
Elucidating the Molecular Determinants of the Binding Modes of a Third-Generation HIV-1 Integrase Strand Transfer Inhibitor: The Importance of Side Chain and Solvent Reorganization


Abstract
:1. Introduction
2. Results
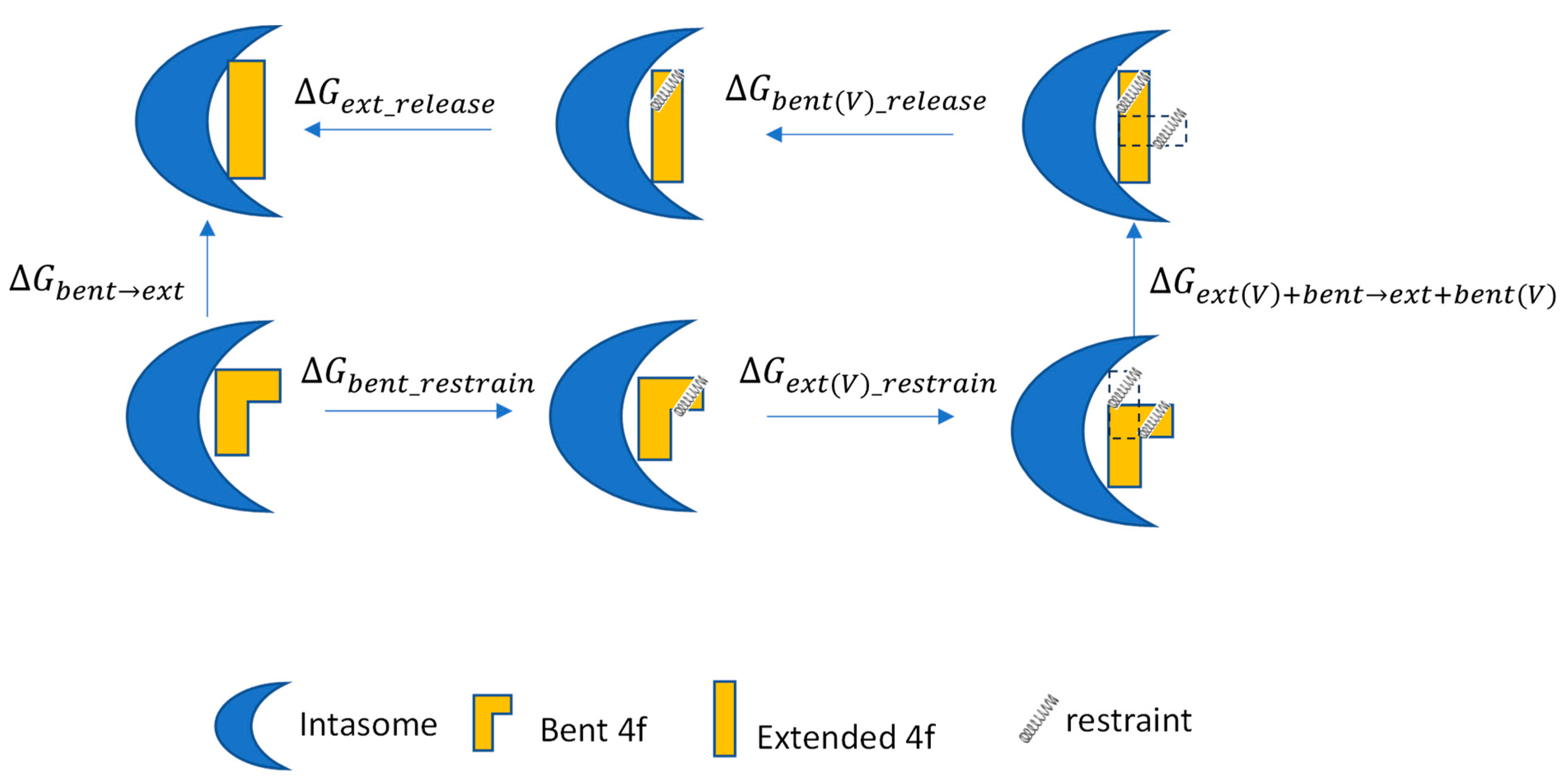
2.1. The Calculated Conformational Free Energies Recapitulate the Preference for the Experimental Binding Modes
2.2. Understanding the Mechanism for the Binding Mode Selectivity in the Two Intasomes: Analysis of MD Structures and Further R-FEP-R Free Energy Calculations
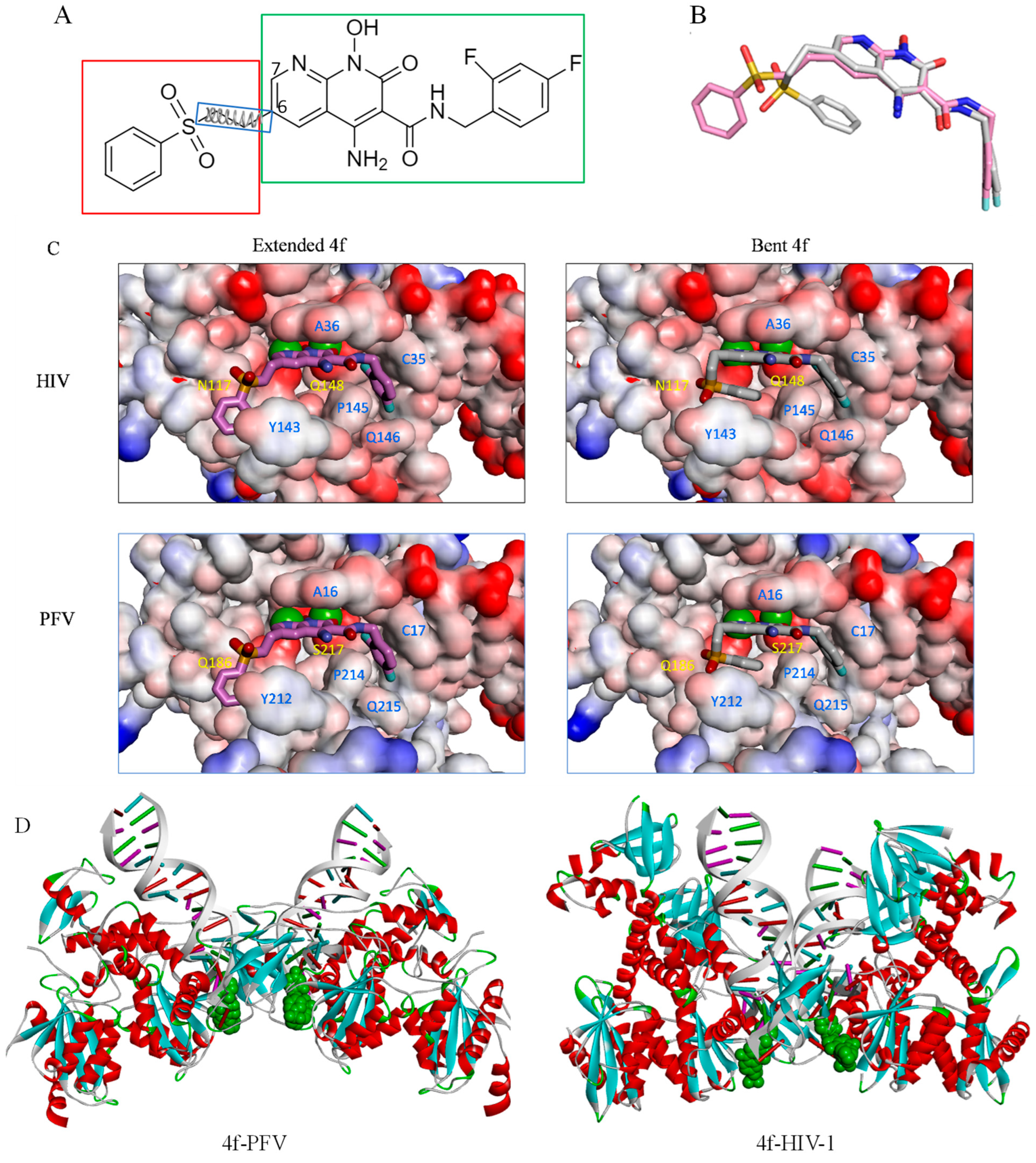
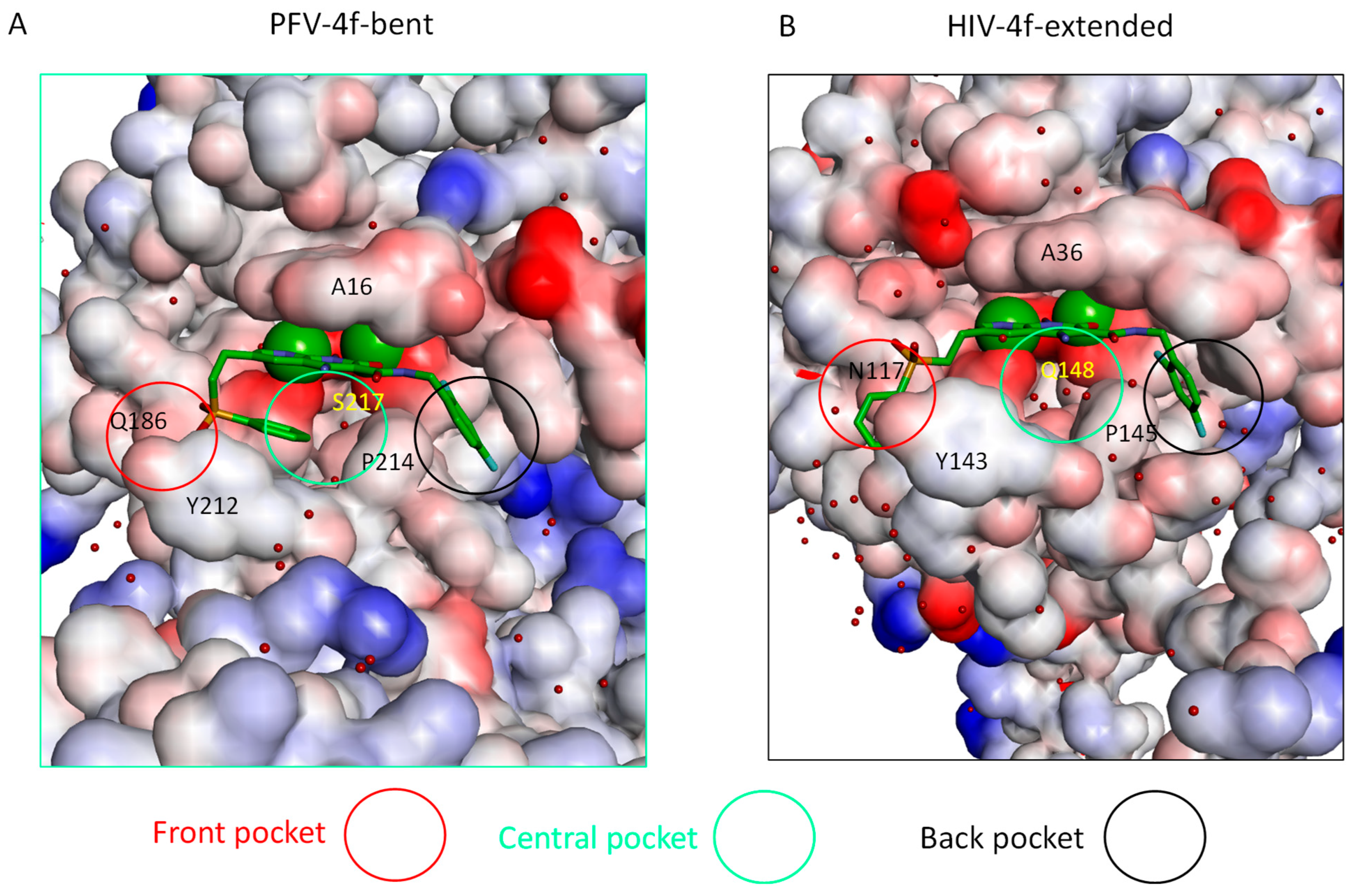
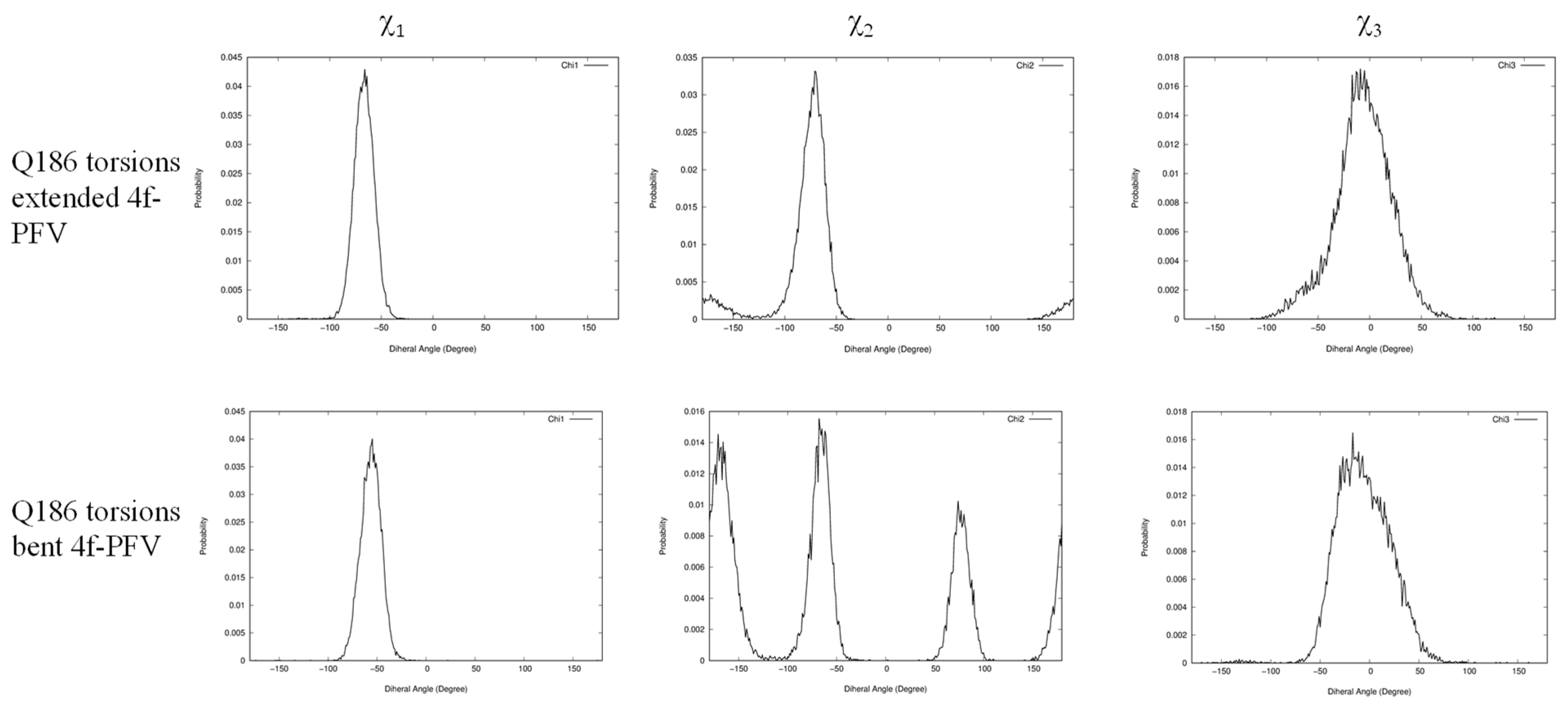
2.3. The Central Sub-Pocket Is Primarily Responsible for the Binding Mode Selection by the Two Intasomes
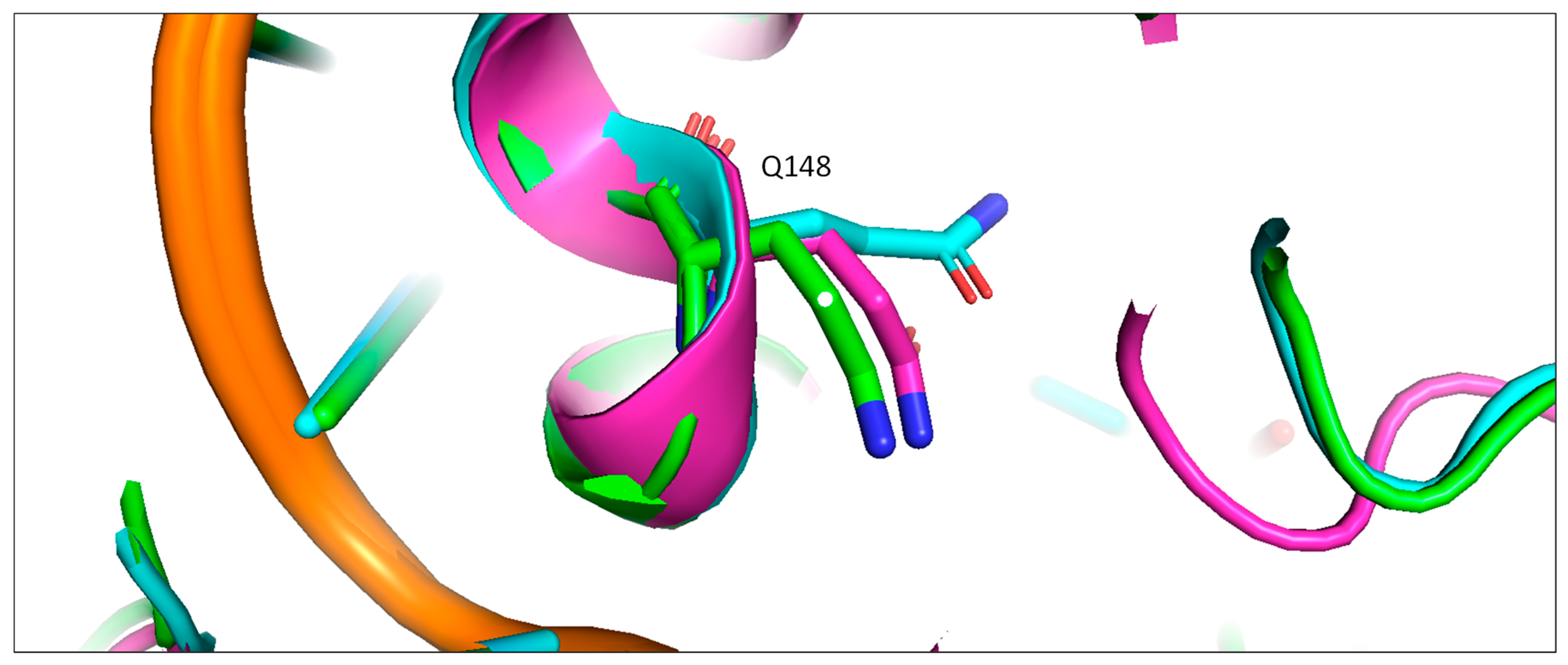
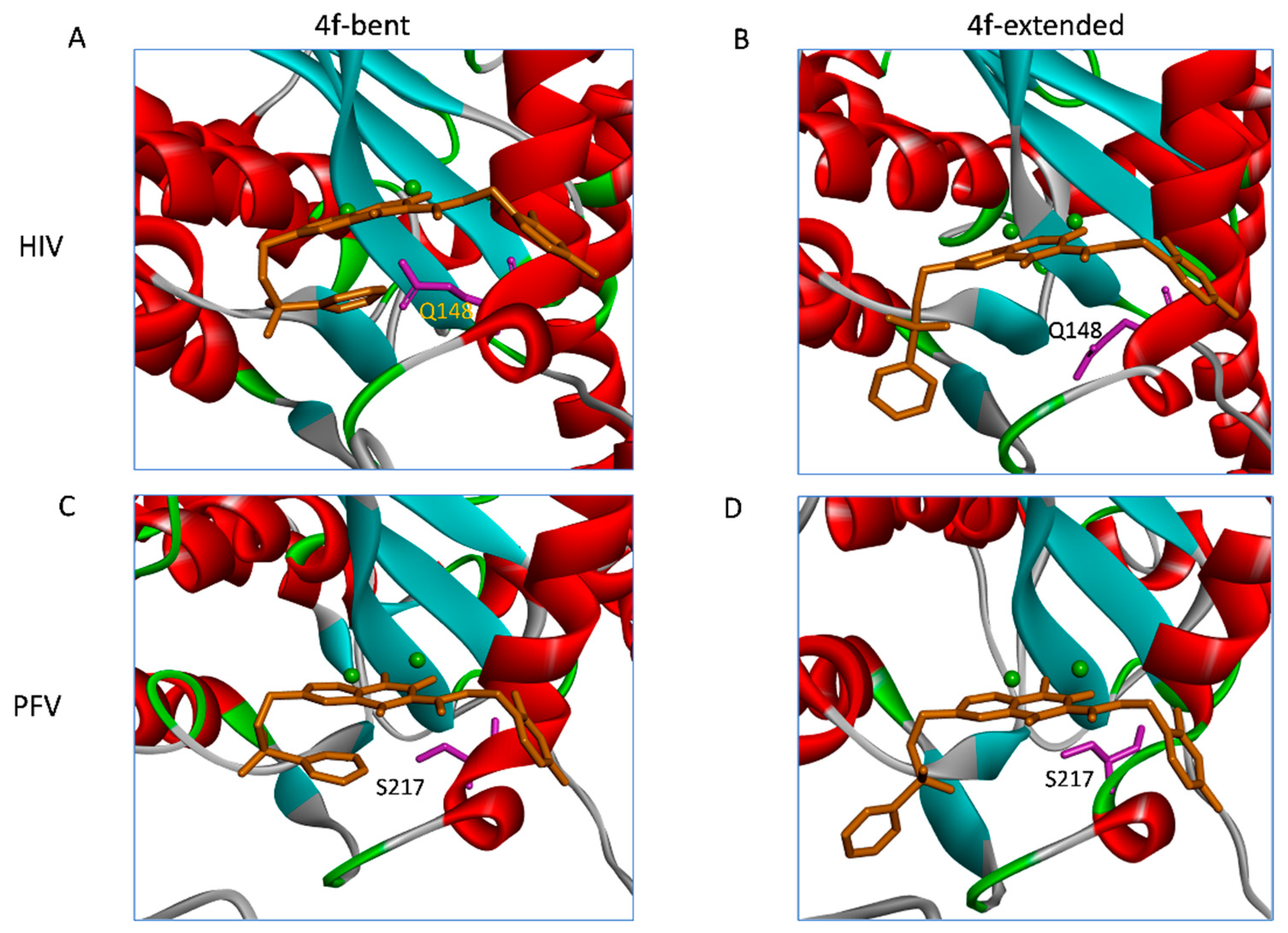
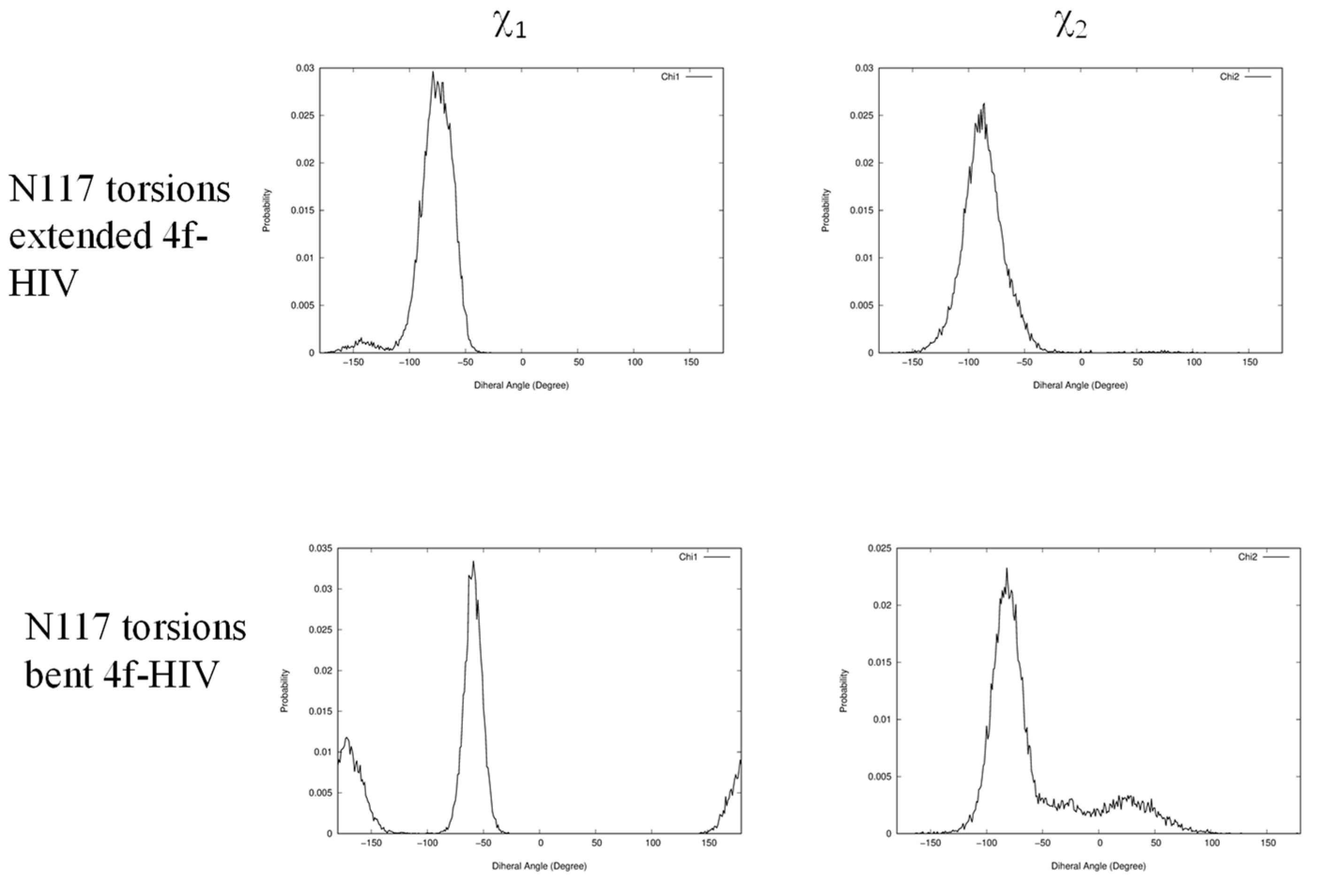
2.4. The Front Sub-Pocket Makes a Comparatively Smaller Contribution to the Binding Mode Selection by the Two Intasomes
3. Discussion
3.1. Implications of the Findings for Drug Resistance and Ligand Binding
3.2. Atomic Models of HIV Intasomes for Studying INSTI Binding and Drug Resistance
3.3. Utility of the Alchemical R-FEP-R for Predicting Ligand Binding Modes in Challenging Environments
4. Conclusions
5. Methods and Materials
System Setup and Simulation Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engelman, A.N. Multifaceted HIV integrase functionalities and therapeutic strategies for their inhibition. J. Biol. Chem. 2019, 294, 15137–15157. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, R. Inhibiting the HIV integration process: Past, present, and the future. J. Med. Chem. 2014, 57, 539–566. [Google Scholar] [CrossRef] [PubMed]
