
Changes in Apoptotic Pathways in MOLM-13 Cell Lines after Induction of Resistance to Hypomethylating Agents

 and
and
Abstract
:1. Introduction
2. Results
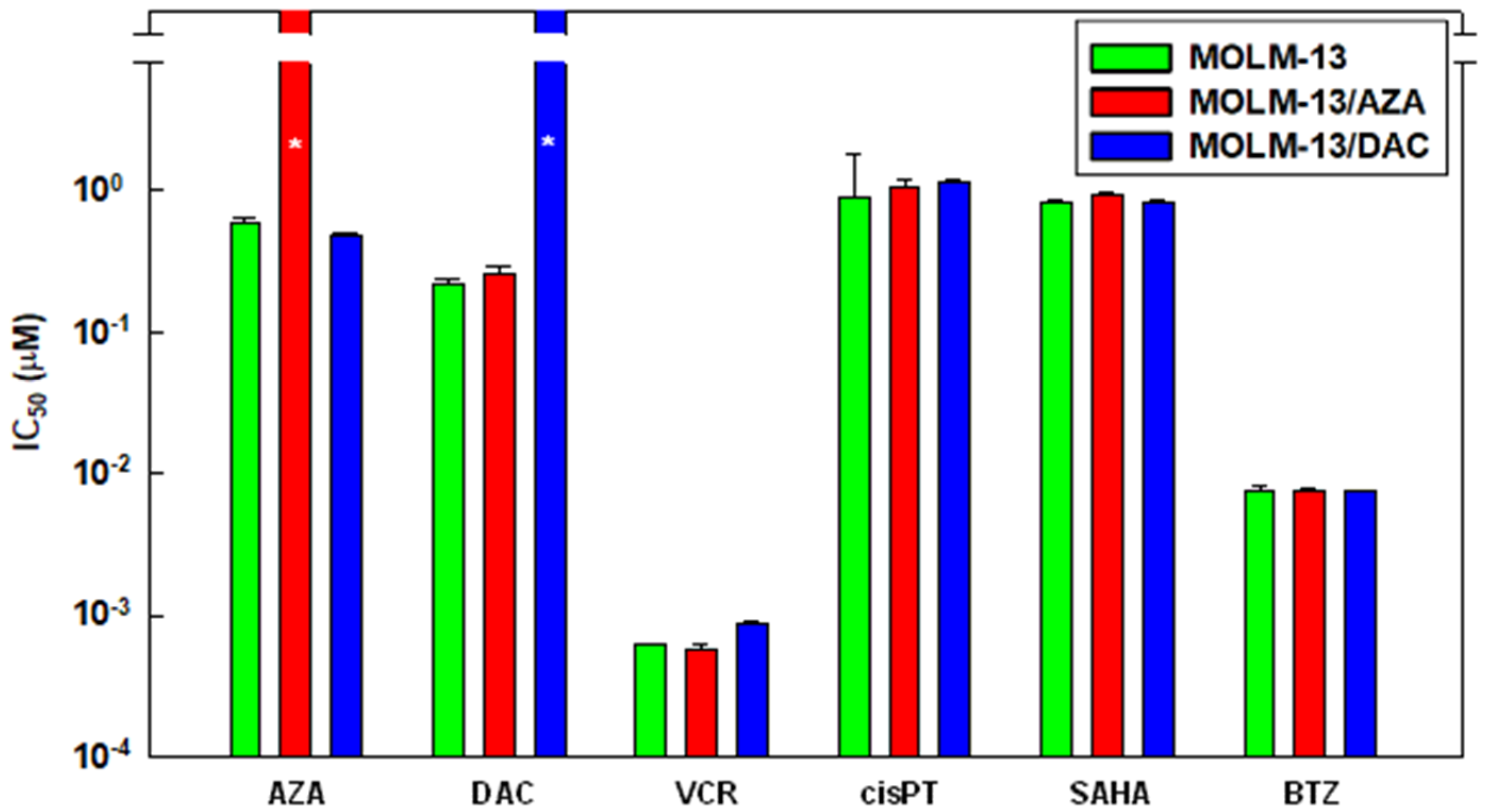
2.1. Introduction of AZA- and DAC-Resistant MOLM-13 Cell Variants
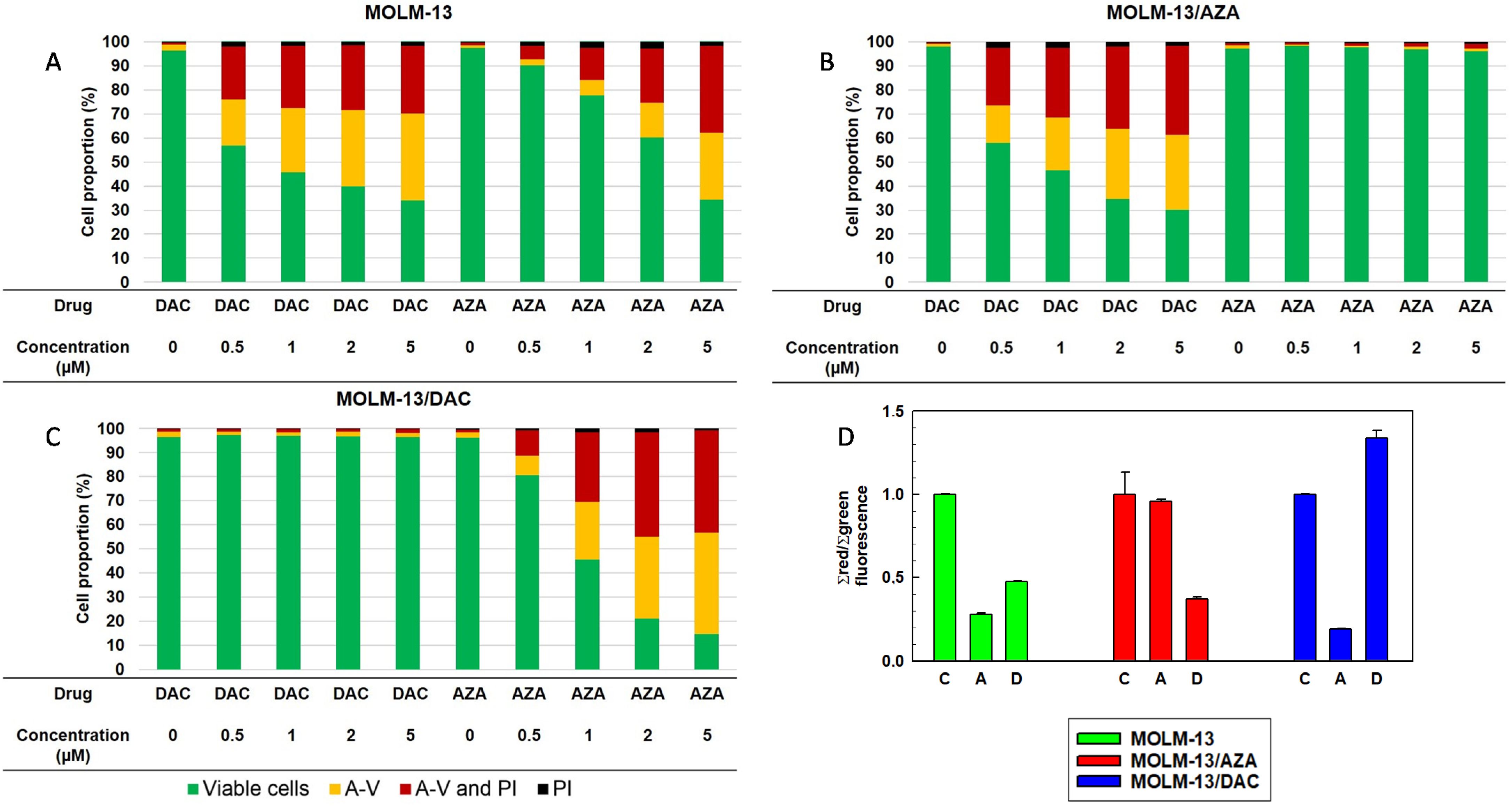
2.2. Effect of AZA and DAC on Apoptosis/Necrosis Progression in the Sensitive MOLM-13 Cell Line and the Resistant MOLM-13/AZA and MOLM-13/DAC Cell Variants
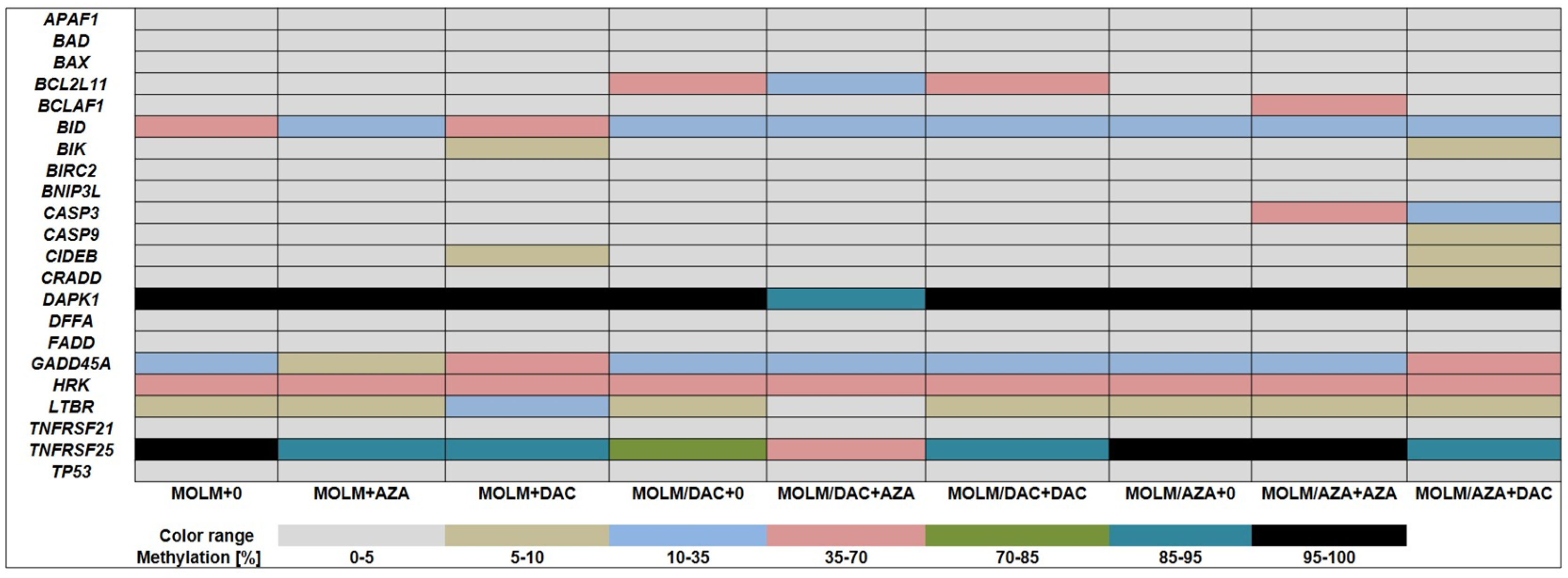
2.3. Methylation Status of Several Genes Involved in the Regulation of Apoptosis of MOLM-13, MOLM-13/AZA and MOLM-13/DAC Cells
2.4. Relative Expression of CASP3, DAPK1, GADD45A, TNFRSF25, BCL2L11, BCLAF1, BID, and BIK in Sensitive and Resistant Cell Lines and Its Relationship to the Methylation Status of the Respective Promoters
2.5. Changes in the Protein Levels of BCL2 and BAX in the MOLM-13, MOLM-13/DAC, MOLM-13/AZA Cells
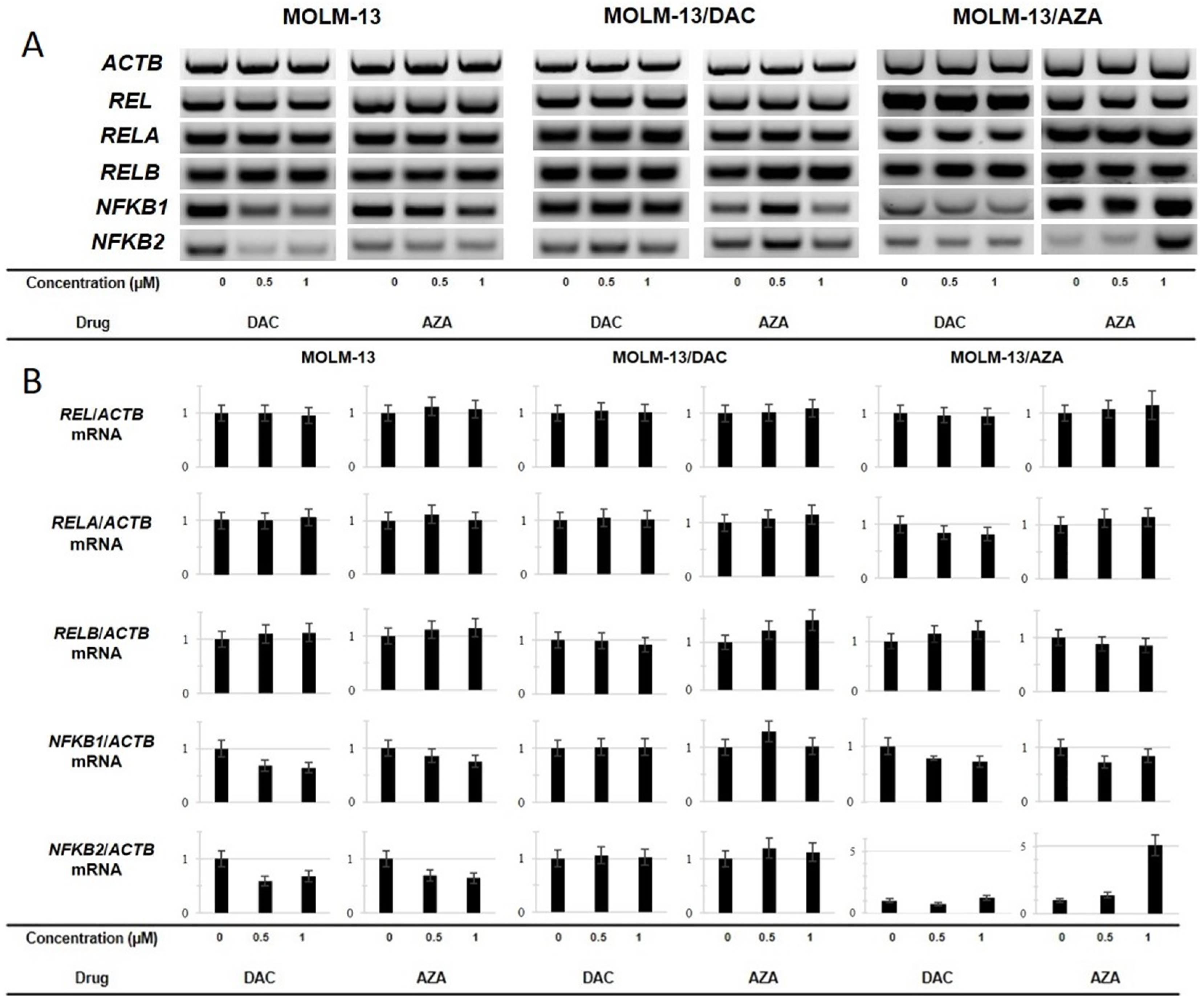
2.6. Expression of the REL, RELA, RELB, NFKB1 and NFKB2 Genes in the Sensitive and Resistant Cell Lines
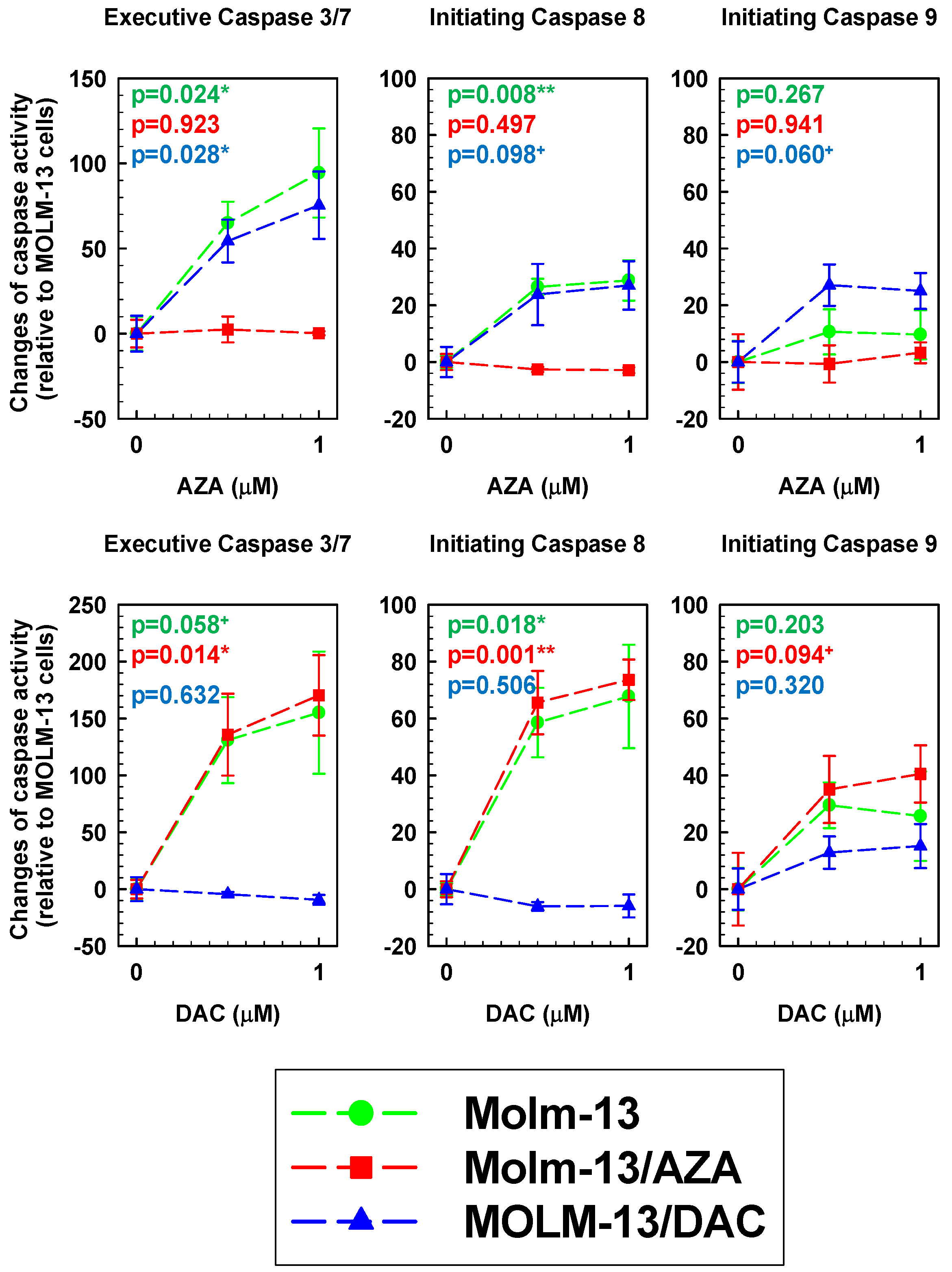
2.7. AZA- and DAC-Induced Activation of Caspase 3/7, 8, and 9 in All Three Types of MOLM-13 Cells
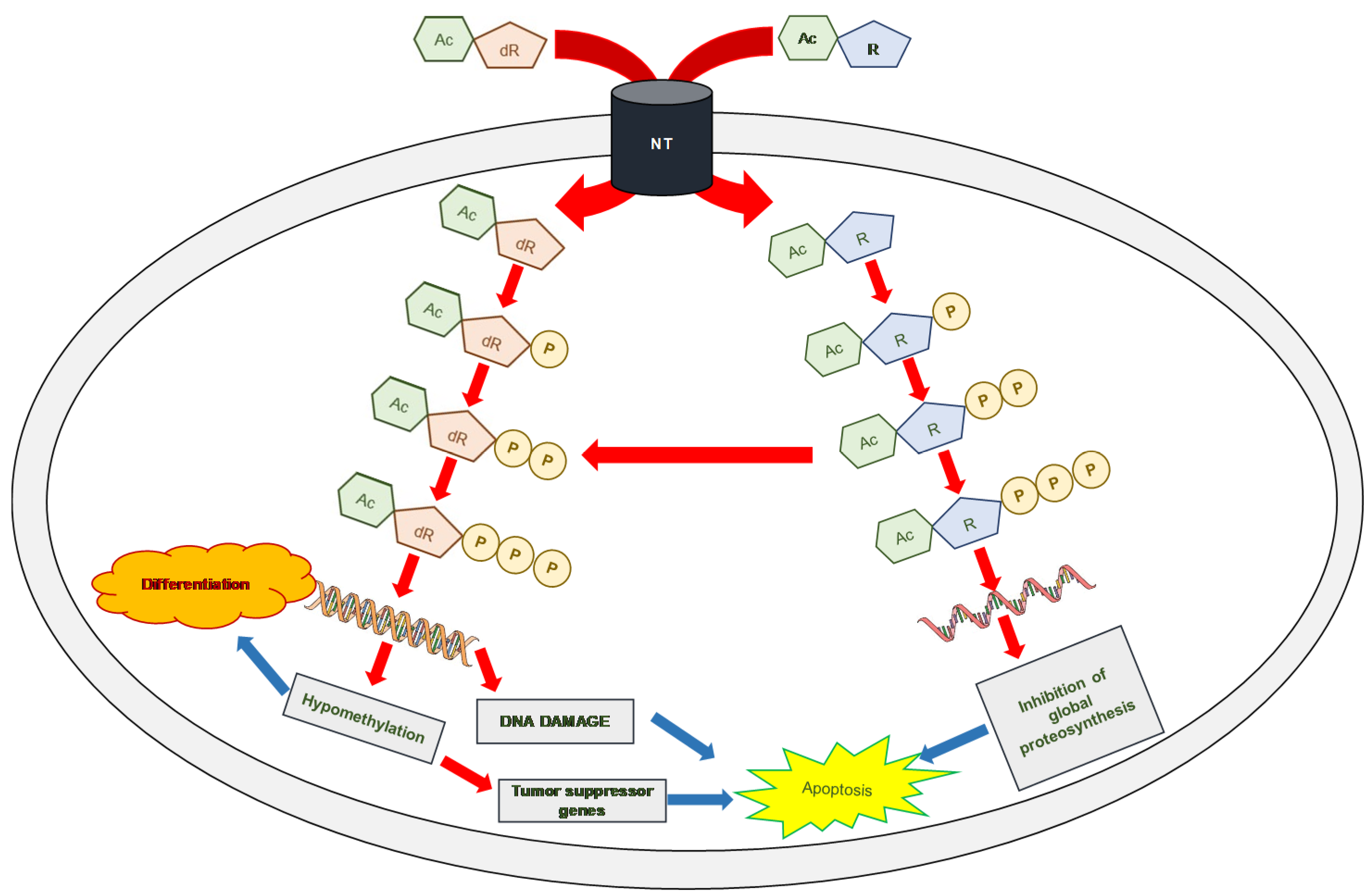
3. Discussion
4. Materials and Methods
4.1. Cell Culture Conditions
4.2. Cell Viability MTS Assay
4.3. Genomic DNA Isolation from Cell Lines
4.4. DNA Methylation Detection on Human Apoptosis Genes
4.5. Detection of BCL2, BAX and GAPDH Protein Levels in MOLM-13, MOLM-13/DAC, and MOLM 13/AZA Cells
4.6. Detection of AZA- and DAC-Induced Apoptosis and Necrosis in Sensitive and Resistant MOLM-13 Cells
4.7. Determination of the REL, NFKB1, NFKB2, RELA, and RELB Transcript Levels in MOLM-13 and MOLM-13/DAC and MOLM-13/AZA Cells
4.8. Measurement of JC-1 Fluorescence in Sensitive and Resistant MOLM-13 Cells by Fluorescence Cytometry
4.9. qRT-PCR
