
Neuroprotective and Anti-Inflammatory Effects of Evernic Acid in an MPTP-Induced Parkinson’s Disease Model

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
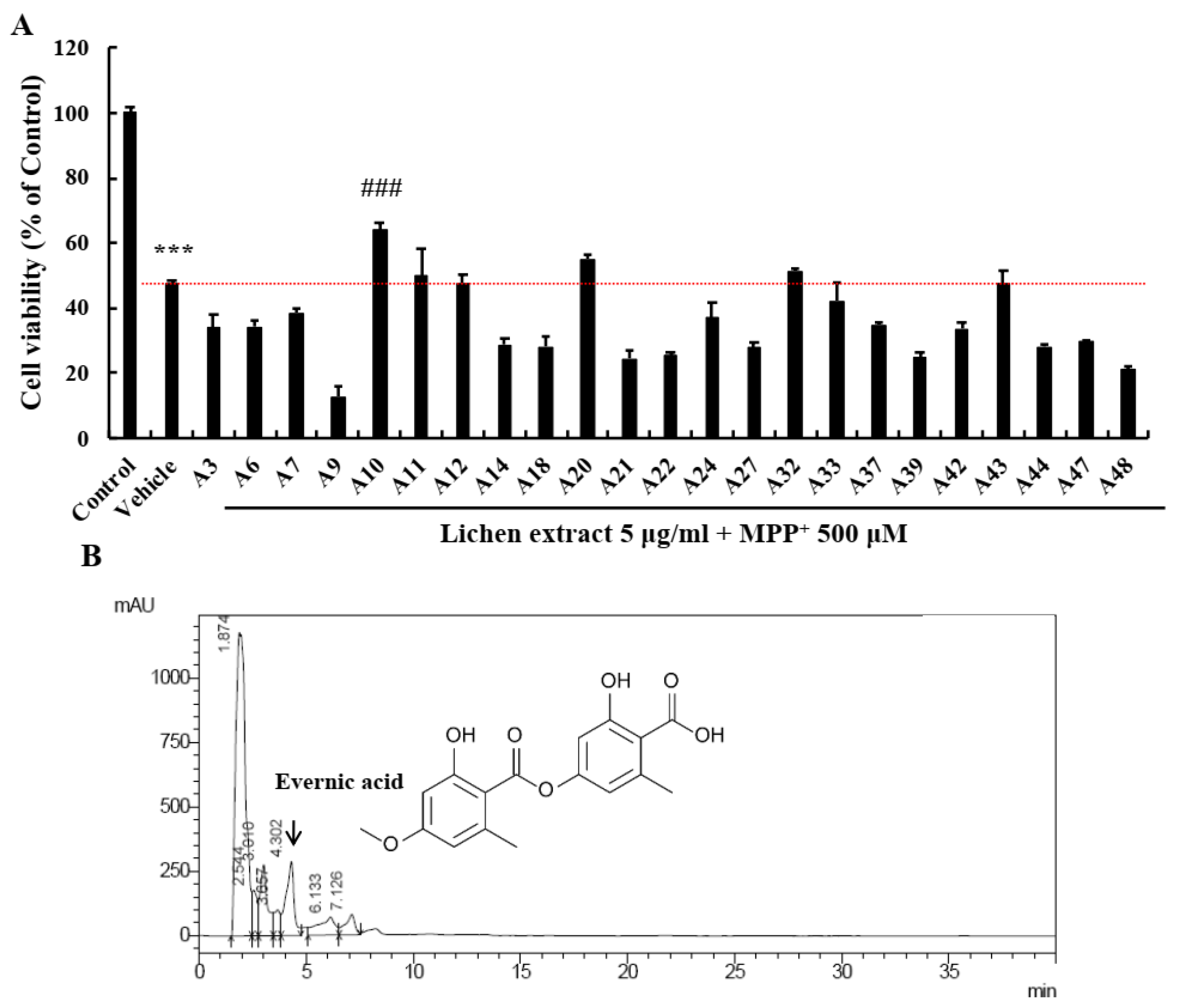
2.1. Screening of EA as a Neuroprotective Candidate in the Lichen Extract Library
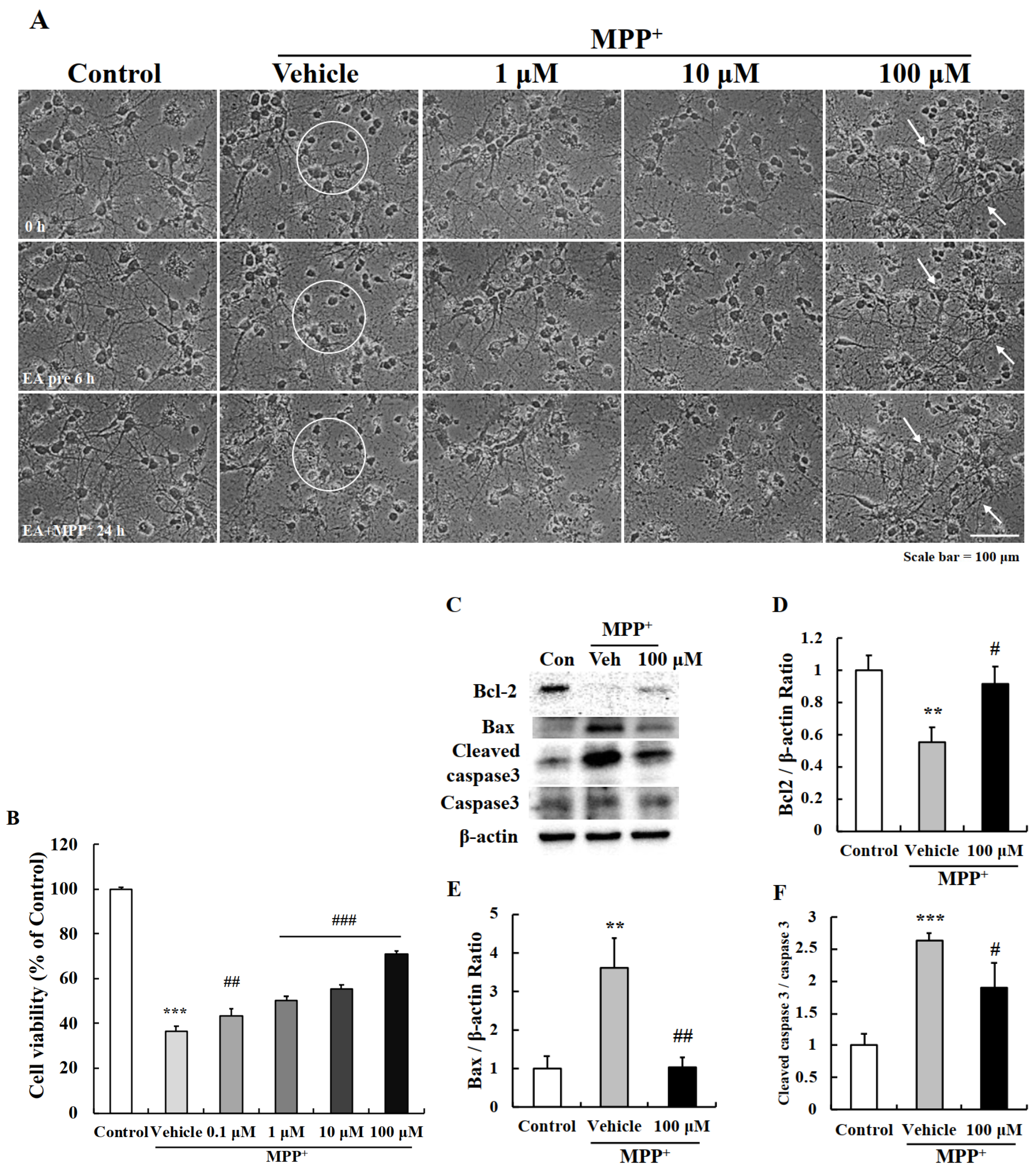
2.2. EA Protected Primary Neurons from MPP+-Induced Apoptosis
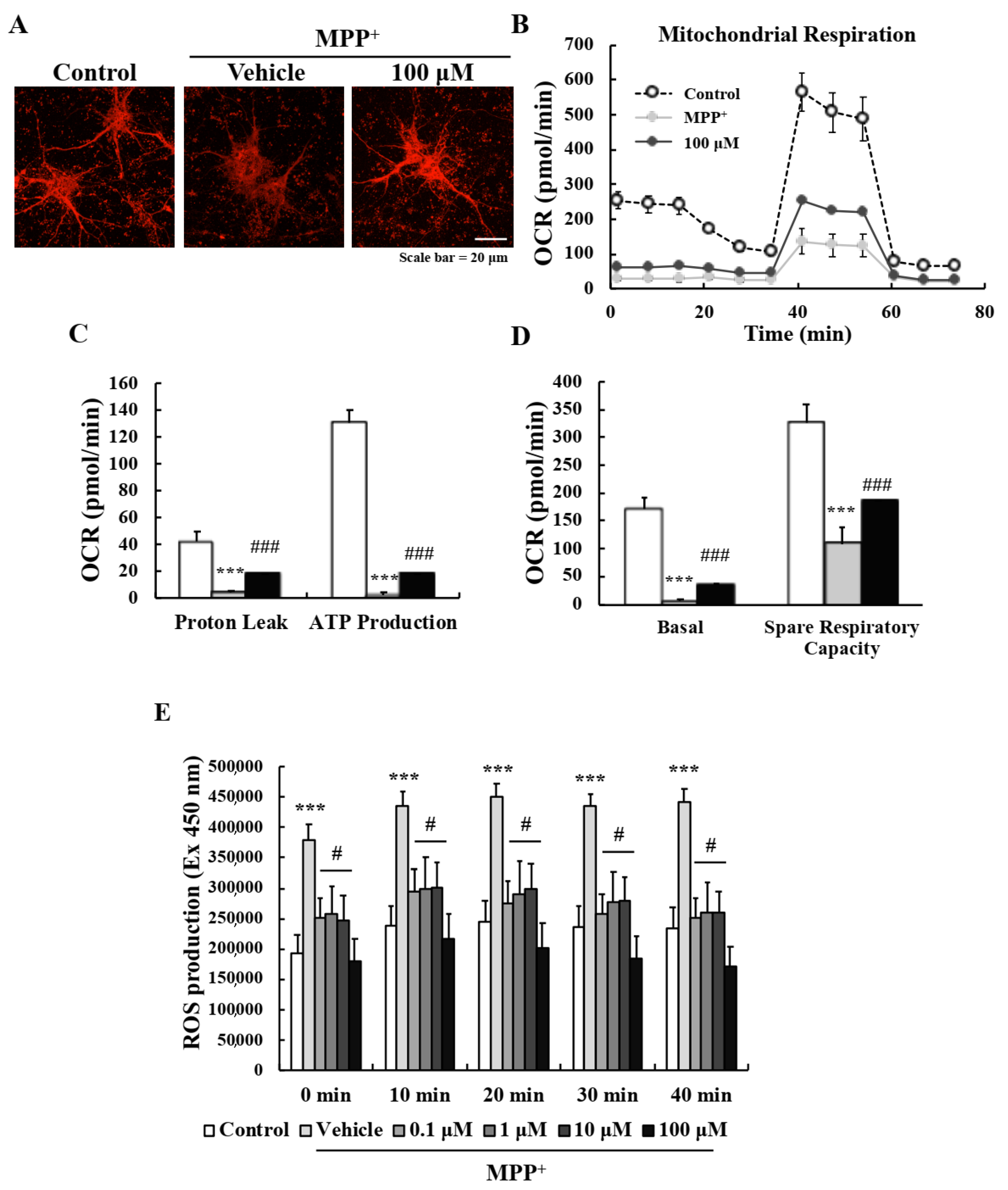
2.3. EA Suppressed MPP+-Induced Mitochondrial Dysfunction in Primary Neurons
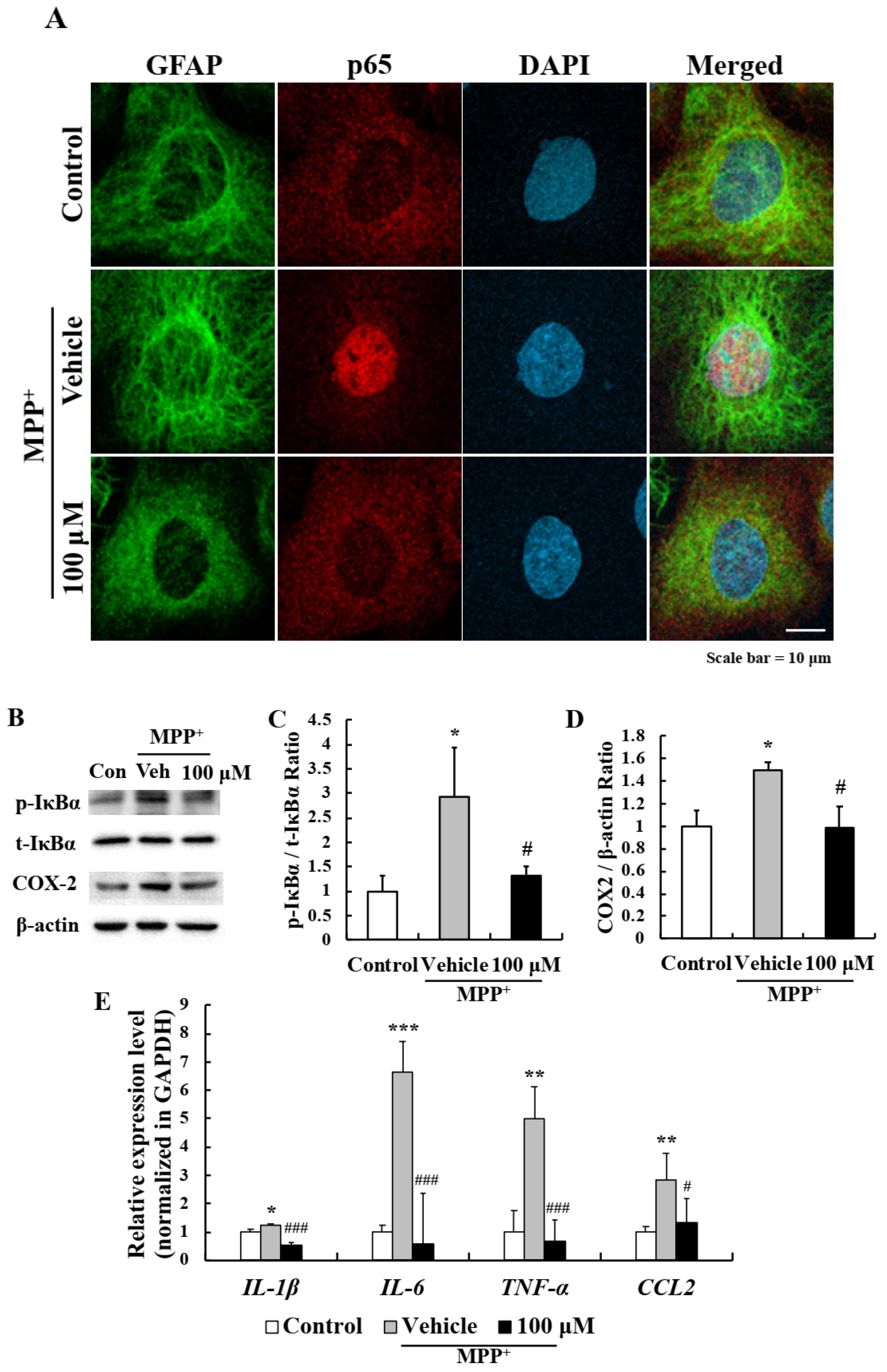
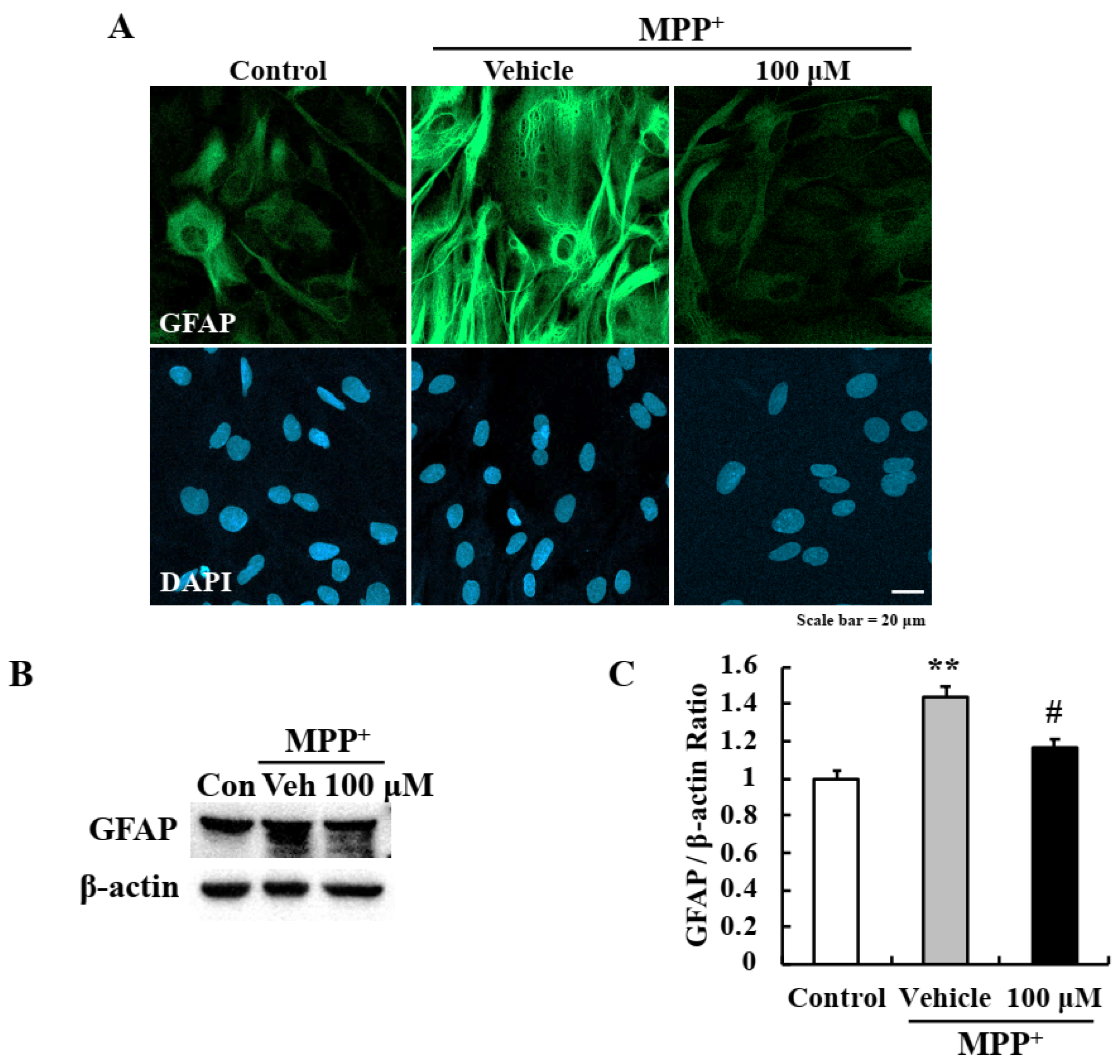
2.4. EA Inhibited MPP+-Induced Astrocyte Activation
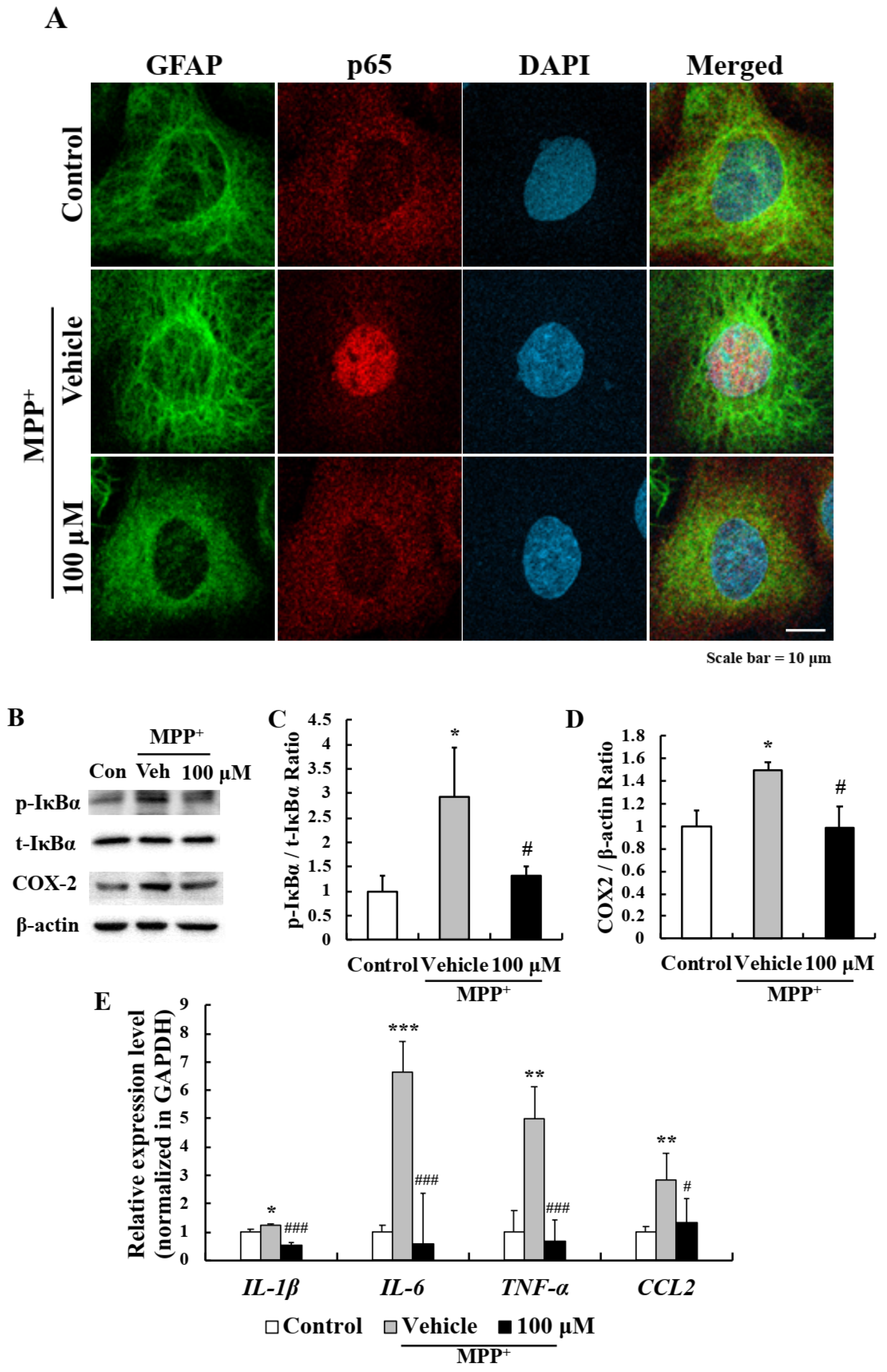
2.5. EA Inhibited the NF-κB Signaling Pathway in Primary Astrocytes
2.6. EA Attenuated Motor Dysfunction in the MPTP-Induced PD Mouse Model
2.7. EA Diminished MPTP-Induced Dopaminergic Neuronal Death in the Mouse Model
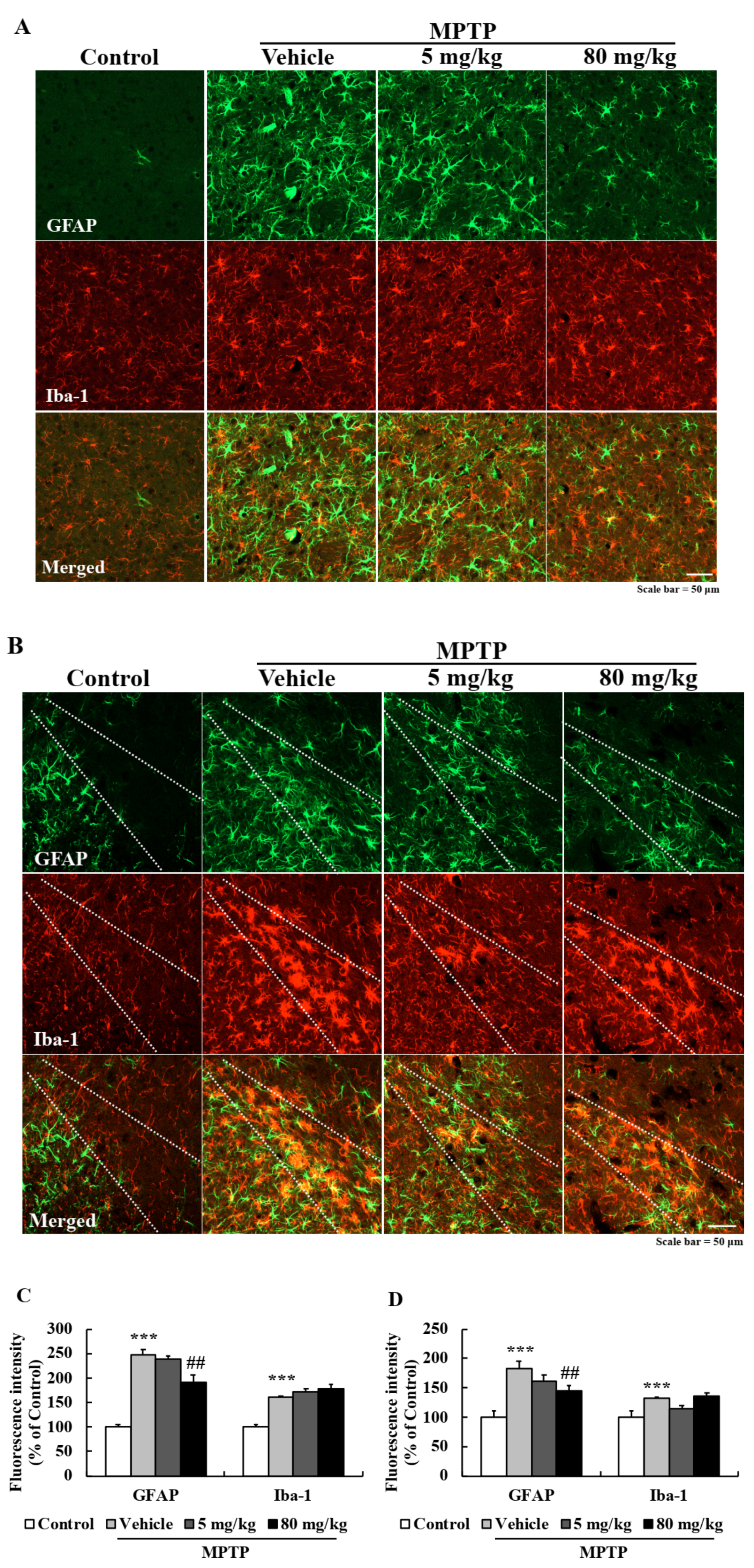
2.8. EA Suppressed Astroglial Activation in the PD Mouse Model
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. High-Performance Liquid Chromatography (HPLC)
4.3. Primary Neuron Culture
4.4. MTT Assay
4.5. Mitochondrial Membrane Potential (MMP) Measurement
4.6. Measurement of Cellular Mitochondrial Respiration
4.7. Reactive Oxygen Species (ROS) Assay
4.8. Western Blot Analysis
4.9. Primary Astrocyte Culture
4.10. Immunocytochemistry