- Puertas, M.C.; Ploumidis, G.; Ploumidis, M.; Fumero, E.; Clotet, B.; Walworth, C.M.; Petropoulos, C.J.; Martinez-Picado, J. Pan-resistant HIV-1 emergence in the era of integrase strand-transfer inhibitors: A case report. Lancet Microbe 2020, 1, e130–e135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Smith, S.J.; Maskell, D.P.; Metifiot, M.; Pye, V.E.; Fesen, K.; Marchand, C.; Pommier, Y.; Cherepanov, P.; Hughes, S.H.; et al. HIV-1 Integrase Strand Transfer Inhibitors with Reduced Susceptibility to Drug Resistant Mutant Integrases. ACS Chem. Biol. 2016, 11, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Smith, S.J.; Metifiot, M.; Marchand, C.; Boyer, P.L.; Pommier, Y.; Hughes, S.H.; Burke, T.R., Jr. 4-amino-1-hydroxy-2-oxo-1,8-naphthyridine-containing compounds having high potency against raltegravir-resistant integrase mutants of HIV-1. J. Med. Chem. 2014, 57, 5190–5202. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef]
- Hare, S.; Smith, S.J.; Metifiot, M.; Jaxa-Chamiec, A.; Pommier, Y.; Hughes, S.H.; Cherepanov, P. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 2011, 80, 565–572. [Google Scholar] [CrossRef]
- Metifiot, M.; Maddali, K.; Johnson, B.C.; Hare, S.; Smith, S.J.; Zhao, X.Z.; Marchand, C.; Burke, T.R., Jr.; Hughes, S.H.; Cherepanov, P.; et al. Activities, crystal structures, and molecular dynamics of dihydro-1H-isoindole derivatives, inhibitors of HIV-1 integrase. ACS Chem. Biol. 2013, 8, 209–217. [Google Scholar] [CrossRef]
- Hare, S.; Vos, A.M.; Clayton, R.F.; Thuring, J.W.; Cummings, M.D.; Cherepanov, P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20057–20062. [Google Scholar] [CrossRef]
