
Whole Blood Thromboelastometry by ROTEM and Thrombin Generation by Genesia According to the Genotype and Clinical Phenotype in Congenital Fibrinogen Disorders

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Thrombin Generation
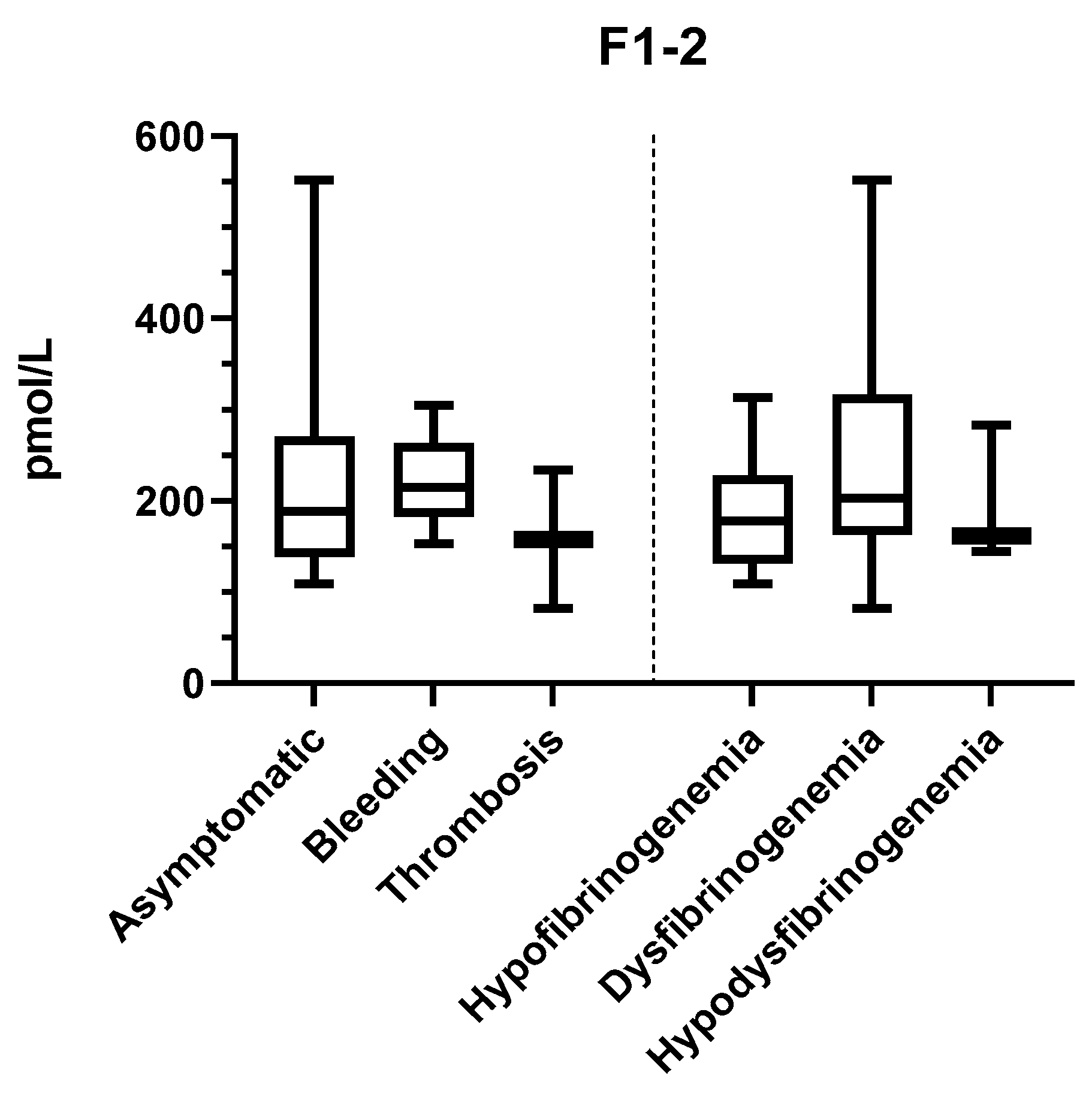
2.2. In Vivo Thrombin Generation
2.3. ROTEM
3. Discussion
4. Materials and Methods
4.1. Blood Sampling
4.2. Coagulation Studies
4.3. DNA Preparation and Genetic Analysis
4.4. Thrombin Generation
4.5. Rotational Thromboelastometry
4.6. Prothrombin Fragments F1+2
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital Fibrinogen Disorders: An Update. Semin. Thromb. Hemost. 2013, 39, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; De Moerloose, P.; Casini, A. Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin. Thromb. Hemost. 2016, 42, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Brungs, T.; Lavenu-Bombled, C.; Vilar, R.; Neerman-Arbez, M.; De Moerloose, P. Genetics, diagnosis and clinical features of congenital hypodysfibrinogenemia: A systematic literature review and report of a novel mutation. J. Thromb. Haemost. 2017, 15, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Palla, R.; Menegatti, M.; Siboni, S.M.; Halimeh, S.; Faeser, B.; Pergantou, H.; Platokouki, H.; Giangrande, P.; Peerlinck, K.; et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: Results from the European Network of Rare Bleeding Disorders. J. Thromb. Haemost. 2012, 10, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; De Moerloose, P. the Congenital Fibrinogen Disorders Group Management of congenital quantitative fibrinogen disorders: A Delphi consensus. Haemophilia 2016, 22, 898–905. [Google Scholar] [CrossRef]
- Krammer, B.; Anders, O.; Nagel, H.-R.; Burstein, C.; Steiner, M. Screening of dysfibrinogenaemia using the fibrinogen function versus antigen concentration ratio. Thromb. Res. 1994, 76, 577–579. [Google Scholar] [CrossRef]
- Cunningham, M.T.; Brandt, J.T.; Laposata, M.; Olson, J.D. Laboratory Diagnosis of Dysfibrinogenemia. Arch. Pathol. Lab. Med. 2002, 126, 499–505. [Google Scholar] [CrossRef]
- Casini, A.; Undas, A.; Palla, R.; Thachil, J.; De Moerloose, P. Diagnosis and classification of congenital fibrinogen disorders: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1887–1890. [Google Scholar] [CrossRef]
- Desborough, M.J.R.; Jairath, V. Thrombelastography-guided blood product use before invasive procedures in cirrhosis with severe coagulopathy. Hepatology 2016, 64, 682–683. [Google Scholar] [CrossRef]
