
Combination of Cyclosporine A and Levosimendan Induces Cardioprotection under Acute Hyperglycemia

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Animal Characteristics
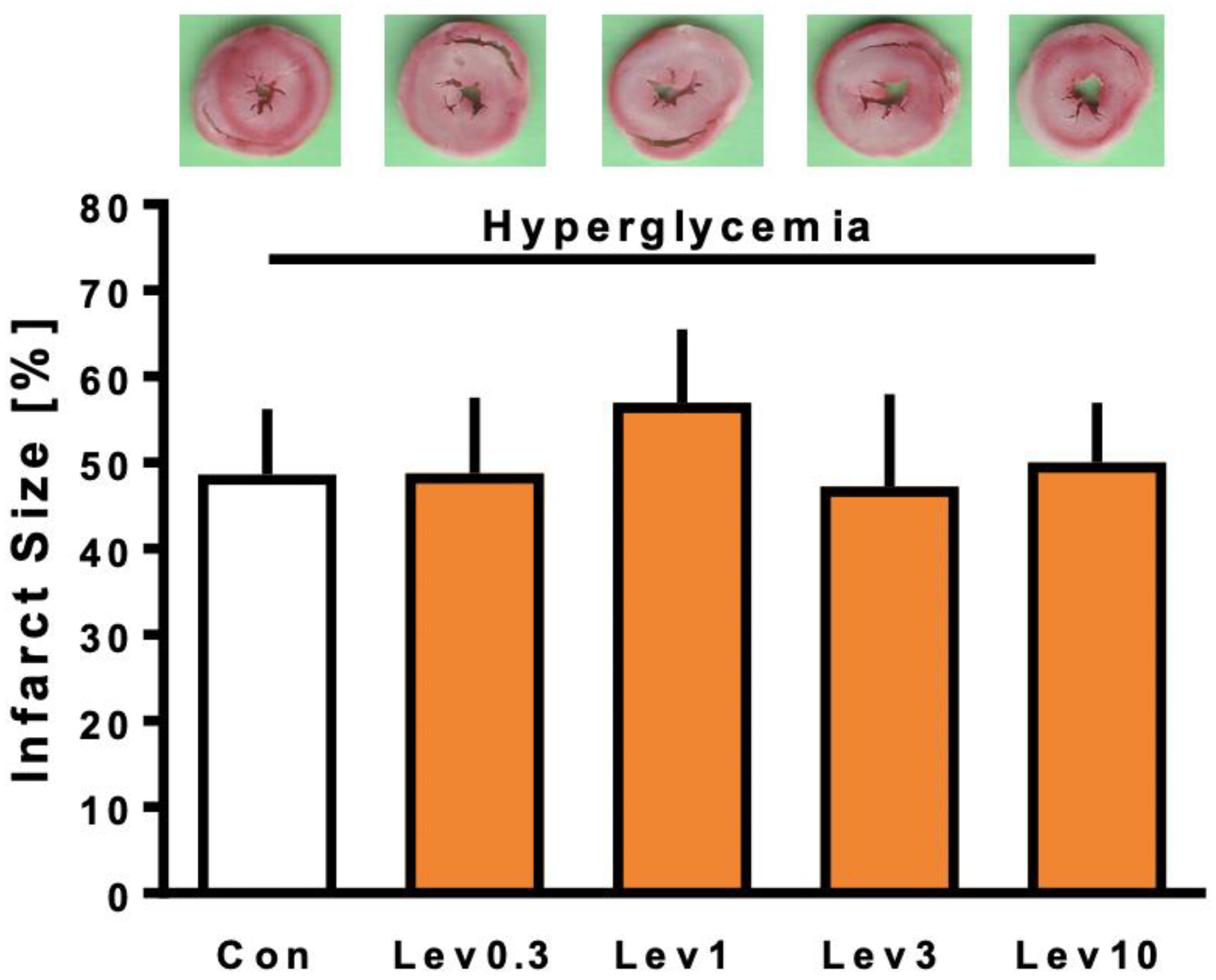
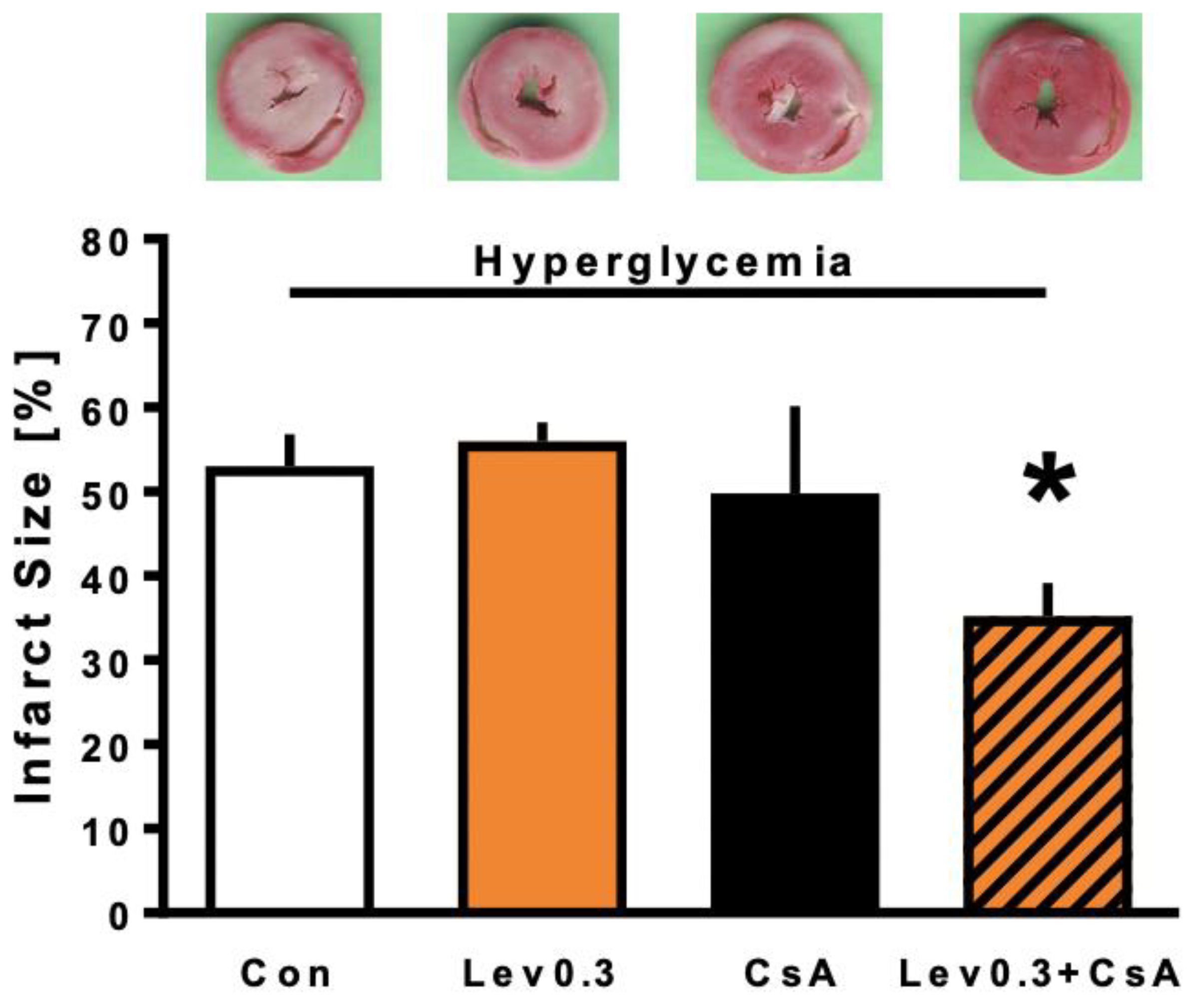
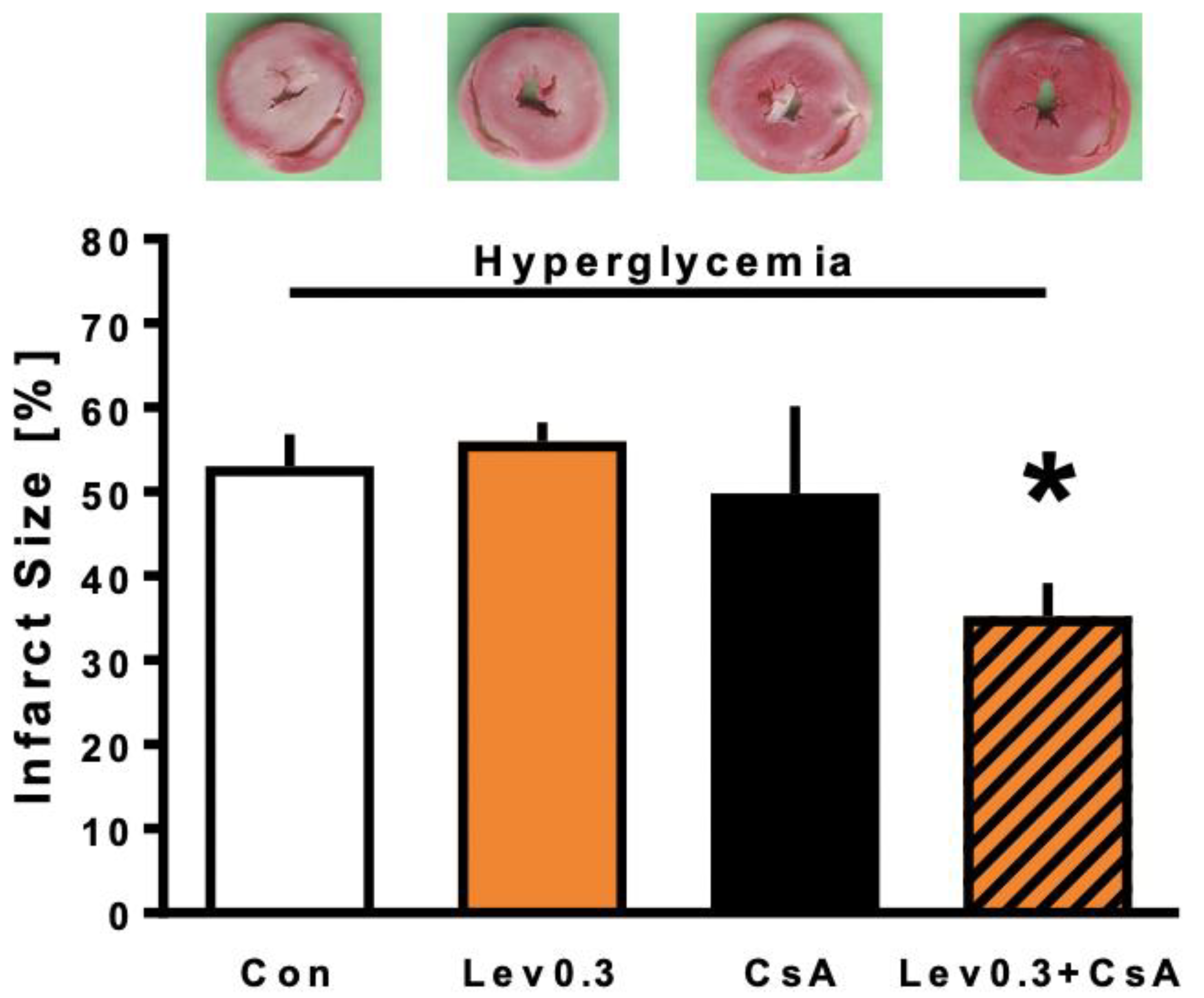
2.2. Infarct Size
2.3. Cardiac Function
2.4. Glucose Levels
3. Discussion
3.1. Influence of Hyperglycemia on Levosimendan-Induced Postconditioning
3.2. Reversing the Loss of Levosimendan-Induced Cardioprotection under Hyperglycemia by Combined Treatment with CsA
3.3. Limitations
4. Materials and Methods
4.1. Surgical Preparation
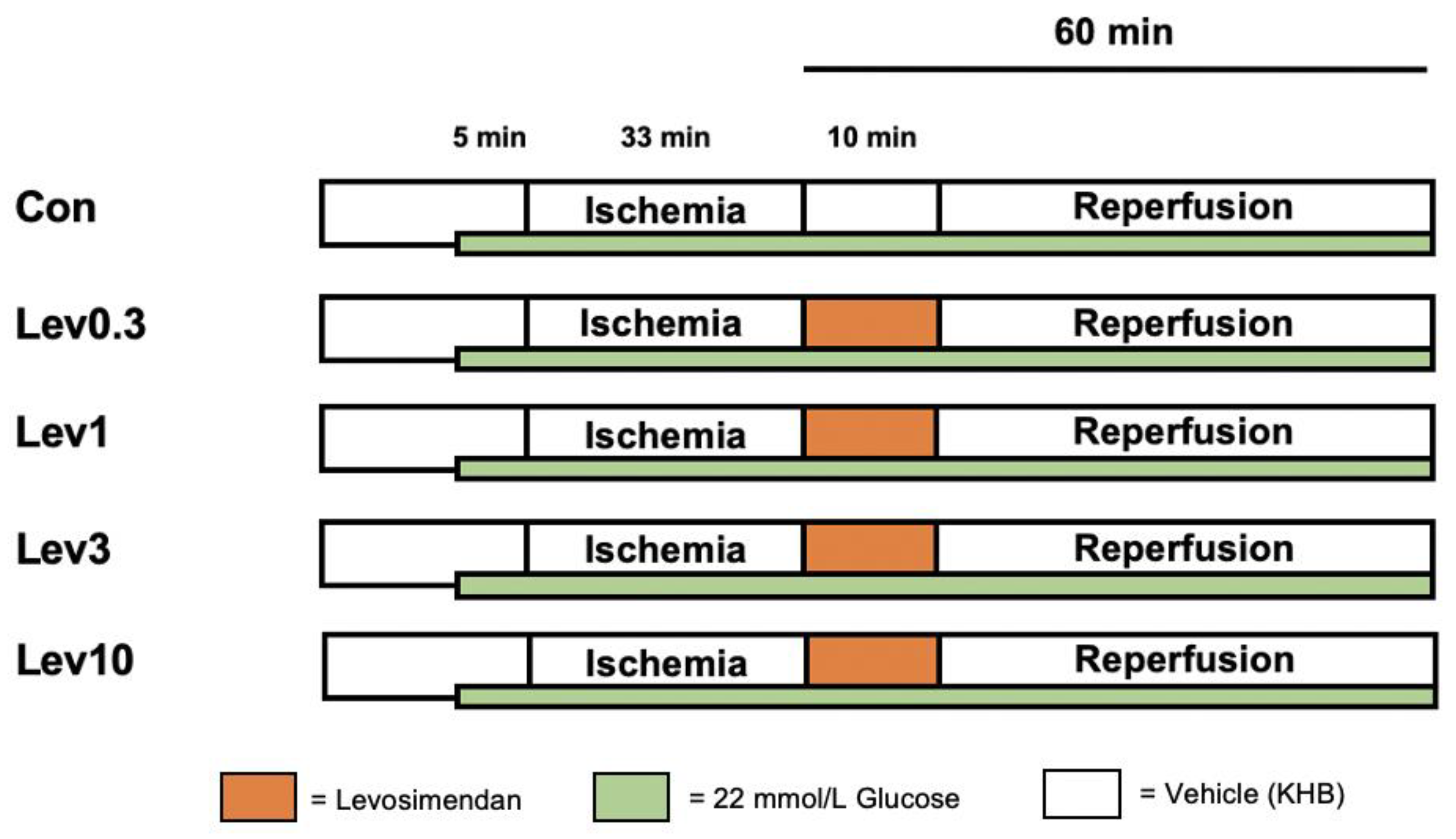
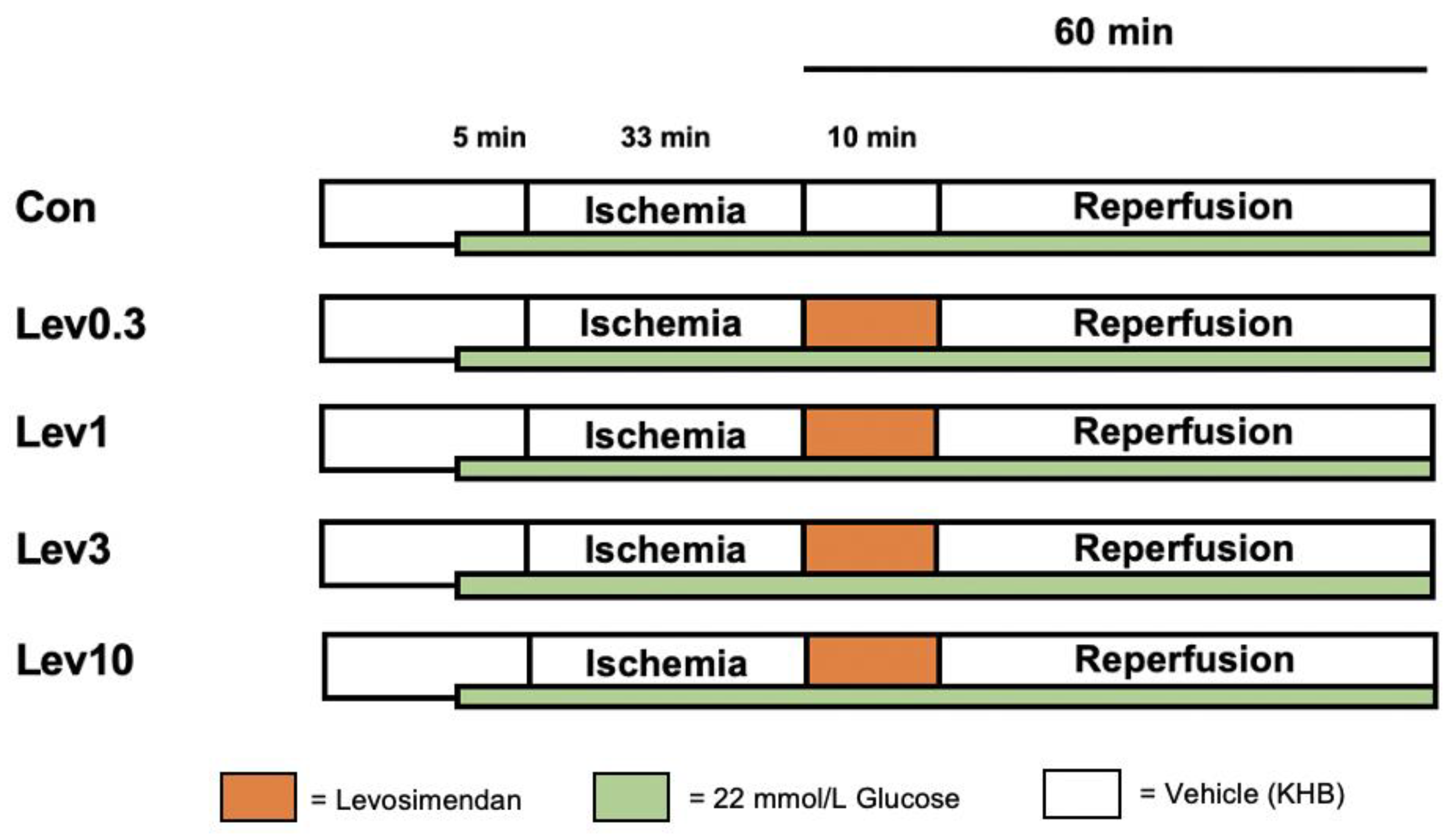
4.2. Experimental Protocol
4.2.1. Part 1: Concentration–Response Relationship of Levosimendan under Hyperglycemia
4.2.2. Part 2: Underlying Mechanisms of Levosimendan-Induced Postconditioning under Hyperglycemia
4.3. Statistical Analysis
4.3.1. Sample Size Analysis
4.3.2. Statistical Approach
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kayani, W.T.; Ballantyne, C.M. Improving Outcomes after Myocardial Infarction in the US Population. J. Am. Heart Assoc. 2018, 7, e008407. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. The 10 Leading Causes of Death in the World, 2000 and 2012. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 9 March 2021).
- Ko, D.T.; Khera, R.; Lau, G.; Qiu, F.; Wang, Y.; Austin, P.C.; Koh, M.; Lin, Z.; Lee, D.S.; Wijeysundera, H.C.; et al. Readmission and Mortality After Hospitalization for Myocardial Infarction and Heart Failure. J. Am. Coll. Cardiol. 2020, 75, 736–746. [Google Scholar] [CrossRef]
- Deckers, J.W.; van Domburg, R.T.; Akkerhuis, M.; Nauta, S.T. Relation of admission glucose levels, short- and long-term (20-year) mortality after acute myocardial infarction. Am. J. Cardiol. 2013, 112, 1306–1310. [Google Scholar] [CrossRef] [PubMed]
- Jelesoff, N.E.; Feinglos, M.; Granger, C.B.; Califf, R.M. Outcomes of diabetic patients following acute myocardial infarction: A review of the major thrombolytic trials. Coron. Artery Dis. 1996, 7, 732–743. [Google Scholar] [CrossRef]
- Bellodi, G.; Manicardi, V.; Malavasi, V.; Veneri, L.; Bernini, G.; Bossini, P.; Distefano, S.; Magnanini, G.; Muratori, L.; Rossi, G.; et al. Hyperglycemia and prognosis of acute myocardial infarction in patients without diabetes mellitus. Am. J. Cardiol. 1989, 64, 885–888. [Google Scholar] [CrossRef]