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R., Jr.; Hughes, S.H. Integrase Strand Transfer Inhibitors Are Effective Anti-HIV Drugs. Viruses 2021, 13, 205. [Google Scholar] [CrossRef]
- Passos, D.O.; Li, M.; Jóźwik, I.K.; Zhao, X.Z.; Santos-Martins, D.; Yang, R.; Smith, S.J.; Jeon, Y.; Forli, S.; Hughes, S.H.; et al. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 2020, 367, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Sakae, Y.; Zhang, B.W.; Levy, R.M.; Deng, N. Absolute Protein Binding Free Energy Simulations for Ligands with Multiple Poses, a Thermodynamic Path That Avoids Exhaustive Enumeration of the Poses. J. Comput. Chem. 2020, 41, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Raniolo, S.; Limongelli, V. Ligand binding free-energy calculations with funnel metadynamics. Nat. Protoc. 2020, 15, 2837–2866. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Shao, X.; Cai, W.; Chipot, C. Taming Rugged Free Energy Landscapes Using an Average Force. Acc. Chem. Res. 2019, 52, 3254–3264. [Google Scholar] [CrossRef] [PubMed]
- Khuttan, S.; Azimi, S.; Wu, J.Z.; Dick, S.; Wu, C.; Xu, H.; Gallicchio, E. Taming Multiple Binding Poses in Alchemical Binding Free Energy Prediction: The β-cyclodextrin Host-Guest SAMPL9 Blinded Challenge. arXiv 2023, arXiv:2302.08620. [Google Scholar] [CrossRef]
- Arasteh, S.; Zhang, B.W.; Levy, R.M. Protein Loop Conformational Free Energy Changes via an Alchemical Path without Reaction Coordinates. J. Phys. Chem. Lett. 2021, 12, 4368–4377. [Google Scholar] [CrossRef]