4.11. RNA Isolation and Real-Time Polymerase Chain Reaction (Real-Time PCR)
4.12. Animals and Treatments
4.13. Motor Performance Testing
4.14. Tissue Preparation
4.15. Diaminobenzidine (DAB) Immunohistochemistry
4.16. Double Fluorescence Immunohistochemistry
4.17. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer’s Res. Ther. 2017, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, S.; Chang, S.C.; Lee, J. Significant roles of neuroinflammation in Parkinson’s disease: Therapeutic targets for PD prevention. Arch. Pharm Res. 2019, 42, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.J.; Bennett, J.P.; Biro, S.M.; Duque-Velasquez, J.C.; Rodriguez, C.M.; Bessen, R.A.; Rocke, T.E. Degradation of the disease-associated prion protein by a serine protease from lichens. PLoS ONE 2011, 6, e19836. [Google Scholar] [CrossRef] [PubMed]
- Kosanic, M.; Manojlovic, N.; Jankovic, S.; Stanojkovic, T.; Rankovic, B. Evernia prunastri and Pseudoevernia furfuraceae lichens and their major metabolites as antioxidant, antimicrobial and anticancer agents. Food Chem. Toxicol. 2013, 53, 112–118. [Google Scholar] [CrossRef]
- Olivier-Jimenez, D.; Chollet-Krugler, M.; Rondeau, D.; Beniddir, M.A.; Ferron, S.; Delhaye, T.; Allard, P.M.; Wolfender, J.L.; Sipman, H.J.M.; Lucking, R.; et al. A database of high-resolution MS/MS spectra for lichen metabolites. Sci. Data 