- Wei, C.H.; Litwin, S.E. Hyperglycemia and Adverse Outcomes in Acute Coronary Syndromes: Is Serum Glucose the Provocateur or Innocent Bystander? Diabetes 2014, 63, 2209–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, A.; Mehta, S.R.; Díaz, R.; Gerstein, H.C.; Afzal, R.; Xavier, D.; Liu, L.; Pais, P.; Yusuf, S. Differential clinical outcomes associated with hypoglycemia and hyperglycemia in acute myocardial infarction. Circulation 2009, 120, 2429–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, M.T.; Messenger, J.C.; Weintraub, W.S.; Cannon, C.P.; Fonarow, G.C.; Dai, D.; Chen, A.Y.; Klein, L.W.; Masoudi, F.A.; McKay, C.; et al. Treatments, trends, and outcomes of acute myocardial infarction and percutaneous coronary intervention. J. Am. Coll. Cardiol. 2010, 56, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef] [PubMed]
- Bunte, S.; Behmenburg, F.; Majewski, N.; Stroethoff, M.; Raupach, A.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R. Characteristics of Dexmedetomidine Postconditioning in the Field of Myocardial Ischemia-Reperfusion Injury. Anesth. Analg. 2019. [Google Scholar] [CrossRef]
- Huhn, R.; Heinen, A.; Weber, N.C.; Schlack, W.; Preckel, B.; Hollmann, M.W. Ischaemic and morphine-induced post-conditioning: Impact of mK(Ca) channels. Br. J. Anaesth. 2010, 105, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Stroethoff, M.; Bunte, S.; Raupach, A.; van de Snepscheut, M.; Torregroza, C.; Heinen, A.; Mathes, A.; Hollmann, M.W.; Huhn, R.; Sixt, S.U. Impact of Ca(2+)-Sensitive Potassium Channels in Levosimendan-Induced Postconditioning. Cardiovasc. Drugs 2019, 33, 581–588. [Google Scholar] [CrossRef]
- Kersten, J.R.; Montgomery, M.W.; Pagel, P.S.; Warltier, D.C. Levosimendan, a new positive inotropic drug, decreases myocardial infarct size via activation of K(ATP) channels. Anesth. Analg. 2000, 90, 5–11. [Google Scholar] [CrossRef]
- Levin, R.; Degrange, M.; Del Mazo, C.; Tanus, E.; Porcile, R. Preoperative levosimendan decreases mortality and the development of low cardiac output in high-risk patients with severe left ventricular dysfunction undergoing coronary artery bypass grafting with cardiopulmonary bypass. Exp. Clin. Cardiol. 2012, 17, 125–130. [Google Scholar]
- Papp, Z.; Édes, I.; Fruhwald, S.; De Hert, S.G.; Salmenperä, M.; Leppikangas, H.; Mebazaa, A.; Landoni, G.; Grossini, E.; Caimmi, P.; et al. Levosimendan: Molecular mechanisms and clinical implications: Consensus of experts on the mechanisms of action of levosimendan. Int. J. Cardiol. 2012, 159, 82–87. [Google Scholar] [CrossRef] [Green Version]