4.10. Detection of Caspase Activity
4.11. Statistical Analysis and Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassette |
| ABCB1 (C1, G2) | ABC transporter B1 (C1, G2) |
| ACTB | Gene for β-actin |
| AML | Acute myeloid leukemia |
| APAF1 | Apoptotic peptidase activating factor 1 |
| AZA | 5-Azacytidine |
| BAD | BCL2-associated agonist of cell death |
| BAX | BCL2-associated X protein |
| BCL2 | B-Cell CLL/Lymphoma 2 |
| BCL2L11 | BCL2-like 11 |
| BCLAF1 | BCL2-associated transcription factor |
| BID | BH3 interacting domain death agonist |
| BIK | BCL2-interacting killer |
| BIRC | Baculoviral IAP repeat-containing 2 |
| BM | Bone marrow |
| BNIPL3 | BCL2-interacting protein 3-like |
| BTZ | Bortezomib |
| CASP3 | Caspase 3 |
| CASP9 | Caspase 9 |
| CCCP | Carbonyl cyanide m-chlorophenylhydrazone |
| CD33 | Cluster of differentiation 33 protein |
| CIDEB | Cell death- inducing DFFA like effector B |
| cisPt | Cisplatin |
| DAC | 5-Aza-2′-deoxycytidine |
| DFFA | DNA fragmentation factor, 45 kDa, alpha polypeptide |
| DNA | Deoxyribonucleic acid |
| DNaseI | Deoxyribonuclease I |
| DR3 | Death receptor 3 |
| FADD | Fas (TNFRSF6)-associated via death domain |
| FAV | FITC-linked annexin V |
| FITC | Fluorescein isothiocyanate |
| GADD45A | Growth arrest and DNA damage-inducible, alpha factor |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GRADO | CASP2 and RIPK1 domain containing adaptor with death domain |
| HMA | Hypomethylating agents |
| HMC-1,2 cells | Human mast cell leukemia cell line |
| HRK | Harakiri, BCL2 Interacting protein |
| JC-1 | 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide |
| LTBR | Lymphotoxin beta receptor (TNFR superfamily, member 3) |
| MDS | Myelodysplastic syndromes |
| MMP | Mitochondrial membrane potential |
| MOLM-13/AZA | AZA resistant MOLM-13 cells |
| MOLM-13/DAC | DAC resistant MOLM-13 cells |
| mPTP | Mitochondrial permeability transition pore |