2019, 6, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugni, T.S.; Andjelic, C.D.; Pole, A.R.; Rai, P.; Ireland, C.M.; Barrows, L.R. Biologically active components of a Papua New Guinea analgesic and anti-inflammatory lichen preparation. Fitoterapia 2009, 80, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanas, S.; Odabasoglu, F.; Halici, Z.; Cakir, A.; Aygun, H.; Aslan, A.; Suleyman, H. Evaluation of anti-inflammatory and antioxidant activities of Peltigera rufescens lichen species in acute and chronic inflammation models. J. Nat. Med. 2010, 64, 42–49. [Google Scholar] [CrossRef]
- Song, Y.; Dai, F.; Zhai, D.; Dong, Y.; Zhang, J.; Lu, B.; Luo, J.; Liu, M.; Yi, Z. Usnic acid inhibits breast tumor angiogenesis and growth by suppressing VEGFR2-mediated AKT and ERK1/2 signaling pathways. Angiogenesis 2012, 15, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Zhang, X.; Zhou, B.; Zhang, C.; Tu, J.; Chen, X.; Wang, J.; Gao, H.; Qin, G.; Pan, W. Usnic Acid Induces Cycle Arrest, Apoptosis, and Autophagy in Gastric Cancer Cells In Vitro and In Vivo. Med. Sci. Monit. 2018, 24, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Moriano, C.; Divakar, P.K.; Crespo, A.; Gomez-Serranillos, M.P. Protective effects of lichen metabolites evernic and usnic acids against redox impairment-mediated cytotoxicity in central nervous system-like cells. Food Chem. Toxicol. 2017, 105, 262–277. [Google Scholar] [CrossRef]