- du Toit, E.F.; Genis, A.; Opie, L.H.; Pollesello, P.; Lochner, A. A role for the RISK pathway and K(ATP) channels in pre- and post-conditioning induced by levosimendan in the isolated guinea pig heart. Br. J. Pharm. 2008, 154, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunte, S.; Behmenburg, F.; Bongartz, A.; Stroethoff, M.; Raupach, A.; Heinen, A.; Minol, J.P.; Hollmann, M.W.; Huhn, R.; Sixt, S.U. Preconditioning by Levosimendan is Mediated by Activation of Mitochondrial Ca(2+)-Sensitive Potassium (mBKCa) Channels. Cardiovasc. Drugs 2018, 32, 427–434. [Google Scholar] [CrossRef]
- Hönisch, A.; Theuring, N.; Ebner, B.; Wagner, C.; Strasser, R.H.; Weinbrenner, C. Postconditioning with levosimendan reduces the infarct size involving the PI3K pathway and KATP-channel activation but is independent of PDE-III inhibition. Basic Res. Cardiol. 2010, 105, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, E.F.; Muller, C.A.; McCarthy, J.; Opie, L.H. Levosimendan: Effects of a calcium sensitizer on function and arrhythmias and cyclic nucleotide levels during ischemia/reperfusion in the Langendorff-perfused guinea pig heart. J. Pharm. Exp. 1999, 290, 505–514. [Google Scholar]
- Huhn, R.; Heinen, A.; Weber, N.C.; Hollmann, M.W.; Schlack, W.; Preckel, B. Hyperglycaemia blocks sevoflurane-induced postconditioning in the rat heart in vivo: Cardioprotection can be restored by blocking the mitochondrial permeability transition pore. Br. J. Anaesth. 2008, 100, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Raphael, J.; Gozal, Y.; Navot, N.; Zuo, Z. Hyperglycemia inhibits anesthetic-induced postconditioning in the rabbit heart via modulation of phosphatidylinositol-3-kinase/Akt and endothelial nitric oxide synthase signaling. J. Cardiovasc. Pharm. 2010, 55, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Torregroza, C.; Feige, K.; Schneider, L.; Bunte, S.; Stroethoff, M.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Raupach, A. Influence of Hyperglycemia on Dexmedetomidine-Induced Cardioprotection in the Isolated Perfused Rat Heart. J. Clin. Med. 2020, 9, 1445. [Google Scholar] [CrossRef]
- Baranyai, T.; Nagy, C.T.; Koncsos, G.; Onodi, Z.; Karolyi-Szabo, M.; Makkos, A.; Varga, Z.V.; Ferdinandy, P.; Giricz, Z. Acute hyperglycemia abolishes cardioprotection by remote ischemic perconditioning. Cardiovasc. Diabetol. 2015, 14, 151. [Google Scholar] [CrossRef] [Green Version]