- Levi, M.; Hunt, B.J. A critical appraisal of point-of-care coagulation testing in critically ill patients. J. Thromb. Haemost. 2015, 13, 1960–1967. [Google Scholar] [CrossRef]
- Galanakis, D.K.; Neerman-Arbez, M.; Brennan, S.; Rafailovich, M.; Hyder, L.; Travlou, O.; Papadakis, E.; Manco-Johnson, M.J.; Henschen, A.; Scharrer, I. Thromboelastographic phenotypes of fibrinogen and its variants: Clinical and non-clinical implications. Thromb. Res. 2014, 133, 1115–1123. [Google Scholar] [CrossRef]
- Zhou, J.; Xin, Y.; Ding, Q.; Jiang, L.; Chen, Y.; Dai, J.; Lu, Y.; Wu, X.; Liang, Q.; Wang, H.; et al. Thromboelastography predicts risks of obstetric complication occurrence in (hypo)dysfibrinogenemia patients under non-pregnant state. Clin. Exp. Pharmacol. Physiol. 2016, 43, 149–156. [Google Scholar] [CrossRef]
- Zhou, J.; Ding, Q.; Chen, Y.; Ouyang, Q.; Jiang, L.; Dai, J.; Lu, Y.; Wu, X.; Liang, Q.; Wang, H.; et al. Clinical features and molecular basis of 102 Chinese patients with congenital dysfibrinogenemia. Blood Cells Mol. Dis. 2015, 55, 308–315. [Google Scholar] [CrossRef]
- Hemker, H.C.; Giesen, P.; Al Dieri, R.; Regnault, V.; De Smedt, E.; Wagenvoord, R.; Lecompte, T.; Béguin, S. Calibrated Automated Thrombin Generation Measurement in Clotting Plasma. Pathophysiol. Haemost. Thromb. 2003, 33, 4–15. [Google Scholar] [CrossRef]
- Bouvier, S.; Chea, M.; Ripart, S.; Hanss, M.; De Mazancourt, P.; Gris, J.-C. Successful Pregnancy under Fibrinogen Substitution with Heparin and Aspirin in a Woman with Dysfibrinogenemia Revealed by Placental Abruption. Thromb. Haemost. 2018, 118, 2006–2008. [Google Scholar] [CrossRef]
- Calzavarini, S.; Brodard, J.; Angelillo-Scherrer, A.; Quarroz, C.; Maire, L.; Nützi, R.; Jankovic, J.; Rotondo, L.C.; Giabbani, E.; Fiedler, G.M.; et al. Thrombin generation measurement using the ST Genesia Thrombin Generation System in a cohort of healthy adults: Normal values and variability. Res. Pract. Thromb. Haemost. 2019, 3, 758–768. [Google Scholar] [CrossRef]
- Rubbia-Brandt, L.; Neerman-Arbez, M.; Rougemont, A.-L.; Malé, P.-J.; Spahr, L. Fibrinogen Gamma375 Arg→Trp Mutation (Fibrinogen Aguadilla) Causes Hereditary Hypofibrinogenemia, Hepatic Endoplasmic Reticulum Storage Disease and Cirrhosis. Am. J. Surg. Pathol. 2006, 30, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Ząbczyk, M.; Undas, A. Plasma fibrin clot structure and thromboembolism: Clinical implications. Pol. Arch. Intern. Med. 2017, 127, 873–881. [Google Scholar] [CrossRef]
- Undas, A.; Zabczyk, M. Antithrombotic medications and their impact on fibrin clot structure and function. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2018, 69. [Google Scholar] [CrossRef]