- Geronimo, I.; De Vivo, M. Alchemical Free-Energy Calculations of Watson-Crick and Hoogsteen Base Pairing Interconversion in DNA. J. Chem. Theory Comput. 2022, 18, 6966–6973. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Wang, F.; Landau, D.P. Efficient, Multiple-Range Random Walk Algorithm to Calculate the Density of States. Phys. Rev. Lett. 2001, 86, 2050–2053. [Google Scholar] [CrossRef]
- Sun, Q.; Levy, R.M.; Kirby, K.A.; Wang, Z.; Sarafianos, S.G.; Deng, N. Molecular Dynamics Free Energy Simulations Reveal the Mechanism for the Antiviral Resistance of the M66I HIV-1 Capsid Mutation. Viruses 2021, 13, 920. [Google Scholar] [CrossRef]
- Gallicchio, E.; Levy, R.M. Recent theoretical and computational advances for modeling protein–ligand binding affinities. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 85, pp. 27–80. [Google Scholar]
- Deng, N.; Forli, S.; He, P.; Perryman, A.; Wickstrom, L.; Vijayan, S.K.V.; Tiefenbrunn, T.; Stout, C.D.; Gallicchio, E.; Olson, A.J.; et al. Distinguishing Binders from False Positives by Free Energy Calculations: Fragment Screening Against the Flap Site of HIV Protease. J. Phys. Chem. B 2014, 119, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, L.; Gallicchio, E.; Chen, L.; Kurtzman, T.; Deng, N. Developing end-point methods for absolute binding free energy calculation using the Boltzmann-quasiharmonic model. Phys. Chem. Chem. Phys. 2022, 24, 6037–6052. [Google Scholar] [CrossRef] [PubMed]
- Gerton, J.L.; Ohgi, S.; Olsen, M.; DeRisi, J.; Brown, P.O. Effects of mutations in residues near the active site of human immunodeficiency virus type 1 integrase on specific enzyme-substrate interactions. J. Virol. 1998, 72, 5046–5055. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Craigie, R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 1992, 66, 6361–6369. [Google Scholar] [CrossRef] [PubMed]