- Goergens, J.I.; Heinen, N.M.; Zoller, J.; Preckel, B.; Bauer, I.; Huhn, R.; Ebel, D.; Raupach, A. Influence of Hyperglycemia During Different Phases of Ischemic Preconditioning on Cardioprotection-A Focus on Apoptosis and Aggregation of Granulocytes. Shock 2019. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, S.Y.; Kwak, Y.L.; Hwang, K.C.; Shim, Y.H. Hyperglycemia attenuates myocardial preconditioning of remifentanil. J. Surg. Res. 2012, 174, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Andreadou, I.; Aragno, M.; Beauloye, C.; Bertrand, L.; Lazou, A.; Falcão-Pires, I.; Bell, R.; Zuurbier, C.J.; Pagliaro, P.; et al. Effect of hyperglycaemia and diabetes on acute myocardial ischaemia–reperfusion injury and cardioprotection by ischaemic conditioning protocols. Br. J. Pharmacol. 2020, 177, 5312–5335. [Google Scholar] [CrossRef]
- Matsumoto, S.; Cho, S.; Tosaka, S.; Higashijima, U.; Maekawa, T.; Hara, T.; Sumikawa, K. Hyperglycemia raises the threshold of levosimendan- but not milrinone-induced postconditioning in rat hearts. Cardiovasc. Diabetol. 2012, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” target of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef] [Green Version]
- Lisa, F.D.; Canton, M.; Menabò, R.; Kaludercic, N.; Bernardi, P. Mitochondria and cardioprotection. Heart Fail. Rev. 2007, 12, 249–260. [Google Scholar] [CrossRef]
- Di Lisa, F.; Carpi, A.; Giorgio, V.; Bernardi, P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 1316–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. The mitochondrial permeability transition pore: Its fundamental role in mediating cell death during ischaemia and reperfusion. J. Mol. Cell. Cardiol. 2003, 35, 339–341. [Google Scholar] [CrossRef] [Green Version]
- Torregroza, C.; Raupach, A.; Feige, K.; Hollmann, M.W.; Huhn, R. Perioperative Cardioprotection: General Mechanisms and Pharmacological Approaches. Anesth. Analg. 2020, 131, 1765–1780. [Google Scholar] [CrossRef]
- Kersten, J.R.; Schmeling, T.J.; Orth, K.G.; Pagel, P.S.; Warltier, D.C. Acute hyperglycemia abolishes ischemic preconditioning in vivo. Am. J. Physiol. 1998, 275, H721–H725. [Google Scholar] [CrossRef]