| mRNA | Messenger RNA |
| NFKB1(2) | Nuclear Factor-Kappa B Subunit 1 (2) |
| NF-κB | Nuclear Factor-Kappa B |
| PCR | Polymerase chain reaction |
| Pgp | P glycoprotein |
| PI | Propidium iodide |
| qRT-PCR | Quantitative reverse transcription—PCR |
| REL | V-Rel Avian Reticuloendotheliosis Viral Oncogene Homolog |
| RELA | V-Rel Avian Reticuloendotheliosis Viral Oncogene Homolog A |
| RELB | V-Rel Avian Reticuloendotheliosis Viral Oncogene Homolog B |
| RNA | Ribonucleic acid |
| RT-PCR | Reverse transcription—PCR |
| SAHA | Suberanilohydroxamic acid |
| TNFR | Tumor necrosis factor receptor |
| TNFRSF21 (25) | Tumor necrosis factor receptor superfamily 21 (25) |
| TP53 | Tumor protein p53 |
| TRADD | TNFR-associated death domain |
References
- Steensma, D.P. Myelodysplastic Syndromes: Diagnosis and Treatment. Mayo. Clin. Proc. 2015, 90, 969–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, J.K.; Wang, J.C.; Hope, K.J.; Jin, L.; Dick, J.E. Concepts of human leukemic development. Oncogene 2004, 23, 7164–7177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P. Myelodysplastic syndromes current treatment algorithm 2018. Blood Cancer J. 2018, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Pískala, A.; Šorm, F. Nucleic acids components and their analogues. LI. Synthesis of 1-glycosyl derivatives of 5-azauracil and 5-azacytosine. Collect. Czechoslov. Chem. Commun. 1964, 29, 2060–2076. [Google Scholar] [CrossRef]
- Pliml, J.; Sorm, F. Synthesis of a 2-deoxy-D-ribofuranosyl-s-azacytosine. Collect. Czechoslov. Chem. Commun. 1964, 29, 2576–2578. [Google Scholar] [CrossRef]
- Diesch, J.; Zwick, A.; Garz, A.K.; Palau, A.; Buschbeck, M.; Gotze, K.S. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenetics 2016, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [Green Version]