- Joulain, D.; Tabacchi, R. Lichen extracts as raw materials in perfumery. Part 1: Oakmoss. Flavour Frag. J. 2009, 24, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Halama, P.; Van Haluwin, C. Antifungal activity of lichen extracts and lichenic acids. Biocontrol 2004, 49, 95–107. [Google Scholar] [CrossRef]
- Sahin, S.; Oran, S.; Sahinturk, P.; Demir, C.; Ozturk, S. Ramalina Lichens and Their Major Metabolites as Possible Natural Antioxidant and Antimicrobial Agents. J. Food Biochem. 2015, 39, 471–477. [Google Scholar] [CrossRef]
- Kizil, H.E.; Agar, G.; Anar, M. Cytotoxic and antiproliferative effects of evernic acid on HeLa cell lines: A candidate anticancer drug. J. Biotechnol. 2014, 185, S29. [Google Scholar] [CrossRef]
- Shcherbakova, A.; Nyugen, L.; Koptina, A.; Backlund, A.; Shurgin, A.; Romanov, E.; Ulrich-Merzenich, G. Screening of compounds of Evernia prunastri (L.) for their antiproliferative activity in glioblastoma cells. Planta Med. 2016, 82, 465. [Google Scholar] [CrossRef]
- Lee, S.; Lee, Y.; Ha, S.; Chung, H.Y.; Kim, H.; Hur, J.S.; Lee, J. Anti-inflammatory effects of usnic acid in an MPTP-induced mouse model of Parkinson’s disease. Brain Res. 2020, 1730, 146642. [Google Scholar] [CrossRef] [PubMed]