- Weber, N.C.; Goletz, C.; Huhn, R.; Grueber, Y.; Preckel, B.; Schlack, W.; Ebel, D. Blockade of anaesthetic-induced preconditioning in the hyperglycaemic myocardium: The regulation of different mitogen-activated protein kinases. Eur. J. Pharm. 2008, 592, 48–54. [Google Scholar] [CrossRef]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Roth, S.; Torregroza, C.; Huhn, R.; Hollmann, M.W.; Preckel, B. Perioperative Cardioprotection: Clinical Implications. Anesth. Analg. 2020, 131, 1751–1764. [Google Scholar] [CrossRef]
- Kersten, J.R.; Montgomery, M.W.; Ghassemi, T.; Gross, E.R.; Toller, W.G.; Pagel, P.S.; Warltier, D.C. Diabetes and hyperglycemia impair activation of mitochondrial K(ATP) channels. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1744–H1750. [Google Scholar] [CrossRef] [Green Version]
- Lei, S.; Su, W.; Xia, Z.Y.; Wang, Y.; Zhou, L.; Qiao, S.; Zhao, B.; Xia, Z.; Irwin, M.G. Hyperglycemia-Induced Oxidative Stress Abrogates Remifentanil Preconditioning-Mediated Cardioprotection in Diabetic Rats by Impairing Caveolin-3-Modulated PI3K/Akt and JAK2/STAT3 Signaling. Oxid. Med. Cell Longev. 2019, 2019, 9836302. [Google Scholar] [CrossRef]
- Tsang, A.; Hausenloy, D.J.; Mocanu, M.M.; Carr, R.D.; Yellon, D.M. Preconditioning the Diabetic Heart: The Importance of Akt Phosphorylation. Diabetes 2005, 54, 2360–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehl, F.; Krolikowski, J.G.; Mraovic, B.; Pagel, P.S.; Warltier, D.C.; Kersten, J.R. Hyperglycemia prevents isoflurane-induced preconditioning against myocardial infarction. Anesthesiology 2002, 96, 183–188. [Google Scholar] [CrossRef]
- Antila, S.; Sundberg, S.; Lehtonen, L.A. Clinical pharmacology of levosimendan. Clin. Pharm. 2007, 46, 535–552. [Google Scholar] [CrossRef]
- Harjola, V.-P.; Giannakoulas, G.; von Lewinski, D.; Matskeplishvili, S.; Mebazaa, A.; Papp, Z.; Schwinger, R.H.G.; Pollesello, P.; Parissis, J.T. Use of levosimendan in acute heart failure. Eur. Heart J. Suppl. 2018, 20, I2–I10. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, E.N.; Antila, S.; McFadyen, L.; Lehtonen, L.; Karlsson, M.O. Population pharmacokinetics of levosimendan in patients with congestive heart failure. Br. J. Clin. Pharm. 2003, 55, 544–551. [Google Scholar] [CrossRef]
- Craig, T.J.; Ashcroft, F.M.; Proks, P. How ATP inhibits the open K(ATP) channel. J. Gen. Physiol. 2008, 132, 131–144. [Google Scholar] [CrossRef]
- Garlid, K.D.; Dos Santos, P.; Xie, Z.-J.; Costa, A.D.T.; Paucek, P. Mitochondrial potassium transport: The role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochim. Biophys. Acta (BBA) Bioenerg. 2003, 1606, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Callaghan, M.J.; Ceradini, D.J.; Gurtner, G.C. Hyperglycemia-induced reactive oxygen species and impaired endothelial progenitor cell function. Antioxid. Redox Signal. 2005, 7, 1476–1482. [Google Scholar] [CrossRef]
