one to verify that the sample has not “escaped” from microstate
m. Notice that in the discussion below we define the loop conformation by the set of angles [α
k] rather than by the positions of the loop atoms,
xloop, which are determined by [α
k].
Each of the configurations (frames)
(denoted
i for brevity) of the sample is reconstructed in two stages, where the biotin structure is reconstructed first followed by the reconstruction of the water configuration
xN. Because the position of atom
k’ is defined by a dihedral and a bond angle one has to calculate their TP simultaneously. Thus, at step
k’ (
k = 2
k’) of stage 1
, the
k−2 angles
have already been reconstructed and the TP density of
,
, is calculated from an MD run, where the entire future of the loop and water is moved [
i.e., the loop’s atoms
k’,
k’ + 1,…
K/2 and their connected hydrogens, or oxygens and the water coordinates
xN] while the past (loop’s atoms 1,2,…,
k’−1 and their connected hydrogens or oxygens) are held fixed at their values in conformation
i. By considering a future conformation every 10 fs, a sample of size
nf is generated. Two small segments (bins) δα
k-1 and δα
k are centered at α
k-1(
i) and α
k(
i), respectively, and the number of simultaneous visits,
nvisit, of the future chain to these two bins during the simulation is calculated; one obtains (see
Figure 1):
where
becomes exact for very large
nf (
nf→∞) and very small bins (δα
k-1, δα
k→0). This means that in practice
will be somewhat approximate due to insufficient future sampling (finite
nf) and relatively large bin sizes. The same treatment can be applied to side chain atoms; however, notice that the sidechain of Arg is treated here approximately, where at each step three consecutive atoms are considered and their positions are defined only by the corresponding dihedral angles. In [
8] we have shown that δα
k and δα
k+1 can be optimized. Notice that with HSMD the future loop conformations generated by MD at each step
k‘ remain in general within the limits of
m, which is represented by the analyzed MD sample. The corresponding probability density related to the loop is:
defines an approximate entropy functional, denoted
, which can be shown (using Jensen’s inequality) to constitute a
rigorous upper bound for
(Equation (9)) [
3,
59]:
(Equation (9)) is the Boltzmann probability density of
in
m. (
is also known as the Gibbs’ inequality). Being an upper bound suggests that
will decrease as the approximation improves. In practice, however, this inequality is satisfied only if the probabilities are well defined (e.g., they are correctly normalized). This is pointed out because
is calculated stochastically and its error (and the error in
, the estimation of
, see Equation (14)) increases with increasing system size (
i.e., increasing the number of TPs; see later discussions in
3.3).
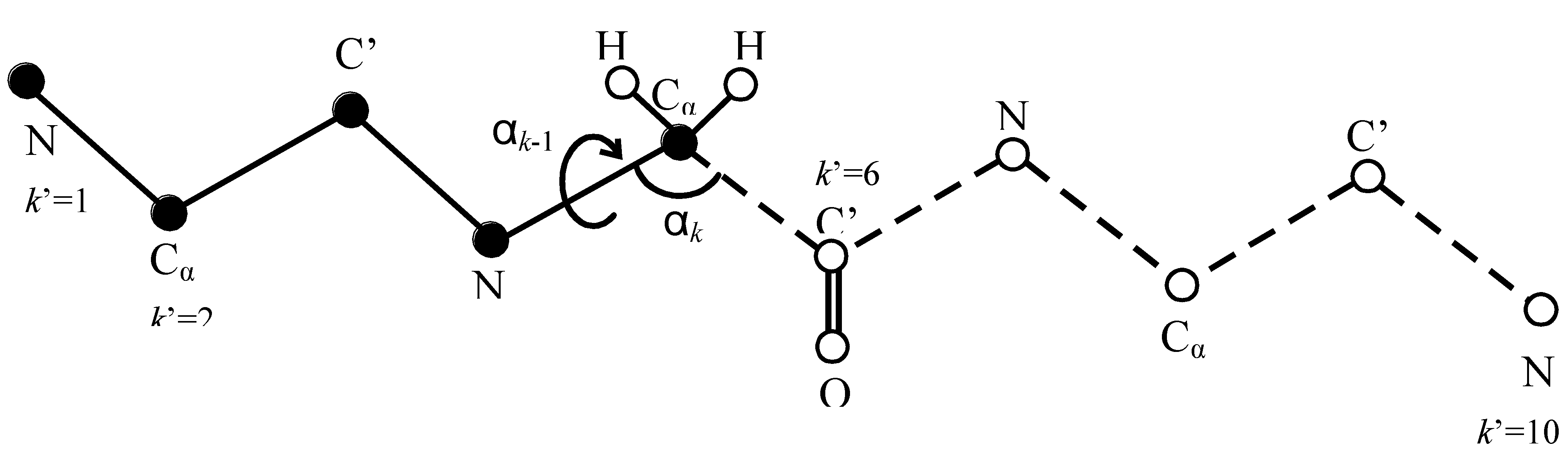
Figure 1.