- Jozwik, I.K.; Passos, D.O.; Lyumkis, D. Structural Biology of HIV Integrase Strand Transfer Inhibitors. Trends Pharmacol. Sci. 2020, 41, 611–626. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Grant, P.M.; Tzou, P.L.; Barrow, G.; Harrigan, P.R.; Ioannidis, J.P.A.; Shafer, R.W. A systematic review of the genetic mechanisms of dolutegravir resistance. J. Antimicrob. Chemother. 2019, 74, 3135–3149. [Google Scholar] [CrossRef]
- McColl, D.J.; Chen, X. Strand transfer inhibitors of HIV-1 integrase: Bringing IN a new era of antiretroviral therapy. Antiviral Res. 2010, 85, 101–118. [Google Scholar] [CrossRef]
- Underwood, M.R.; Johns, B.A.; Sato, A.; Martin, J.N.; Deeks, S.G.; Fujiwara, T. The activity of the integrase inhibitor dolutegravir against HIV-1 variants isolated from raltegravir-treated adults. J. Acquir. Immune Defic. Syndr. 2012, 61, 297–301. [Google Scholar] [CrossRef]
- Santoro, M.M.; Fornabaio, C.; Malena, M.; Galli, L.; Poli, A.; Menozzi, M.; Zazzi, M.; White, K.L.; Castagna, A.; Group, P.S. Susceptibility to HIV-1 integrase strand transfer inhibitors (INSTIs) in highly treatment-experienced patients who failed an INSTI-based regimen. Int. J. Antimicrob. Agents 2020, 56, 106027. [Google Scholar] [CrossRef]
- Cui, D.; Zhang, B.W.; Matubayasi, N.; Levy, R.M. The Role of Interfacial Water in Protein–Ligand Binding: Insights from the Indirect Solvent Mediated Potential of Mean Force. J. Chem. Theory Comput. 2018, 14, 512–526. [Google Scholar] [CrossRef]