- Costa, A.D.; Garlid, K.D. Intramitochondrial signaling: Interactions among mitoKATP, PKCepsilon, ROS, and MPT. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H874–H882. [Google Scholar] [CrossRef] [Green Version]
- Kehl, F.; Krolikowski, J.G.; Weihrauch, D.; Pagel, P.S.; Warltier, D.C.; Kersten, J.R. N-acetylcysteine restores isoflurane-induced preconditioning against myocardial infarction during hyperglycemia. Anesthesiology 2003, 98, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Nakadate, Y.; Sato, H.; Oguchi, T.; Sato, T.; Kawakami, A.; Ishiyama, T.; Matsukawa, T.; Schricker, T. Glycemia and the cardioprotective effects of insulin pre-conditioning in the isolated rat heart. Cardiovasc. Diabetol. 2017, 16, 43. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Argaud, L.; Gateau-Roesch, O.; Muntean, D.; Chalabreysse, L.; Loufouat, J.; Robert, D.; Ovize, M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Maddock, H.L.; Baxter, G.F.; Yellon, D.M. Inhibiting mitochondrial permeability transition pore opening: A new paradigm for myocardial preconditioning? Cardiovasc. Res. 2002, 55, 534–543. [Google Scholar] [CrossRef]
- Watanabe, M.; Okada, T. Langendorff Perfusion Method as an Ex Vivo Model to Evaluate Heart Function in Rats. Methods Mol. Biol. 2018, 1816, 107–116. [Google Scholar] [CrossRef]
- Ferrera, R.; Benhabbouche, S.; Bopassa, J.C.; Li, B.; Ovize, M. One hour reperfusion is enough to assess function and infarct size with TTC staining in Langendorff rat model. Cardiovasc. Drugs 2009, 23, 327–331. [Google Scholar] [CrossRef]
- Feige, K.; Rubbert, J.; Raupach, A.; Stroethoff, M.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Torregroza, C. Cardioprotective Properties of Mannitol-Involvement of Mitochondrial Potassium Channels. Int. J. Mol. Sci. 2021, 22, 2395. [Google Scholar] [CrossRef]
- Behmenburg, F.; Dorsch, M.; Huhn, R.; Mally, D.; Heinen, A.; Hollmann, M.W.; Berger, M.M. Impact of Mitochondrial Ca2+-Sensitive Potassium (mBKCa) Channels in Sildenafil-Induced Cardioprotection in Rats. PLoS ONE 2015, 10, e0144737. [Google Scholar] [CrossRef]