- Messingerova, L.; Imrichova, D.; Coculova, M.; Zelina, M.; Pavlikova, L.; Kavcova, H.; Seres, M.; Bohacova, V.; Lakatos, B.; Sulova, Z.; et al. Different Mechanisms of Drug Resistance in Myelodysplastic Syndromes and Acute Myeloid Leukemia. In Myelodysplastic Syndromes; Fusch, O., Ed.; Intech: Rijeka, Croatia, 2016; Chapter 7; pp. 181–200. [Google Scholar] [CrossRef] [Green Version]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Licht, J.D. DNA Methylation Inhibitors in Cancer Therapy: The Immunity Dimension. Cell 2015, 162, 938–939. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Issa, J.P.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 2006, 106, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Oki, Y.; Garcia-Manero, G.; Huang, X.; O’Brien, S.; Cortes, J.; Faderl, S.; Bueso-Ramos, C.; Ravandi, F.; Estrov, Z.; et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 2007, 109, 52–57. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; O’Brien, S.; Huang, X.; Garcia-Manero, G.; Ravandi, F.; Cortes, J.; Shan, J.; Davisson, J.; Bueso-Ramos, C.E.; Issa, J.P. Survival advantage with decitabine versus intensive chemotherapy in patients with higher risk myelodysplastic syndrome: Comparison with historical experience. Cancer 2007, 109, 1133–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Garcia-Manero, G.; Batty, N.; Shan, J.; O’Brien, S.; Cortes, J.; Ravandi, F.; Issa, J.P.; Kantarjian, H. Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy. Cancer 2010, 116, 3830–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prebet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Ades, L.; et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef]
- Cluzeau, T.; Robert, G.; Mounier, N.; Karsenti, J.M.; Dufies, M.; Puissant, A.; Jacquel, A.; Renneville, A.; Preudhomme, C.; Cassuto, J.P.; et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget 2012, 3, 490–501. [Google Scholar] [CrossRef] [Green Version]