An illustration of the HSMD reconstruction process of conformation i of a peptide consisting of three glycine residues, where for simplicity the oxygens and most of the hydrogens are discarded. At step k’ = 6 the TPs related to the “past” atoms k’ = 1…5 (depicted by full spheres) have already been determined and these atoms are kept fixed in their positions at conformation i. At this step (6) one calculates the TPs of bond angle αk (defined by C’-Cα-N) and dihedral angle αk−1 (defined by C’-Cα-N-C’) which are related to C’, where k = 2k’ and thus k = 12. The TPs are obtained from an MD simulation where the as yet unreconstructed atoms k’ = 6…10 (the “future” atoms) are moved (depicted by empty spheres connected by dashed lines) while the past atoms k’ = 1…5 are kept fixed; notice that the future part should remain within the limits of the microstate and future-past interactions are taken into account. Small bins δαk−1 and δαk are centered at the values αk−1 and αk in i. The TP is calculated from the number of visits (nvisit Equation 11) of the future part to δαk−1 and δαk simultaneously during the simulation. After (Equation 11) has been determined the coordinates of C’ (and O) are fixed at their positions in i, i.e., they become “past” atoms and the process continues.
Figure 1.
An illustration of the HSMD reconstruction process of conformation i of a peptide consisting of three glycine residues, where for simplicity the oxygens and most of the hydrogens are discarded. At step k’ = 6 the TPs related to the “past” atoms k’ = 1…5 (depicted by full spheres) have already been determined and these atoms are kept fixed in their positions at conformation i. At this step (6) one calculates the TPs of bond angle αk (defined by C’-Cα-N) and dihedral angle αk−1 (defined by C’-Cα-N-C’) which are related to C’, where k = 2k’ and thus k = 12. The TPs are obtained from an MD simulation where the as yet unreconstructed atoms k’ = 6…10 (the “future” atoms) are moved (depicted by empty spheres connected by dashed lines) while the past atoms k’ = 1…5 are kept fixed; notice that the future part should remain within the limits of the microstate and future-past interactions are taken into account. Small bins δαk−1 and δαk are centered at the values αk−1 and αk in i. The TP is calculated from the number of visits (nvisit Equation 11) of the future part to δαk−1 and δαk simultaneously during the simulation. After (Equation 11) has been determined the coordinates of C’ (and O) are fixed at their positions in i, i.e., they become “past” atoms and the process continues.
![Entropy 12 01946 g001]()
can be estimated (for microstate
m) from a Boltzmann sample (of size
ns) generated by MD using the arithmetic average:
where
is the value of
obtained for configuration
t of the sample of
m. (with the bar) is an estimation of the ensemble average
(Equation (13)); correspondingly, the ensemble averages of the energy are estimated from a sample of size
ns and should appear with a bar as well. However, from now on only estimations will be considered and for simplicity, all of them will appear without the bar, like the energies defined in Equation (1).