- Pitkänen, H.H.; Jouppila, A.; Lemponen, M.; Ilmakunnas, M.; Ahonen, J.; Lassila, R. Factor XIII deficiency enhances thrombin generation due to impaired fibrin polymerization—An effect corrected by Factor XIII replacement. Thromb. Res. 2017, 149, 56–61. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pitkänen, H.H.; Kärki, M.; Lassila, R.; Niinikoski, H.; Tanner, L.; Näntö-Salonen, K.; Pikta, M.; Kopatz, W.F.; Zuurveld, M.; Meijers, J.C.M.; et al. Abnormal coagulation and enhanced fibrinolysis due to lysinuric protein intolerance associates with bleeds and renal impairment. Haemophilia 2018, 24, e312–e321. [Google Scholar] [CrossRef]
- Treliński, J.; Pachniewska, K.; Matczak, J.; Nowak, P.; Robak, M.; Chojnowski, K. Assessment of Selected ROTEM Parameters, Kinetics of Fibrinogen Polymerization and Plasmin Amidolytic Activity in Patients with Congenital Fibrinogen Defects. Adv. Clin. Exp. Med. 2016, 25, 1255–1263. [Google Scholar] [CrossRef]
- Dupuy, E.; Soria, C.; Molho, P.; Zini, J.-M.; Rosenstingl, S.; Laurian, C.; Bruneval, P.; Tobelem, G. Embolized ischemic lesions of toes in an afibrinogenemic patient: Possible relevance to in vivo circulating thrombin. Thromb. Res. 2001, 102, 211–219. [Google Scholar] [CrossRef]
- Solomon, C.; Rahe-Meyer, N.; Schöchl, H.; Ranucci, M.; Görlinger, K. Effect of haematocrit on fibrin-based clot firmness in the FIBTEM test. Blood Transfus. 2013, 11, 412–418. [Google Scholar]
- Haverkate, F.; Samama, M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost. 1995, 73, 151–161. [Google Scholar]
- Callea, F.; Giovannoni, I.; Sari, S.; Guldal, E.; Dalgic, B.; Akyol, G.; Sogo, T.; Al-Hussaini, A.; Maggiore, G.; Bartuli, A.; et al. Fibrinogen Gamma Chain Mutations Provoke Fibrinogen and Apolipoprotein B Plasma Deficiency and Liver Storage. Int. J. Mol. Sci. 2017, 18, 2717. [Google Scholar] [CrossRef] [PubMed]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.C.; Arnold, D.M.; Coller, B.S.; James, P.D.; Neunert, C.E.; Lillicrap, D.; ISTH/SSC joint VWF; Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J. Thromb. Haemost. 2010, 8, 2063–2065. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; De Moerloose, B.C.; Honsberger, A.; Schönbörner, A.; Rossier, C.; Peerlinck, K.; Claeyssens, S.; Di Michele, D.; D’Oiron, R.; Dreyfus, M.; et al. Mutations in the fibrinogen Aα gene account for the majority of cases of congenital afibrinogenemia. Blood 2000, 96, 149–152. [Google Scholar] [CrossRef]
- Dunnen, J.T.D.; Antonarakis, S.E. Nomenclature for the description of human sequence variations. Hum. Genet. 2001, 109, 121–124. [Google Scholar] [CrossRef]