- Zhang, B.W.; Cui, D.; Matubayasi, N.; Levy, R.M. The Excess Chemical Potential of Water at the Interface with a Protein from End Point Simulations. J. Phys. Chem. B 2018, 122, 4700–4707. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.J.; Li, W.; Berta, D.; Badaoui, M.; Ballandras-Colas, A.; Nans, A.; Kotecha, A.; Rosta, E.; Engelman, A.N.; Cherepanov, P. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 2020, 367, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Gonzalez Paz, O.; Hazuda, D.J.; et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Li, X.; Naraharisetty, H.L.; Hare, S.; Cherepanov, P.; Engelman, A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 15910–15915. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Smith, S.J.; Maskell, D.P.; Metifiot, M.; Pye, V.E.; Fesen, K.; Marchand, C.; Pommier, Y.; Cherepanov, P.; Hughes, S.H.; et al. Structure-Guided Optimization of HIV Integrase Strand Transfer Inhibitors. J. Med. Chem. 2017, 60, 7315–7332. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Oliveira Passos, D.; Shan, Z.; Smith, S.J.; Sun, Q.; Biswas, A.; Choudhuri, I.; Strutzenberg, T.S.; Haldane, A.; Deng, N.; et al. Mechanisms of HIV-1 integrase resistance to dolutegravir and potent inhibition of drug-resistant variants. Sci. Adv. 2023, 9, eadg5953. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Sun, S.; Huggins, D.J. Assessing the effect of forcefield parameter sets on the accuracy of relative binding free energy calculations. Front. Mol. Biosci. 2022, 9, 972162. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, J.Z.H. Thermodynamic Insights of Base Flipping in TNA Duplex: Force Fields, Salt Concentrations, and Free-Energy Simulation Methods. CCS Chem. 2021, 3, 1026–1039. [Google Scholar] [CrossRef]