- de Necochea-Campion, R.; Shouse, G.P.; Zhou, Q.; Mirshahidi, S.; Chen, C.S. Aberrant splicing and drug resistance in AML. J. Hematol. Oncol. 2016, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Masala, E.; Valencia, A.; Brogi, A.; Sanna, A.; Torres Martìn, M.; Figueroa, M.; Santini, v. Resistance to Azacitidine is Determined at Cellular Level by Lower Expression of Nucleoside Metabolizing Enzymes. Leuk. Res. 2017, 55 (Suppl. 1), S90–S91. [Google Scholar] [CrossRef]
- Sripayap, P.; Nagai, T.; Uesawa, M.; Kobayashi, H.; Tsukahara, T.; Ohmine, K.; Muroi, K.; Ozawa, K. Mechanisms of resistance to azacitidine in human leukemia cell lines. Exp. Hematol. 2014, 42, 294–306.e292. [Google Scholar] [CrossRef]
- Wu, P.; Geng, S.; Weng, J.; Deng, C.; Lu, Z.; Luo, C.; Du, X. The hENT1 and DCK genes underlie the decitabine response in patients with myelodysplastic syndrome. Leuk. Res. 2015, 39, 216–220. [Google Scholar] [CrossRef]
- Ghanim, V.; Herrmann, H.; Heller, G.; Peter, B.; Hadzijusufovic, E.; Blatt, K.; Schuch, K.; Cerny-Reiterer, S.; Mirkina, I.; Karlic, H.; et al. 5-azacytidine and decitabine exert proapoptotic effects on neoplastic mast cells: Role of FAS-demethylation and FAS re-expression, and synergism with FAS-ligand. Blood 2012, 119, 4242–4252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, T.H.; Podack, E.R. Immunobiology of TNFSF15 and TNFRSF25. Immunol. Res. 2013, 57, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Messingerova, L.; Imrichova, D.; Kavcova, H.; Seres, M.; Sulova, Z.; Breier, A. A decrease in cellular microRNA-27a content is involved in azacytidine-induced P-glycoprotein expression in SKM-1 cells. Toxicol. In Vitro 2016, 36, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Messingerova, L.; Imrichova, D.; Kavcova, H.; Turakova, K.; Breier, A.; Sulova, Z. Acute myeloid leukemia cells MOLM-13 and SKM-1 established for resistance by azacytidine are