- Stroethoff, M.; Goetze, L.; Torregroza, C.; Bunte, S.; Raupach, A.; Heinen, A.; Mathes, A.; Hollmann, M.W.; Huhn, R. The Melatonin Receptor Agonist Ramelteon Induces Cardioprotection that Requires MT2 Receptor Activation and Release of Reactive Oxygen Species. Cardiovasc. Drugs Ther. 2020, 34, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Raupach, A.; Reinle, J.; Stroethoff, M.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Bunte, S. Milrinone-Induced Pharmacological Preconditioning in Cardioprotection: Hints for a Role of Mitochondrial Mechanisms. J. Clin. Med. 2019, 8, 507. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Body Weight (g) | Heart Weight Wet (g) | Heart Weight Dry (g) | Time of Max. Ischemic Contracture (min) | Level of Max. Ischemic Contracture (mmHg) | ||
|---|---|---|---|---|---|---|---|
| Part 1 | |||||||
| HG | Con | 7 | 298 ± 8 | 1.21 ± 0.10 | 0.13 ± 0.01 | 14 ± 1 | 85 ± 11 |
| Lev0.3 | 7 | 312 ± 7 | 1.22 ± 0.07 | 0.14 ± 0.01 | 15 ± 2 | 82 ± 9 | |
| Lev1 | 7 | 311 ± 14 | 1.28 ± 0.08 | 0.14 ± 0.01 | 15 ± 1 | 75 ± 11 | |
| Lev3 | 7 | 313 ± 17 | 1.32 ± 0.07 | 0.14 ± 0.02 | 15 ± 1 | 82 ± 10 | |
| Lev10 | 7 | 308 ± 13 | 1.22 ± 0.05 | 0.14 ± 0.02 | 16 ± 1 | 65 ± 8 * | |
| Part 2 | |||||||
| HG | Con | 5 | 316 ± 11 | 1.27 ± 0.04 | 0.14 ± 0.00 | 16 ± 2 | 75 ± 15 |
| Lev0.3 | 5 | 288 ± 21 | 1.24 ± 0.07 | 0.12 ± 0.01 | 15 ± 2 | 79 ± 10 | |
| CsA | 5 | 294 ± 24 | 1.15 ± 0.08 | 0.12 ± 0.01 | 15 ± 2 | 77 ± 7 | |
| Lev0.3+CsA | 5 | 307 ± 11 | 1.19 ± 0.04 | 0.12 ± 0.01 | 15 ± 2 | 68 ± 8 | |
| Baseline | Reperfusion | ||||
|---|---|---|---|---|---|
| 30 | 45 | 60 | |||
| Heart Rate (bpm) | |||||
| HG | Con | 316 ± 43 | 242 ± 41 | 219 ± 57 | 207 ± 69 |
| Lev0.3 | 307 ± 36 | 255 ± 32 | 228 ± 52 | 267 ± 43 | |
| Lev1 | 288 ± 38 | 263 ± 109 | 271 ± 83 | 292 ± 40 | |
| Lev3 | 278 ± 20 | 231 ± 48 | 208 ± 65 | 214 ± 45 | |
| Lev10 | 282 ± 20 | 222 ± 64 | 248 ± 86 | 239 ± 79 | |
| Left Ventricular Developed Pressure (mmHg) | |||||
| HG | Con | 109 ± 9 | 27 ± 11 * | 35 ± 12 * | 33 ± 6 * |
| Lev0.3 | 107 ± 16 | 22 ± 14 * | 25 ± 9 * | 21 ± 10 * | |
| Lev1 | 117 ± 17 | 13 ± 10 * | 20 ± 9 * | 20 ± 11 * | |
| Lev3 | 114 ± 13 | 12 ± 12 * | 20 ± 14 * | 26 ± 7 * | |
| Lev10 | 122 ± 11 | 12 ± 10 * | 19 ± 9 * | 25 ± 10 * | |
| Coronary flow (mL/min) | |||||
| HG | Con | 12 ± 2 | 6 ± 1 * | 6 ± 1 * | 6 ± 1 * |
| Lev0.3 | 12 ± 3 | 6 ± 1 * | 6 ± 1 * | 6 ± 1 * | |
| Lev1 | 13 ± 2 | 6 ± 1 * | 6 ± 1 * | 6 ± 1 * | |
| Lev3 | 13 ± 2 | 6 ± 2 * | 7 ± 2 * | 6 ± 2 * | |
| Lev10 | 14 ± 3 | 8 ± 3 * | 8 ± 3 * | 8 ± 3 * | |
| Baseline | Reperfusion | ||||
|---|---|---|---|---|---|
| 30 | 45 | 60 | |||
| Heart Rate (bpm) | |||||
