
Immunogenic Cell Death by the Novel Topoisomerase I Inhibitor TLC388 Enhances the Therapeutic Efficacy of Radiotherapy

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Methods and Materials
2.1. Cell Culture
2.2. Antibodies and Reagents
2.3. Western Blot Analysis
2.4. Evaluation of the Immunogenic TME Induced by Radiotherapy and Chemotherapeutic Drugs
2.5. Detection of the Surface CRT, MHC Class I, and CD80 Levels on Tumor Cells
2.6. Assessment of Cell Growth and Apoptosis
2.7. Immunohistochemistry
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Statistical Analysis
3. Results
3.1. The Novel Chemotherapeutic Drug Lipotecan Can Elicit Surface Exposure of Calreticulin via Endoplasmic Reticulum Stress
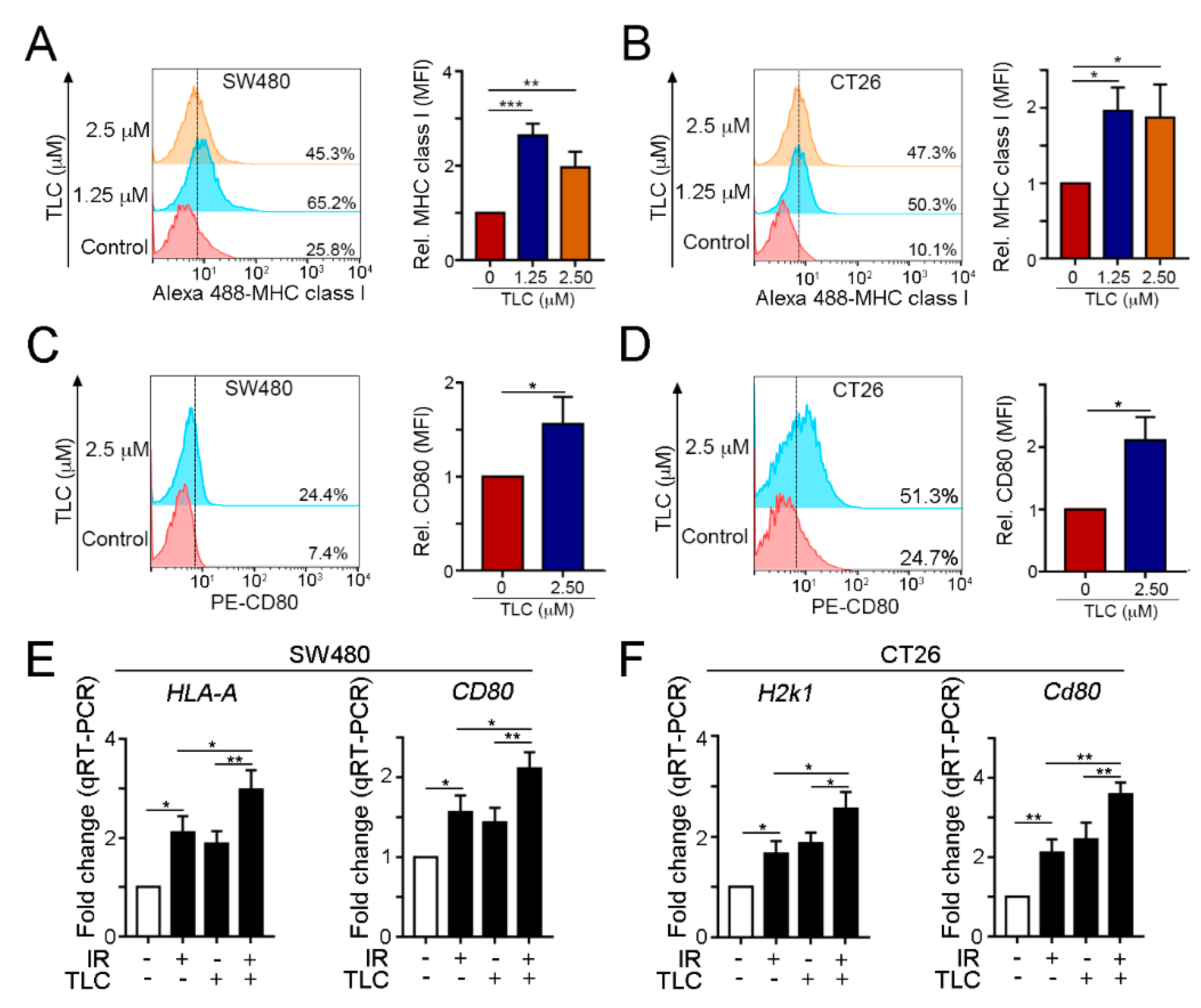
3.2. Lipotecan Remarkably Induces Immunogenic Cell Death (ICD) to Release HMGB1 and ANXA1 and Increase Cancer Immunogenicity
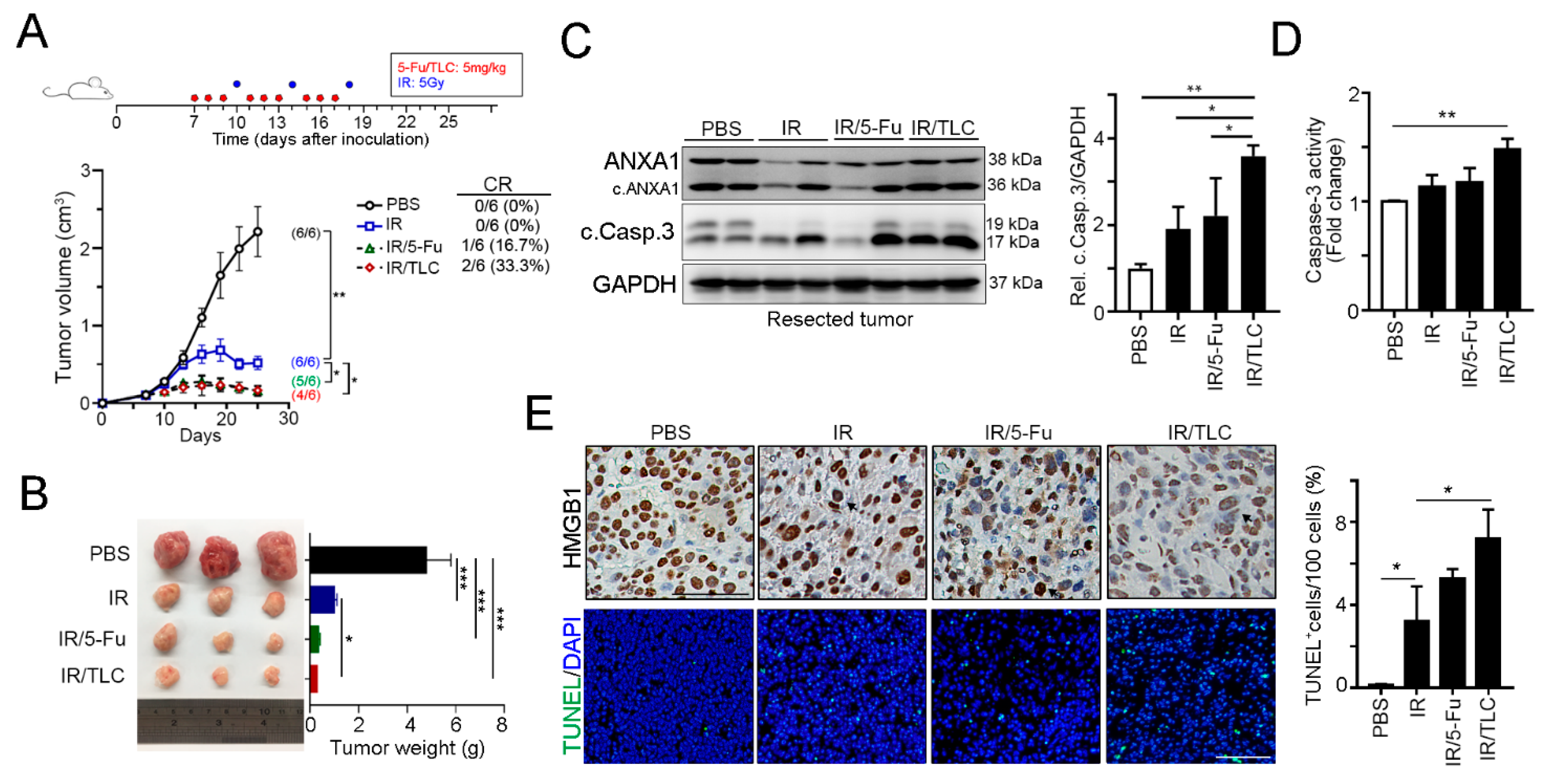
3.3. Lipotecan Remarkably Enhanced the Therapeutic Efficacy of Radiotherapy In Vivo
3.4. Lipotecan-Based CRT Provoked Antitumor Immunity to Enhance Therapeutic Efficacy In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Conde-Muino, R.; Cuadros, M.; Zambudio, N.; Segura-Jimenez, I.; Cano, C.; Palma, P. Predictive Biomarkers to Chemoradiation in Locally Advanced Rectal Cancer. Biomed. Res. Int. 2015, 2015, 921435. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Yoon, W.H.; Kim, H.J.; Kim, C.H.; Joo, J.K.; Kim, Y.J.; Kim, H.R. Oncologic impact of pathologic response on clinical outcome after preoperative chemoradiotherapy in locally advanced rectal cancer. Ann. Surg. Treat. Res. 2015, 88, 15–20. [Google Scholar] [CrossRef][Green Version]
- Deng, Y.; Chi, P.; Lan, P.; Wang, L.; Cui, L.; Chen, D.; Cao, J.; Wei, H.; Peng, X.; Huang, Z.; et al. A multi-center randomized controlled trial of mFOLFOX6 with or without radiation in neoadjuvant treatment of local advanced rectal cancer (FOWARC study): Preliminary results. J. Clin. Oncol. 2015, 33, 3500. [Google Scholar] [CrossRef]