crossresistant to P-glycoprotein substrates. Toxicol. In Vitro 2015, 29, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocibalova, Z.; Guzyova, M.; Imrichova, D.; Sulova, Z.; Breier, A. Overexpression of the ABCB1 drug transporter in acute myeloid leukemia cells is associated with downregulation of latrophilin-1. Gen. Physiol. Biophys 2018, 37, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Elefantova, K.; Lakatos, B.; Kubickova, J.; Sulova, Z.; Breier, A. Detection of the Mitochondrial Membrane Potential by the Cationic Dye JC-1 in L1210 Cells with Massive Overexpression of the Plasma Membrane ABCB1 Drug Transporter. Int. J. Mol. Sci. 2018, 19, 1985. [Google Scholar] [CrossRef] [Green Version]
- Burke, C. JC-1 Mitochondrial Membrane Potential; Accuri Cytometers Inc: Ann Arbor, MI, USA, 2009; pp. 1–6. [Google Scholar]
- Cande, C.; Vahsen, N.; Metivier, D.; Tourriere, H.; Chebli, K.; Garrido, C.; Tazi, J.; Kroemer, G. Regulation of cytoplasmic stress granules by apoptosis-inducing factor. J. Cell Sci. 2004, 117, 4461–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakke, A.C. The Principles of Flow Cytometry. Lab. Med. 2001, 32, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Webster, G.A.; Bowles, M.J.; Karim, M.S.; Wood, R.F.; Pockley, A.G. Flow cytometric analysis of peripheral blood lymphocyte subset light scatter characteristics as a means of monitoring the development of rat small bowel allograft rejection. Clin. Exp. Immunol. 1995, 100, 536–542. [Google Scholar] [CrossRef]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and beyond: Cytometry in studies of programmed cell death. Methods Cell Biol. 2011, 103, 55–98. [Google Scholar] [CrossRef] [Green Version]
- QIAGEN. EpiTect® Methyl II PCR Array Handbook. 2019. Available online: https://www.qiagen.com/de/resources/resourcedetail?id=b466ccad-9550-471a-914f-e512d49f609e&lang=en (accessed on 8 March 2019).