- Einarsdottir, E.; Groeneweg, J.; Bjornsdottir, G.G.; Harethardottir, G.; Omarsdottir, S.; Ingolfsdottir, K.; Ogmundsdottir, H.M. Cellular mechanisms of the anticancer effects of the lichen compound usnic acid. Planta Med. 2010, 76, 969–974. [Google Scholar] [CrossRef]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Dunnett, S.B.; Bjorklund, A. Prospects for new restorative and neuroprotective treatments in Parkinson’s disease. Nature 1999, 399, A32–A39. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.; Lebiedzinska, M.; Machado, N.G.; Oliveira, P.J.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Mitochondrial Tolerance to Drugs and Toxic Agents in Ageing and Disease. Curr. Drug Targets 2011, 12, 827–849. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Choi, W.S.; Klintworth, H.M.; Xia, Z. JNK3-mediated apoptotic cell death in primary dopaminergic neurons. Methods Mol. Biol. 2011, 758, 279–292. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.M.; Ries, V.; Oo, T.F.; Yarygina, O.; Jackson-Lewis, V.; Ryu, E.J.; Lu, P.D.; Marciniak, S.J.; Ron, D.; Przedborski, S.; et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem. 2005, 95, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Lev, N.; Melamed, E.; Offen, D. Apoptosis and Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 245–250. [Google Scholar] [CrossRef]