- Aklilu, M.; Eng, C. The current landscape of locally advanced rectal cancer. Nat. Rev. Clin. Oncol. 2011, 8, 649–659. [Google Scholar] [CrossRef]
- Gerard, J.P.; Azria, D.; Gourgou-Bourgade, S.; Martel-Lafay, I.; Hennequin, C.; Etienne, P.L.; Vendrely, V.; Francois, E.; de La Roche, G.; Bouche, O.; et al. Clinical outcome of the ACCORD 12/0405 PRODIGE 2 randomized trial in rectal cancer. J. Clin. Oncol. 2012, 30, 4558–4565. [Google Scholar] [CrossRef] [PubMed]
- Bosset, J.F.; Calais, G.; Mineur, L.; Maingon, P.; Stojanovic-Rundic, S.; Bensadoun, R.J.; Bardet, E.; Beny, A.; Ollier, J.C.; Bolla, M.; et al. Fluorouracil-based adjuvant chemotherapy after preoperative chemoradiotherapy in rectal cancer: Long-term results of the EORTC 22921 randomised study. Lancet Oncol. 2014, 15, 184–190. [Google Scholar] [CrossRef]
- de Wilt, J.H.; Vermaas, M.; Ferenschild, F.T.; Verhoef, C. Management of locally advanced primary and recurrent rectal cancer. Clin. Colon Rectal Surg. 2007, 20, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Sinukumar, S.; Patil, P.; Engineer, R.; Desouza, A.; Saklani, A. Clinical outcome of patients with complete pathological response to neoadjuvant chemoradiotherapy for locally advanced rectal cancers: The Indian scenario. Gastroenterol. Res. Pract. 2014, 2014, 867841. [Google Scholar] [CrossRef] [PubMed]
- Breugom, A.J.; Swets, M.; Bosset, J.F.; Collette, L.; Sainato, A.; Cionini, L.; Glynne-Jones, R.; Counsell, N.; Bastiaannet, E.; van den Broek, C.B.; et al. Adjuvant chemotherapy after preoperative (chemo)radiotherapy and surgery for patients with rectal cancer: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015, 16, 200–207. [Google Scholar] [CrossRef]
- O’Connell, M.J.; Colangelo, L.H.; Beart, R.W.; Petrelli, N.J.; Allegra, C.J.; Sharif, S.; Pitot, H.C.; Shields, A.F.; Landry, J.C.; Ryan, D.P.; et al. Capecitabine and Oxaliplatin in the Preoperative Multimodality Treatment of Rectal Cancer: Surgical End Points From National Surgical Adjuvant Breast and Bowel Project Trial R-04. J. Clin. Oncol. 2014, 32, 1927–1934. [Google Scholar] [CrossRef]