- Sai, T.; Goto, Y.; Yoshioka, R.; Maeda, A.; Matsuda, F.; Sugimoto, M.; Wongpanit, K.; Jin, H.Z.; Li, J.Y.; Manabe, N. Bid and Bax are involved in granulosa cell apoptosis during follicular atresia in porcine ovaries. J. Reprod. Dev. 2011, 57, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Huang, K.; O’Neill, K.L.; Pang, X.; Luo, X. Bax/Bak activation in the absence of Bid, Bim, Puma, and p53. Cell Death Dis. 2016, 7, e2266. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Caspase-3 and prostaglandins signal for tumor regrowth in cancer therapy. Oncogene 2012, 31, 2805–2808. [Google Scholar] [CrossRef]
- Farag, A.K.; Roh, E.J. Death-associated protein kinase (DAPK) family modulators: Current and future therapeutic outcomes. Med. Res. Rev. 2019, 39, 349–385. [Google Scholar] [CrossRef] [Green Version]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Jacobson, K.N.; McDermott, K.M.; Reddy, S.P.; Cress, A.E.; Tang, H.; Dudek, S.M.; Black, S.M.; Garcia, J.G.; Makino, A.; et al. ATP promotes cell survival via regulation of cytosolic [Ca2+] and Bcl-2/Bax ratio in lung cancer cells. Am. J. Physiol. Cell Physiol. 2016, 310, C99–C114. [Google Scholar] [CrossRef] [Green Version]
- AAT Bioquest. Cell Meter™ Multiplexing Caspase 3/7, 8 and 9 Activity Assay Kit “Triple Fluorescence Colors”. 2020. Available online: https://www.aatbio.com/products/cell-meter-multiplexing-caspase-3-7-8-and-9-activity-assay-kit-triple-fluorescence-colors (accessed on 8 February 2020).
- Hano, M.; Tomasova, L.; Seres, M.; Pavlikova, L.; Breier, A.; Sulova, Z. Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors. Molecules 2018, 23, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imrichova, D.; Messingerova, L.; Seres, M.; Kavcova, H.; Pavlikova, L.; Coculova, M.; Breier, A.; Sulova, Z. Selection of resistant acute myeloid leukemia SKM-1 and MOLM-13 cells by vincristine-, mitoxantrone- and lenalidomide-induced upregulation of P-glycoprotein activity and downregulation of CD33 cell surface exposure. Eur. J. Pharm. Sci. 2015, 77, 29–39. [Google Scholar] [CrossRef]
- Coculova, M.; Imrichova, D.; Seres, M.; Messingerova, L.; Bohacova, V.; Sulova, Z.; Breier, A. The expression of P-glycoprotein in leukemia cells is associated with the upregulated expression of nestin, a class 6 filament protein. Leuk. Res. 2016, 48, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Imrichova, D.; Coculova, M.; Messingerova, L.; Sulova, Z.; Breier, A. Vincristine-induced expression of P-glycoprotein in MOLM-13 and SKM-1 acute myeloid leukemia cell lines is associated with coexpression of nestin transcript. Gen. Physiol. Biophys. 2014, 33, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into p-glycoprotein as a drug target. Anticancer. Agents Med. Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Duchmann, M.; Itzykson, R. Clinical update on hypomethylating agents. Int. J. Hematol. 2019, 110, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Borthakur, G.; Ahdab, S.E.; Ravandi, F.; Faderl, S.; Ferrajoli, A.; Newman, B.; Issa, J.P.; Kantarjian, H. Activity of decitabine in patients with myelodysplastic syndrome previously treated with azacitidine. Leuk. Lymphoma 2008, 49, 690–695. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.H.; Jung, S.H.; Goo, B.K.; Moon, J.; Choi, Y.; Choi, D.R.; Chung, Y.J.; Lee, J.H. Establishment and characterization of hypomethylating agent-resistant cell lines, MOLM/AZA-1 and MOLM/DEC-5. Oncotarget 2017, 8, 11748–11762. [Google Scholar] [CrossRef] [Green Version]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, H.C.; Giedl, J.; Wild, P.J.; Fine, S.W.; Humphrey, P.A.; Dehner, L.P.; Amin, M.B.; Epstein, J.I.; Blaszyk, H.; Hughes, D.; et al. Low frequency of epigenetic events in urothelial tumors in young patients. J. Urol. 2010, 184, 459–463. [Google Scholar] [CrossRef]
- Seelan, R.S.; Mukhopadhyay, P.; Warner, D.R.; Smolenkova, I.A.; Pisano, M.M.; Greene, R.M. Determinants of orofacial clefting II: Effects of 5-Aza-2’-deoxycytidine on gene methylation during development of the first branchial arch. Reprod. Toxicol. 2017, 67, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Tobiasson, M.; Abdulkadir, H.; Lennartsson, A.; Katayama, S.; Marabita, F.; De Paepe, A.; Karimi, M.; Krjutskov, K.; Einarsdottir, E.; Grovdal, M.; et al. Comprehensive mapping of the effects of azacitidine on DNA methylation, repressive/permissive histone marks and gene expression in primary cells from patients with MDS and MDS-related disease. Oncotarget 2017, 8, 28812–28825. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.E.; de Vasconcellos, J.F.; Sarkar, D.; Libermann, T.A.; Fisher, P.B.; Zerbini, L.F. GADD45 proteins: Central players in tumorigenesis. Curr. Mol. Med. 2012, 12, 634–651. [Google Scholar] [CrossRef] [PubMed]