- Wickstrom, L.; Deng, N.; He, P.; Mentes, A.; Nguyen, C.; Gilson, M.K.; Kurtzman, T.; Gallicchio, E.; Levy, R.M. Parameterization of an effective potential for protein-ligand binding from host-guest affinity data: Force Field Optimization with Host-Guest Systems. J. Mol. Recognit. 2016, 29, 10–21. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M., Jr. Metal Ion Modeling Using Classical Mechanics. Chem. Rev. 2017, 117, 1564–1686. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M., Jr. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Vedani, A.; Huhta, D.W. A new force field for modeling metalloproteins. J. Am. Chem. Soc. 1990, 112, 4759–4767. [Google Scholar] [CrossRef]
- Lin, F.; Wang, R. Systematic Derivation of AMBER Force Field Parameters Applicable to Zinc-Containing Systems. J. Chem. Theory Comput. 2010, 6, 1852–1870. [Google Scholar] [CrossRef]
- Seminario, J.M. Calculation of intramolecular force fields from second-derivative tensors. Int. J. Quantum Chem. 1996, 60, 1271–1277. [Google Scholar] [CrossRef]
- Melse, O.; Antes, I.; Kaila, V.R.I.; Zacharias, M. Benchmarking biomolecular force field-based Zn2+ for mono- and bimetallic ligand binding sites. J. Comput. Chem. 2023, 44, 912–926. [Google Scholar] [CrossRef]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [PubMed]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhang, B.W.; Arasteh, S.; Wang, L.; Abel, R.; Levy, R.M. Conformational Free Energy Changes via an Alchemical Path without Reaction Coordinates. J. Phys. Chem. Lett. 2018, 9, 4428–4435. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Postma, J.V.; Van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Klimovich, P.V.; Shirts, M.R.; Mobley, D.L. Guidelines for the analysis of free energy calculations. J. Comput. Aided Mol. Des. 2015, 29, 397–411. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | (kcal/mol) | Predicted Binding Mode | |