- Nilsson, P.J.; van Etten, B.; Hospers, G.A.; Pahlman, L.; van de Velde, C.J.; Beets-Tan, R.G.; Blomqvist, L.; Beukema, J.C.; Kapiteijn, E.; Marijnen, C.A.; et al. Short-course radiotherapy followed by neo-adjuvant chemotherapy in locally advanced rectal cancer--the RAPIDO trial. BMC Cancer 2013, 13, 279. [Google Scholar] [CrossRef]
- Dewdney, A.; Cunningham, D.; Tabernero, J.; Capdevila, J.; Glimelius, B.; Cervantes, A.; Tait, D.; Brown, G.; Wotherspoon, A.; Gonzalez de Castro, D.; et al. Multicenter randomized phase II clinical trial comparing neoadjuvant oxaliplatin, capecitabine, and preoperative radiotherapy with or without cetuximab followed by total mesorectal excision in patients with high-risk rectal cancer (EXPERT-C). J. Clin. Oncol. 2012, 30, 1620–1627. [Google Scholar] [CrossRef]
- Gollins, S.; West, N.; Sebag-Montefiore, D.; Myint, A.S.; Saunders, M.; Susnerwala, S.; Quirke, P.; Essapen, S.; Samuel, L.; Sizer, B.; et al. Preoperative chemoradiation with capecitabine, irinotecan and cetuximab in rectal cancer: Significance of pre-treatment and post-resection RAS mutations. Br. J. Cancer 2017, 117, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Bains, S.J.; Abrahamsson, H.; Flatmark, K.; Dueland, S.; Hole, K.H.; Seierstad, T.; Redalen, K.R.; Meltzer, S.; Ree, A.H. Immunogenic cell death by neoadjuvant oxaliplatin and radiation protects against metastatic failure in high-risk rectal cancer. Cancer Immunol. Immunother. 2020, 69, 355–364. [Google Scholar] [CrossRef]
- Rödel, C.; Graeven, U.; Fietkau, R.; Hohenberger, W.; Hothorn, T.; Arnold, D.; Hofheinz, R.-D.; Ghadimi, M.; Wolff, H.A.; Lang-Welzenbach, M.; et al. Oxaliplatin added to fluorouracil-based preoperative chemoradiotherapy and postoperative chemotherapy of locally advanced rectal cancer (the German CAO/ARO/AIO-04 study): Final results of the multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2015, 16, 979–989. [Google Scholar] [CrossRef]