- Chitlur, M.; Rivard, G.E.; Lillicrap, D.; Mann, K.; Shima, M.; Young, G.; on the behalf of the Factor VIII, Factor IX, and Rare Coagulation Disorders Subcommittee of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Recommendations for performing thromboelastography/thromboelastometry in hemophilia: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2014, 12, 103–106. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Gender | Phenotypes | Fg Act | Fg Ag | Type | Gene | Mutation | Family Members | |
|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | HEL 1.1 | 72 | Female | Asymptomatic | 1.3 | 1.3 | Hypo 1 | FGG, exon 8 | het Thr303Pro and het Asp327His | Thrombosis |
| HEL 1.2 | 24 | Male | Asymptomatic | 0.7 | 0.7 | Hypo 1 | FGG, exon 8 | het Thr303Pro and het Asp327His | ||
| HEL 1.3 | 48 | Female | Thrombosis | 1.0 | 1.2 | Hypo 1 | FGG, exon 8 | het Thr303Pro and het Asp327His | ||
| Family 2 | HEL 2.1 | 82 | Male | Thrombosis | 1.2 | 2.5 | Dys 2 | FGG, exon 8 | het Tyr306Cys | Thrombosis |
| HEL 2.2 | 58 | Male | Asymptomatic | 1.6 | 2.5 | Dys 2 | FGG, exon 8 | homozygous Tyr306Cys | ||
| HEL 2.3 | 83 | Male | Asymptomatic | 1.7 | 3.5 | Dys 2 | FGG, exon 8 | het Tyr306Cys | ||
| Family 3 | HEL 3.1 | 40 | Female | Bleeding and thrombosis | 1.0 | 1.7 | Hypo 1 | FGG, exon 8 | het Thr303Pro and het Asp327His | Bleeding and thrombosis * |
| HEL 3.2 | 30 | Female | Bleeding | <1 | 0.7 | Hypo 1 | FGG, exon 8 | het Thr303Pro and het Asp327His | ||
| Not related | HEL 4 | 35 | Female | Asymptomatic | 0.8 | 1.8 | Hypodys 3 | FGG, exon 9 | het Trp395Leu | Thrombosis |
| HEL 5 | 38 | Female | Asymptomatic | <1 | 2.6 | Dys 2 | FGG, exon 9 | het c.1283-1284 del TG | Thrombosis | |
| HEL 6 | 43 | Female | Asymptomatic | 1.9 | 2.4 | Dys 2 | FGA, exon 2 | het Leu28Pro | NA | |
| HEL 7 | 47 | Female | Bleeding | <1 | 1.0 | Hypo 1 | FGG, exon 9 | het Thr397Ile | Bleeding | |
| GEN 1 | 37 | Female | Bleeding | 0.6 | 2.4 | Dys 2 | FGG | Arg301His | Bleeding | |
| GEN 2 | 39 | Female | Asymptomatic | 0.5 | 1.8 | Dys 2 | FGB | c.402_410 del GGAAGCTGT | NA | |
| GEN 3 | 49 | Female | Bleeding | 0.7 | 2.1 | Dys 2 | FGA | Arg35His | Bleeding | |
| GEN 4 | 30 | Male | Asymptomatic | 0.7 | 1.4 | Hypo 1 | FGB | Thr407Met | Bleeding | |
| GEN 5 | 51 | Male | Asymptomatic | 0.5 | 2.8 | Dys 2 | FGA | Arg38Gly | Asymptomatic | |
| GEN 6 | 44 | Male | Asymptomatic | 1.1 | 1.2 | Hypo 1 | FGG | Arg375Trp | Asymptomatic | |
| GEN 7 | 57 | Female | Bleeding and thrombosis | 1.1 | 1.8 | Hypodys 3 | FGB | Arg285Leu | Bleeding | |
| GEN 8 | 31 | Male | Asymptomatic | 0.4 | 0.4 | Hypo 1 | FGA | 11 kb del | Asymptomatic | |