|---|---|---|---|
| HIV-1 Intasome | Wild Type | −2.9 ± 0.4 | Extended |
| N117Q | −2.5 ± 0.4 | Extended | |
| Q148S | −0.7 ± 0.3 | Mixed | |
| PFV Intasome | Wild Type | 2.2 ± 0.4 | Bent |
| Q186N | 1.4 ± 0.4 | Bent | |
| S217Q | −0.4 ± 0.2 | Mixed | |
| Structure | χ1 N-CA-CB-CG | χ2 CA-CB-CG-CD | χ3 CB-CG-CD-OE1 |
|---|---|---|---|
| 4f-bound (extended) | −66.0° | 91.5° | 42.8° |
| 4f-bound (bent) | −59.7° | 145.7° | −120.1° |
| Apo | −49.3° | 103.4° | 29.5° |
| Intasome | Extended 4f | Bent 4f |
|---|---|---|
| HIV | 5–6 | 0 |
| PFV | 6–7 | 1–2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Q.; Biswas, A.; Lyumkis, D.; Levy, R.; Deng, N. Elucidating the Molecular Determinants of the Binding Modes of a Third-Generation HIV-1 Integrase Strand Transfer Inhibitor: The Importance of Side Chain and Solvent Reorganization. Viruses 2024, 16, 76. https://doi.org/10.3390/v16010076
Sun Q, Biswas A, Lyumkis D, Levy R, Deng N. Elucidating the Molecular Determinants of the Binding Modes of a Third-Generation HIV-1 Integrase Strand Transfer Inhibitor: The Importance of Side Chain and Solvent Reorganization. Viruses. 2024; 16(1):76. https://doi.org/10.3390/v16010076
Chicago/Turabian StyleSun, Qinfang, Avik Biswas, Dmitry Lyumkis, Ronald Levy, and Nanjie Deng. 2024. "Elucidating the Molecular Determinants of the Binding Modes of a Third-Generation HIV-1 Integrase Strand Transfer Inhibitor: The Importance of Side Chain and Solvent Reorganization" Viruses 16, no. 1: 76. https://doi.org/10.3390/v16010076