- Cheng, E.H.; Kirsch, D.G.; Clem, R.J.; Ravi, R.; Kastan, M.B.; Bedi, A.; Ueno, K.; Hardwick, J.M. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 1997, 278, 1966–1968. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.; Caretto, A.; Boido, M.; Vercelli, A. Mitochondrial Dysfunctions: A Red Thread across Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3719. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Kosanic, M.; Rankovic, B.; Vukojevic, J. Antioxidant properties of some lichen species. J. Food Sci. Technol. 2011, 48, 584–590. [Google Scholar] [CrossRef] [Green Version]
- Suwalsky, M.; Jemiola-Rzeminska, M.; Astudillo, C.; Gallardo, M.J.; Staforelli, J.P.; Villena, F.; Strzalka, K. An in vitro study on the antioxidant capacity of usnic acid on human erythrocytes and molecular models of its membrane. Biochim. Biophys. Acta 2015, 1848, 2829–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Moriano, C.; Gomez-Serranillos, M.P.; Crespo, A. Antioxidant potential of lichen species and their secondary metabolites. A systematic review. Pharm. Biol. 2016, 54, 1–17. [Google Scholar] [CrossRef]
- Ebadi, M.; Srinivasan, S.K.; Baxi, M.D. Oxidative stress and antioxidant therapy in Parkinson’s disease. Prog. Neurobiol. 1996, 48, 1–19. [Google Scholar] [CrossRef]