| GEN 9 | 53 | Female | Asymptomatic | 0.6 | 3.1 | Dys 2 | FGA | Arg35His | NA | |
| GEN 10 | 21 | Female | Bleeding | 0.9 | 1.0 | Hypo 1 | FGB | Arg294Ser | Asymptomatic |
| STG-BLS Normalized | Lag Time, Ratio (Range) | ETP, % (Range) | Peak Height, % (Range) | Time to Peak, Ratio (Range) |
| Hypofibrinogenemia (n = 8) | 1.2 (0.9–1.4) | 82.7 (41.6–101.5) | 88,5 (30.6–113.2) | 1.1 (1.0–1.4) |
| Dysfibrinogenemia (n = 8) | 1.1 (0.8–2.0) | 80.4 (49.2–111.2) | 81.4 (46.6–104.5) | 1.1 (0.9–1.7) |
| Hypodys-fibrinogenemia (n = 3) | 1.0 (1.0–1.6) | 83.0 (49.2–105.0) | 81.4 (39.2–121.7) | 1.1 (1.0–1.3) |
| Reference range * | 0.9–1.3 | 57–114 | 51–99 | 0.9–1.3 |
| STG-TS-TM Normalized | Lag Time, Ratio (Range) | ETP, % (Range) | Peak Height, % (Range) | Time to Peak, Ratio (Range) |
| Hypofibrinogenemia (n = 8) | 1.1 (0.9–1.3) | 79.0 (46.3–90.5) | 75.1 (32.7–90.0) | 1.2 (1.1–1.7) |
| Dysfibrinogenemia (n = 8) | 1.1 (0.8–2.2) | 65.0 (48.1–96.5) | 62.2 (33.7–78.8) | 1.2 (0.9–2.0) |
| Hypodys-fibrinogenemia (n = 3) | 1.1 (1.1–1.5) | 80.8 (52.9–83.1) | 65.5 (28.8–89.1) | 1.2 (1.1–1.6) |
| Reference range * | 1.0–1.3 | 59–99 | 45–102 | 1.0–1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szanto, T.; Lassila, R.; Lemponen, M.; Lehtinen, E.; Neerman-Arbez, M.; Casini, A. Whole Blood Thromboelastometry by ROTEM and Thrombin Generation by Genesia According to the Genotype and Clinical Phenotype in Congenital Fibrinogen Disorders. Int. J. Mol. Sci. 2021, 22, 2286. https://doi.org/10.3390/ijms22052286
Szanto T, Lassila R, Lemponen M, Lehtinen E, Neerman-Arbez M, Casini A. Whole Blood Thromboelastometry by ROTEM and Thrombin Generation by Genesia According to the Genotype and Clinical Phenotype in Congenital Fibrinogen Disorders. International Journal of Molecular Sciences. 2021; 22(5):2286. https://doi.org/10.3390/ijms22052286
Chicago/Turabian StyleSzanto, Timea, Riitta Lassila, Marja Lemponen, Elina Lehtinen, Marguerite Neerman-Arbez, and Alessandro Casini. 2021. "Whole Blood Thromboelastometry by ROTEM and Thrombin Generation by Genesia According to the Genotype and Clinical Phenotype in Congenital Fibrinogen Disorders" International Journal of Molecular Sciences 22, no. 5: 2286. https://doi.org/10.3390/ijms22052286
APA StyleSzanto, T., Lassila, R., Lemponen, M., Lehtinen, E., Neerman-Arbez, M., & Casini, A. (2021). Whole Blood Thromboelastometry by ROTEM and Thrombin Generation by Genesia According to the Genotype and Clinical Phenotype in Congenital Fibrinogen Disorders. International Journal of Molecular Sciences, 22(5), 2286. https://doi.org/10.3390/ijms22052286