(Equations (13) and (14)) constitutes a measure of a pure geometrical character for the loop flexibility,
i.e., with no
direct dependence on the interaction energy. When the
converged or the
best value of
is considered, it will be denoted by
Sloop; thus,
Floop =
Eloop−TSloop is defined as the loop’s contribution to the total free energy, where
Eloop is defined in Equation (1). In the same way, the difference in the loop entropies between the free and bound microstates obtained for a specific set of parameters is denoted by
while the converged difference is denoted by
:
where:
One can define a free energy difference for the loop, Δ
Floop = Δ
Eloop−TΔ
Sloop, where
is obtained from Equation (1). Notice that (unlike in [
10]) Δ
Sloop and Δ
Floop are obtained for different templates; for simplicity their dependence on the templates is omitted.
2.6. Thermodynamic Integration of Water
To reconstruct the water configuration one can use, in principle, the HSMC(D) procedure for fluids mentioned previously [
3], which for a fixed loop structure, [α
k] would lead to
(as an approximation to
; see discussion preceding Equation (9)) and then to the contribution of the water configuration to the free energy
, which depends on the free or bound template (tmpl). However, this procedure for fluids has not been optimized yet and it is relatively time consuming.
Alternatively, as in [
9,
10], one can obtain
(defined up to an additive constant) by a thermodynamic integration (TI) procedure based on the same reference state for the free and bound structures (
i.e., the above constant is the same for both structures). In this state only the water-water interactions and the harmonic restraining force that keeps the waters in the spherical volume are preserved, while the water-template and water-loop interactions [electrostatic and Lennard Jones (LJ)] are switched off,
i.e., the fixed loop structure
and the fixed template do not “see” the surrounding waters. Thus, Δ
Fwater (our main interest) can be obtained by a TI procedure (applied to each system) where these interactions are gradually increased (from zero) during an MD simulation of water (while the loop structure and template remain fixed). (Notice that while the templates are structurally different, the template-template interactions are ignored, therefore the templates are considered to be in the same state.)
In practice, however, it is more convenient to carry out the integration in an opposite direction, starting from the current water configuration with full water-loop and water-template interactions, which are gradually decreased to zero. More specifically, the water-loop interactions are integrated (eliminated) first (in the presence of intact water-template interactions), which leads to
. The subsequent template–water integration (that depends only on the template) is denoted
, where
m stands for “free” or “bound”. Each of the free and bound integrations are carried out in two stages, where in the first stage the electrostatic interactions (
i.e., the charges) are decreased to zero followed by decreasing to zero the Lennard Jones (LJ) interactions, where the latter is performed by decreasing a parameter λ from 1 to zero (see
section 3.3 and
Appendix).
The water-loop integration is carried out
ns times for each of the loop structures
of the sample of size
ns. Denoting these
of the sample by
t and keeping for brevity the letter
m (“free” or “bound”) only on the left side of the equation, leads to:
Unlike the different
in the sample, the template remains constant and therefore, in principle, only a single integration is needed. However, to decrease the statistical fluctuations,
ns’ integrations (denoted by
t) are carried out, each starts from a different water configuration and a set of velocities (denoted
t) thus:
It should be pointed out again that the template-template energy is not considered in our model, therefore the two templates only differ by their geometry and the corresponding reference states are thus the same.
2.7. The Reconstruction Procedure with HSMD
The HSMD reconstruction procedure needs further discussions. Thus, the MD simulation of the future chain at step k’ starts from the reconstructed conformation i, and every g = 10 fs the current conformation is considered, where the ninit initial considered conformations are discarded for equilibration. For each of the next nf (considered) future conformations the pair (αk-1,αk) (k = 2k’) are calculated and their contribution to nvisit (Equation (11)) is calculated. An essential issue is how to guarantee an adequate coverage of microstate m, i.e., that the future chains will span its entire region (in particular the side chain rotamers) while avoiding their “overflow” to neighboring microstates, conditions that will occur for a too small and a too large nf, respectively. [Note that even at step k’, where the “past” of the loop (atoms 1…k’−1) is kept fixed, the (future) unfixed part (atoms k’, k’ + 1…) can leave the microstate during long MD simulations. Such an “overflow” is more likely to happen for small residues such as Gly and for small k’.]