- Filograna, R.; Beltramini, M.; Bubacco, L.; Bisaglia, M. Anti-Oxidants in Parkinson’s Disease Therapy: A Critical Point of View. Curr. Neuropharmacol. 2016, 14, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation-An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8412. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Lee, K.M.; Lee, Y.; Chun, H.J.; Kim, A.H.; Kim, J.Y.; Lee, J.Y.; Ishigami, A.; Lee, J. Neuroprotective and anti-inflammatory effects of morin in a murine model of Parkinson’s disease. J. Neurosci. Res. 2016, 94, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.F.; Wang, W.P.; Zheng, X.F.; Chen, Z.; Chen, T.; Huang, Z.Y.; Jia, L.J.; Lei, W.L. Characteristic response of striatal astrocytes to dopamine depletion. Neural. Regen. Res. 2020, 15, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadiere, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Invest. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.H. CD4+ T cells mediate cytotoxicity in neurodegenerative diseases. J. Clin. Invest. 2009, 119, 13–15. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E. Drug delivery systems-associated with pediatric endocrinology. J. Korean Soc. Pediatr. Endocrinol. 2011, 16, 7–12. [Google Scholar] [CrossRef]
- Patel, R.; Barker, J.; ElShaer, A. Pharmaceutical Excipients and Drug Metabolism: A Mini-Review. Int. J. Mol. Sci. 2020, 21, 8224. [Google Scholar] [CrossRef] [PubMed]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef]