| HG | Con | 311 ± 54 | 205 ± 42 * | 220 ± 69 | 192 ± 16 * |
| Lev0.3 | 297 ± 30 | 245 ± 87 | 259 ± 45 | 216 ± 52 | |
| CsA | 292 ± 23 | 215 ± 63 | 214 ± 67 | 229 ± 47 | |
| Lev0.3 + CsA | 306 ± 29 | 297 ± 43 | 267 ± 20 | 265 ± 23 | |
| Left Ventricular Developed Pressure (mmHg) | |||||
| HG | Con | 117 ± 19 | 20 ± 16 * | 28 ± 16 * | 28 ± 11 * |
| Lev0.3 | 106 ± 14 | 15 ± 13 * | 20 ± 19 * | 25 ± 16 * | |
| CsA | 123 ± 26 | 35 ± 8 * | 37 ± 13 * | 35 ± 10 * | |
| Lev0.3 + CsA | 112 ± 21 | 23 ± 18 * | 24 ± 13 * | 24 ± 12 * | |
| Coronary flow (mL/min) | |||||
| HG | Con | 15 ± 3 | 7 ± 3 * | 6 ± 1 * | 6 ± 2 * |
| Lev0.3 | 13 ± 2 | 6 ± 1 * | 6 ± 1 * | 5 ± 1 * | |
| CsA | 15 ± 2 | 5 ± 1 * | 6 ± 1 * | 6 ± 1 * | |
| Lev0.3 + CsA | 14 ± 2 | 7 ± 2 * | 7 ± 2 * | 6 ± 1 * | |
| Baseline | Pre Ischemia | Reperfusion 15 | Reperfusion 60 | ||
|---|---|---|---|---|---|
| Part 1 | |||||
| HG | Con | 196 ± 5 | 370 ± 10 * | 394 ± 18 * | 425 ± 44 * |
| Lev0.3 | 197 ± 6 | 374 ± 37 * | 393 ± 11 * | 425 ± 28 * | |
| Lev1 | 197 ± 2 | 360 ± 46 * | 389 ± 13 * | 409 ± 33 * | |
| Lev3 | 197 ± 4 | 377 ± 10 * | 389 ± 32 * | 393 ± 41 * | |
| Lev10 | 198 ± 3 | 372 ± 16 * | 377 ± 13 * | 400 ± 32 * | |
| Part 2 | |||||
| HG | Con | 203 ± 3 | 384 ± 6 * | 389 ± 12 * | 418 ± 35 * |
| Lev0.3 | 199 ± 9 | 373 ± 16 * | 391 ± 9 * | 441 ± 18 * | |
| CsA | 204 ± 10 | 377 ± 9 * | 390 ± 10 * | 425 ± 20 * | |
| Lev0.3 + CsA | 203 ± 6 | 389 ± 8 * | 398 ± 5 * | 448 ± 35 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torregroza, C.; Yueksel, B.; Ruske, R.; Stroethoff, M.; Raupach, A.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Feige, K. Combination of Cyclosporine A and Levosimendan Induces Cardioprotection under Acute Hyperglycemia. Int. J. Mol. Sci. 2021, 22, 4517. https://doi.org/10.3390/ijms22094517
Torregroza C, Yueksel B, Ruske R, Stroethoff M, Raupach A, Heinen A, Hollmann MW, Huhn R, Feige K. Combination of Cyclosporine A and Levosimendan Induces Cardioprotection under Acute Hyperglycemia. International Journal of Molecular Sciences. 2021; 22(9):4517. https://doi.org/10.3390/ijms22094517
Chicago/Turabian StyleTorregroza, Carolin, Birce Yueksel, Raphael Ruske, Martin Stroethoff, Annika Raupach, André Heinen, Markus W. Hollmann, Ragnar Huhn, and Katharina Feige. 2021. "Combination of Cyclosporine A and Levosimendan Induces Cardioprotection under Acute Hyperglycemia" International Journal of Molecular Sciences 22, no. 9: 4517. https://doi.org/10.3390/ijms22094517