- Azimian, H.; Dayyani, M.; Toossi, M.T.B.; Mahmoudi, M. Bax/Bcl-2 expression ratio in prediction of response to breast cancer radiotherapy. Iran. J. Basic Med. Sci. 2018, 21, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Kulsoom, B.; Shamsi, T.S.; Afsar, N.A.; Memon, Z.; Ahmed, N.; Hasnain, S.N. Bax, Bcl-2, and Bax/Bcl-2 as prognostic markers in acute myeloid leukemia: Are we ready for Bcl-2-directed therapy? Cancer Manag. Res. 2018, 10, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Perlman, H.; Zhang, X.; Chen, M.W.; Walsh, K.; Buttyan, R. An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis. Cell Death Differ. 1999, 6, 48–54. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Nylander, K.D.; Yan, C.; Schor, N.F. Role of caspase 3-dependent Bcl-2 cleavage in potentiation of apoptosis by Bcl-2. Mol. Pharmacol. 2002, 61, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Vardaki, I.; Sanchez, C.; Fonseca, P.; Olsson, M.; Chioureas, D.; Rassidakis, G.; Ullen, A.; Zhivotovsky, B.; Bjorkholm, M.; Panaretakis, T. Caspase-3-dependent cleavage of Bcl-xL in the stroma exosomes is required for their uptake by hematological malignant cells. Blood 2016, 128, 2655–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabacaoglu, D.; Ruess, D.A.; Ai, J.; Algul, H. NF-kappaB/Rel Transcription Factors in Pancreatic Cancer: Focusing on RelA, c-Rel, and RelB. Cancers (Basel) 2019, 11, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene No. | Gene Abbreviation | Gene Description | Gene No. | Gene abbreviation | Gene Description |
|---|---|---|---|---|---|
| Intrinsic apoptotic pathway | 12 | CIDEB | Cell death-inducing DFFA-like effector B | ||
| 1 | APAF1 | Apoptotic peptidase-activating factor 1 | 13 | DFFA | DNA fragmentation factor, 45 kDa, alpha polypeptide |
| 2 | BAD | BCL2-associated agonist of cell death | 14 | GADD45A | Growth arrest and DNA damage-inducible, alpha factor |
| 3 | BAX | BCL2-associated X protein | 15 | HRK | Harakiri, BCL2-interacting protein |
| 4 | BCL2L11 | BCL2-Iike 11 | 16 | TP53 | Tumor protein p53 |
| 5 | BCLAF1 | BCL2-associated transcription factor | Extrinsic apoptotic pathway | ||
| 6 | BID | BH3-interacting domain death agonist | 17 | CRADD | CASP2- and RIPK1-domain containing adaptor with death domain |
| 7 | BIK | BCL2-interacting killer | 18 | DAPK1 | Death-associated protein kinase 1 |
| 8 | BIRC2 | Baculoviral IAP repeat-containing 2 | 19 | FADD | Fas(TNFRSF6)-associated via death domain |
| 9 | BNIP3L | BCL2-interacting protein 3-like | 20 | LTBR | Lymphotoxin beta receptor (TNFR superfamily, member 3) |
| 10 | CASP3 | Caspase3, apoptosis-related cysteine peptidase | 21 | TNFRSF21 | Tumor necrosis tactor receptor superfamily, member 21 |
| 11 | CASP9 | Caspase 9, apoptosis-related cysteine peptidase | 22 | TNFRSF25 | Tumor necrosis tactor receptor superfamily, member 25 |
| MOLM-13 a | MOLM-13/DAC b | MOLM-13/AZA c | a vs. b p | a vs. c p | |
|---|---|---|---|---|---|
| (Activity in MOLM-13 Was Taken as 100%) | |||||
| Caspase 3/7 | 100.0 ± 9.9 | 96.9 ± 3.4 | 89.8 ± 3.5 | 0.78 | 0.39 |
| Caspase 8 | 100.0 ± 2.0 | 103.8 ± 4.3 | 101.1 ± 3.8 | 0.47 | 0.80 |
| Caspase 9 | 100.0 ± 7.4 | 90.3 ± 2.6 | 105.7 ± 6.9 | 0.28 | 0.60 |
| Cells | MOLM-13 | MOLM-13/DAC | MOLM-13/AZA | |||
|---|---|---|---|---|---|---|
| Treatment | DAC | AZA | DAC | AZA | DAC | AZA |
| Gene/Protein | Antiapoptotic BCL2 and proapoptotic BAX proteins | |||||
| BCL2 protein | ↓ | ↓ | 0 | 0 | ↓ | ↓ |
| BAX protein | ↑ | ↓ | ↑ | ↓ | 0 | 0 |
| BCL2/BAX ratio | ↓ | 0/↓ * | ↑/0 * | ↑ | ↓ | ↓ |
| Caspases | ||||||
| CASP 3mRNA | ↓ | ↑ | ↓ | ↑ | ↑ | 0 |
| Casp-3/7 act. | ↑ | ↑ | 0 | ↑ | ↑ | 0 |
| Casp-8 act. | ↑ | ↑ | 0 | ↑ | ↑ | 0 |
| Casp-9 act. | 0 | 0 | 0 | ↑ | ↑ | 0 |
| NF-κB | ||||||
| RELmRNA | 0 | 0 | 0 | 0 | 0 | 0 |
| RELAmRNA | 0 | ↑/0 * | 0 | ↑ | ↓ | ↑ |
| NFKB1mRNA | ↓ | ↓ | 0 | ↑/0 * | ↓ | ↓ |
| RELBmRNA | ↑ | ↑ | 0 | ↑ | ↑ | 0 |
| NFKB2mRNA | ↓ | ↓ | 0 | ↑ | 0 | ↑/↑ * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janotka, Ľ.; Messingerová, L.; Šimoničová, K.; Kavcová, H.; Elefantová, K.; Sulová, Z.; Breier, A. Changes in Apoptotic Pathways in MOLM-13 Cell Lines after Induction of Resistance to Hypomethylating Agents. Int. J. Mol. Sci. 2021, 22, 2076. https://doi.org/10.3390/ijms22042076
Janotka Ľ, Messingerová L, Šimoničová K, Kavcová H, Elefantová K, Sulová Z, Breier A. Changes in Apoptotic Pathways in MOLM-13 Cell Lines after Induction of Resistance to Hypomethylating Agents. International Journal of Molecular Sciences. 2021; 22(4):2076. https://doi.org/10.3390/ijms22042076
Chicago/Turabian StyleJanotka, Ľuboš, Lucia Messingerová, Kristína Šimoničová, Helena Kavcová, Katarína Elefantová, Zdena Sulová, and Albert Breier. 2021. "Changes in Apoptotic Pathways in MOLM-13 Cell Lines after Induction of Resistance to Hypomethylating Agents" International Journal of Molecular Sciences 22, no. 4: 2076. https://doi.org/10.3390/ijms22042076