In previous work [
6,
7,
8,
9] we developed procedures for keeping the loop in its original microstate and measures for estimating the extent of coverage of the microstate by the reconstructed samples. In this paper these procedures have not been applied because the maximal
nf values used are not large and the microstates are concentrated (
i.e., the Δα
k values of Equation (10) are relatively small; see discussions in
3.2).
4. Summary and Conclusions
This is an additional paper in a series of papers where our HSMD-TI method has been extended systematically for mobile loops in proteins. The method provides the absolute entropy and free energy and thus enables one calculating these quantities for microstates m and n with a large conformational variance without the need to carry out a complex thermodynamic integration from m to n. Mobile loops in enzymes can be crucial for the extent of binding, where their open conformation is moved to form a “lid” over the bound active site.
Thus far we have studied the 7-residue mobile loop, 304–310 (Gly-His-Gly-Ala-Gly-Gly-Ser) of the enzyme porcine pancreatic α-amylase, where the system was modeled by the AMBER96 force field alone and by AMBER96 with the GB/SA implicit solvent [
8]. Later the implicit solvent was replaced by explicit solvent,
i.e., the α-amylase loop was capped with 70 TIP3P [
9] water molecules. To determine the minimum size of the template and the minimum number of water molecules required to cap a loop, we have recently carried out a systematic study of the loop, 287–290 (Ile, Phe, Arg, and Phe) of AChE [
10]. The conclusion is that to recover the experimental free energy difference the water density should be close to that of bulk water, and the minimum template size is of 1,000 atoms for this loop. In all these studies and in a recent study of a mobile loop of the protein streptavidin [
62] the free and bound loop structures were attached to a common template that was “cut” from the crystal structure of the free loop. Obviously, one would seek to treat each loop attached to its own template. This has been the objective of the present paper.
As a first step of this study the heavy atoms of the two crystal structures were harmonically restrained (with a force constant of 1 kcal/Å
2) and the energy was minimized. Afterwards the loop and (TIP3P) water atoms were allowed to relax in the presence of a fixed template. The relatively weak force constant (1 kcal/Å
2) (as compared to 100 kcal/Å
2 used in [
10] for the pdb structure 2ace) was chosen to allow more extensive changes in the bound structure due to its relatively large B-factors and the chemical change imposed on it by removing the phosphate group; correspondingly, larger structural changes were also allowed to occur in the free structure. Then two cuts from each crystal structures were performed, which contain exactly the same atoms; these templates and the attached loops were subjected to further optimization steps as described earlier.
The new computational component in this study is the annihilation (for each template) of the water-template interactions using a TI procedure. Unlike the loop-water TI, one would expect the accuracy of the water–template TI to depend strongly on the system size. Thus, for Nwater ≤ 140 the errors are relatively small and the results for the free-bound differences in the total free energy and energy, ΔFsum and ΔEtotal are comparable, while for Nwater = 160 the errors become larger (in particular for the Lennard-Jones part of the water-template TI) and a significant increase in ΔFsum and ΔEtotal occurs. We attribute this jump in the results to changes in the configuration of water stemming from the increase in water pressure in the sphere as the number of waters grows.
Finally, calculation of ΔFloop based on separate templates which are cut from the free and bound crystal structures is more realistic than using a single common template for both loop conformations. ΔFloop should also be considered in the calculation of the absolute free energy of binding when the conformational change of the loop is due to ligand binding is of an induced fit type. Thus, while the present calculations show that recovering the experimental result, ΔFloop = Ffree − Fbound ~ −4 kcal/mol is not straightforward, to achieve improvement more research is needed, which will be based on the conclusions of the initial work carried out here, as specified in the summary of the previous section.


{kind=link}