- Menard, C.; Martin, F.; Apetoh, L.; Bouyer, F.; Ghiringhelli, F. Cancer chemotherapy: Not only a direct cytotoxic effect, but also an adjuvant for antitumor immunity. Cancer Immunol. Immunother. 2008, 57, 1579–1587. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Kitai, Y.; Kawasaki, T.; Sueyoshi, T.; Kobiyama, K.; Ishii, K.J.; Zou, J.; Akira, S.; Matsuda, T.; Kawai, T. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J. Immunol. 2017, 198, 1649–1659. [Google Scholar] [CrossRef]
- Tesniere, A.; Apetoh, L.; Ghiringhelli, F.; Joza, N.; Panaretakis, T.; Kepp, O.; Schlemmer, F.; Zitvogel, L.; Kroemer, G. Immunogenic cancer cell death: A key-lock paradigm. Curr. Opin. Immunol. 2008, 20, 504–511. [Google Scholar] [CrossRef]
- Kusume, A.; Sasahira, T.; Luo, Y.; Isobe, M.; Nakagawa, N.; Tatsumoto, N.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Suppression of dendritic cells by HMGB1 is associated with lymph node metastasis of human colon cancer. Pathobiology 2009, 76, 155–162. [Google Scholar] [CrossRef]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef]
- Teng, F.; Mu, D.; Meng, X.; Kong, L.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor infiltrating lymphocytes (TILs) before and after neoadjuvant chemoradiotherapy and its clinical utility for rectal cancer. Am. J. Cancer Res. 2015, 5, 2064–2074. [Google Scholar] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Galon, J.; Pages, F.; Tartour, E.; Sautes-Fridman, C.; Kroemer, G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res. 2011, 71, 5601–5605. [Google Scholar] [CrossRef]
- Dieci, M.V.; Criscitiello, C.; Goubar, A.; Viale, G.; Conte, P.; Guarneri, V.; Ficarra, G.; Mathieu, M.C.; Delaloge, S.; Curigliano, G.; et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann. Oncol. 2014, 25, 611–618. [Google Scholar] [CrossRef]
- Pages, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef]
- Ohtani, H. Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in human colorectal cancer. Cancer Immun. 2007, 7, 4. [Google Scholar] [PubMed]
- Pages, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011, 71, 5670–5677. [Google Scholar] [CrossRef]
- Huang, G.; Wang, H.; Yang, L.X. Enhancement of radiation-induced DNA damage and inhibition of its repair by a novel camptothecin analog. Anticancer Res. 2010, 30, 937–944. [Google Scholar]