- Lee, S.; Park, H.R.; Lee, J.Y.; Cho, J.H.; Song, H.M.; Kim, A.H.; Lee, W.; Lee, Y.; Chang, S.C.; Kim, H.S.; et al. Learning, memory deficits, and impaired neuronal maturation attributed to acrylamide. J. Toxicol. Environ. Health 2018, 81, 254–265. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Koutouzis, T.K.; Freeman, T.B.; Cahill, D.W.; Sanberg, P.R. Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of Huntington’s disease. Brain Res. 1995, 697, 254–257. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Suh, Y.J.; Yang, S.; Hong, D.G.; Ishigami, A.; Kim, H.; Hur, J.-S.; Chang, S.-C.; Lee, J. Neuroprotective and Anti-Inflammatory Effects of Evernic Acid in an MPTP-Induced Parkinson’s Disease Model. Int. J. Mol. Sci. 2021, 22, 2098. https://doi.org/10.3390/ijms22042098
Lee S, Suh YJ, Yang S, Hong DG, Ishigami A, Kim H, Hur J-S, Chang S-C, Lee J. Neuroprotective and Anti-Inflammatory Effects of Evernic Acid in an MPTP-Induced Parkinson’s Disease Model. International Journal of Molecular Sciences. 2021; 22(4):2098. https://doi.org/10.3390/ijms22042098
Chicago/Turabian StyleLee, Seulah, Yeon Ji Suh, Seonguk Yang, Dong Geun Hong, Akihito Ishigami, Hangun Kim, Jae-Seoun Hur, Seung-Cheol Chang, and Jaewon Lee. 2021. "Neuroprotective and Anti-Inflammatory Effects of Evernic Acid in an MPTP-Induced Parkinson’s Disease Model" International Journal of Molecular Sciences 22, no. 4: 2098. https://doi.org/10.3390/ijms22042098