- Ghamande, S.; Lin, C.-C.; Cho, D.C.; Shapiro, G.I.; Kwak, E.L.; Silverman, M.H.; Tseng, Y.; Kuo, M.-W.; Mach, W.B.; Hsu, S.-C.; et al. A phase 1 open-label, sequential dose-escalation study investigating the safety, tolerability, and pharmacokinetics of intravenous TLC388 administered to patients with advanced solid tumors. Investig. New Drugs 2013, 32, 445–451. [Google Scholar] [CrossRef]
- Fumet, J.-D.; Limagne, E.; Thibaudin, M.; Ghiringhelli, F. Immunogenic Cell Death and Elimination of Immunosuppressive Cells: A Double-Edged Sword of Chemotherapy. Cancers 2020, 12, 2637. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chiang, S.F.; Ke, T.W.; Chen, T.W.; Lan, Y.C.; You, Y.S.; Shiau, A.C.; Chen, W.T.; Chao, K.S.C. Cytosolic high-mobility group box protein 1 (HMGB1) and/or PD-1+ TILs in the tumor microenvironment may be contributing prognostic biomarkers for patients with locally advanced rectal cancer who have undergone neoadjuvant chemoradiotherapy. Cancer Immunol. Immunother. 2018, 67, 551–562. [Google Scholar] [CrossRef]
- Lin, T.Y.; Fan, C.W.; Maa, M.C.; Leu, T.H. Lipopolysaccharide-promoted proliferation of Caco-2 cells is mediated by c-Src induction and ERK activation. Biomedicine 2015, 5, 5. [Google Scholar] [CrossRef]
- Huang, K.C.; Chiang, S.F.; Chen, W.T.; Chen, T.W.; Hu, C.H.; Yang, P.C.; Ke, T.W.; Chao, K.S.C. Decitabine Augments Chemotherapy-Induced PD-L1 Upregulation for PD-L1 Blockade in Colorectal Cancer. Cancers 2020, 12, 462. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Song, C.; Chung, J.H.; Kang, S.B.; Kim, D.W.; Oh, H.K.; Lee, H.S.; Kim, J.W.; Lee, K.W.; Kim, J.S. Impact of tumor regression grade as a major prognostic factor in locally advanced rectal cancer after neoadjuvant chemoradiotherapy: A proposal for a modified staging system. Cancers 2018, 10, 319. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chiang, S.F.; Ke, T.W.; Chen, T.W.; You, Y.S.; Chen, W.T.; Chao, K.S.C. Clinical significance of programmed death 1 ligand-1 (CD274/PD-L1) and intra-tumoral CD8+ T-cell infiltration in stage II-III colorectal cancer. Sci. Rep. 2018, 8, 15658. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Huang, K.C.; Chiang, S.F.; Chen, W.T.; Ke, T.W.; Chao, K.S.C. Prognostic relevance of programmed cell death-ligand 1 expression and CD8+ TILs in rectal cancer patients before and after neoadjuvant chemoradiotherapy. J. Cancer Res. Clin. Oncol. 2019, 145, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.A.; Mbofung, R.M.; Malu, S.; Zhang, M.; Ashkin, E.; Devi, S.; Williams, L.; Tieu, T.; Peng, W.; Pradeep, S.; et al. The Effect of Topoisomerase I Inhibitors on the Efficacy of T-Cell-Based Cancer Immunotherapy. J. Natl. Cancer Inst. 2018, 110, 777–786. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, K.C.-Y.; Chiang, S.-F.; Yang, P.-C.; Ke, T.-W.; Chen, T.-W.; Hu, C.-H.; Huang, Y.-W.; Chang, H.-Y.; Chen, W.T.-L.; Chao, K.S.C. Immunogenic Cell Death by the Novel Topoisomerase I Inhibitor TLC388 Enhances the Therapeutic Efficacy of Radiotherapy. Cancers 2021, 13, 1218. https://doi.org/10.3390/cancers13061218
Huang KC-Y, Chiang S-F, Yang P-C, Ke T-W, Chen T-W, Hu C-H, Huang Y-W, Chang H-Y, Chen WT-L, Chao KSC. Immunogenic Cell Death by the Novel Topoisomerase I Inhibitor TLC388 Enhances the Therapeutic Efficacy of Radiotherapy. Cancers. 2021; 13(6):1218. https://doi.org/10.3390/cancers13061218
Chicago/Turabian StyleHuang, Kevin Chih-Yang, Shu-Fen Chiang, Pei-Chen Yang, Tao-Wei Ke, Tsung-Wei Chen, Ching-Han Hu, Yi-Wen Huang, Hsin-Yu Chang, William Tzu-Liang Chen, and K. S. Clifford Chao. 2021. "Immunogenic Cell Death by the Novel Topoisomerase I Inhibitor TLC388 Enhances the Therapeutic Efficacy of Radiotherapy" Cancers 13, no. 6: 1218. https://doi.org/10.3390/cancers13061218
APA StyleHuang, K. C.-Y., Chiang, S.-F., Yang, P.-C., Ke, T.-W., Chen, T.-W., Hu, C.-H., Huang, Y.-W., Chang, H.-Y., Chen, W. T.-L., & Chao, K. S. C. (2021). Immunogenic Cell Death by the Novel Topoisomerase I Inhibitor TLC388 Enhances the Therapeutic Efficacy of Radiotherapy. Cancers, 13(6), 1218. https://doi.org/10.3390/cancers13061218