
Fluorescence Properties of Novel Multiresonant Indolocarbazole Derivatives for Deep-Blue OLEDs from Multiscale Computer Modelling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
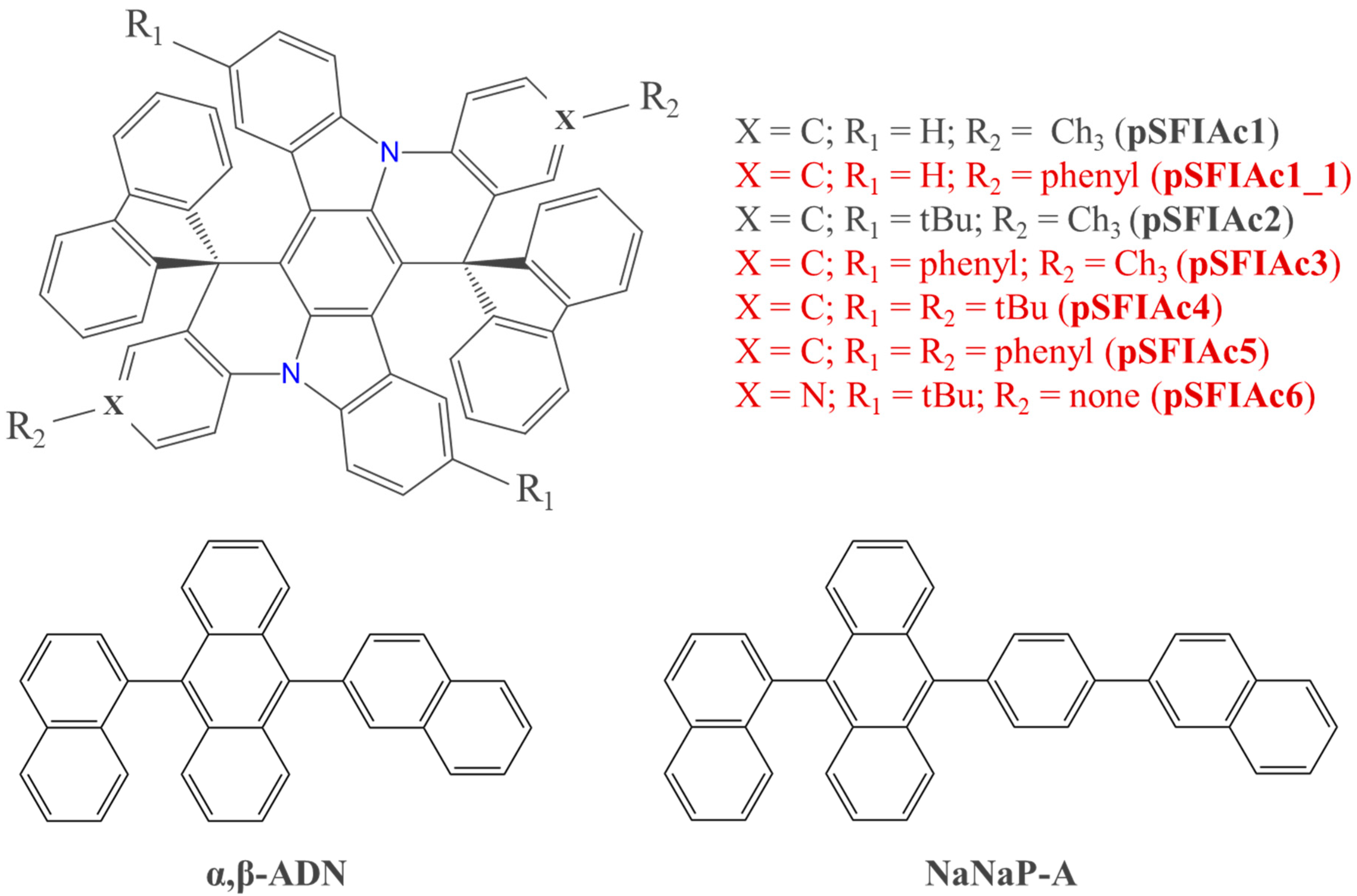
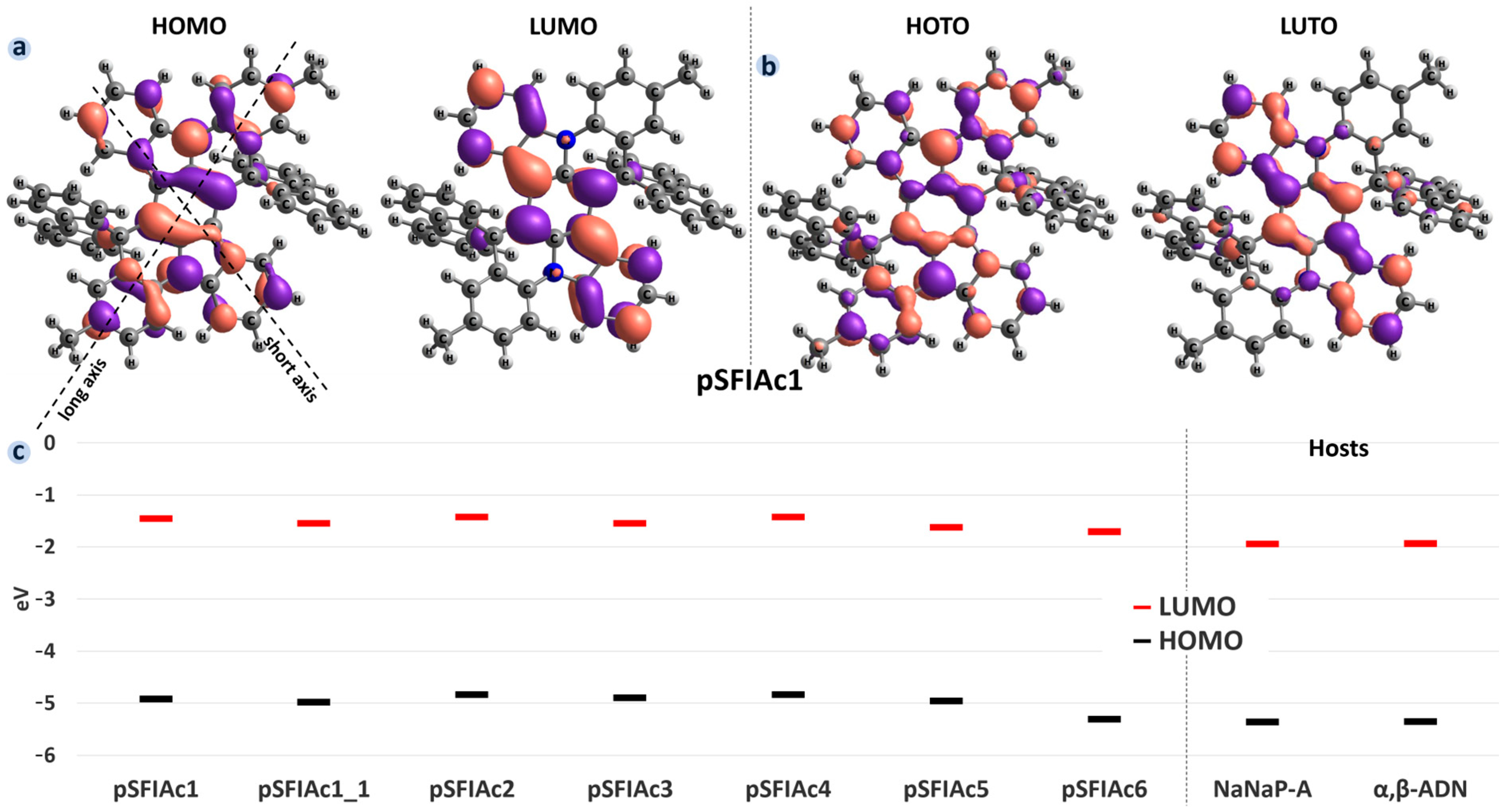
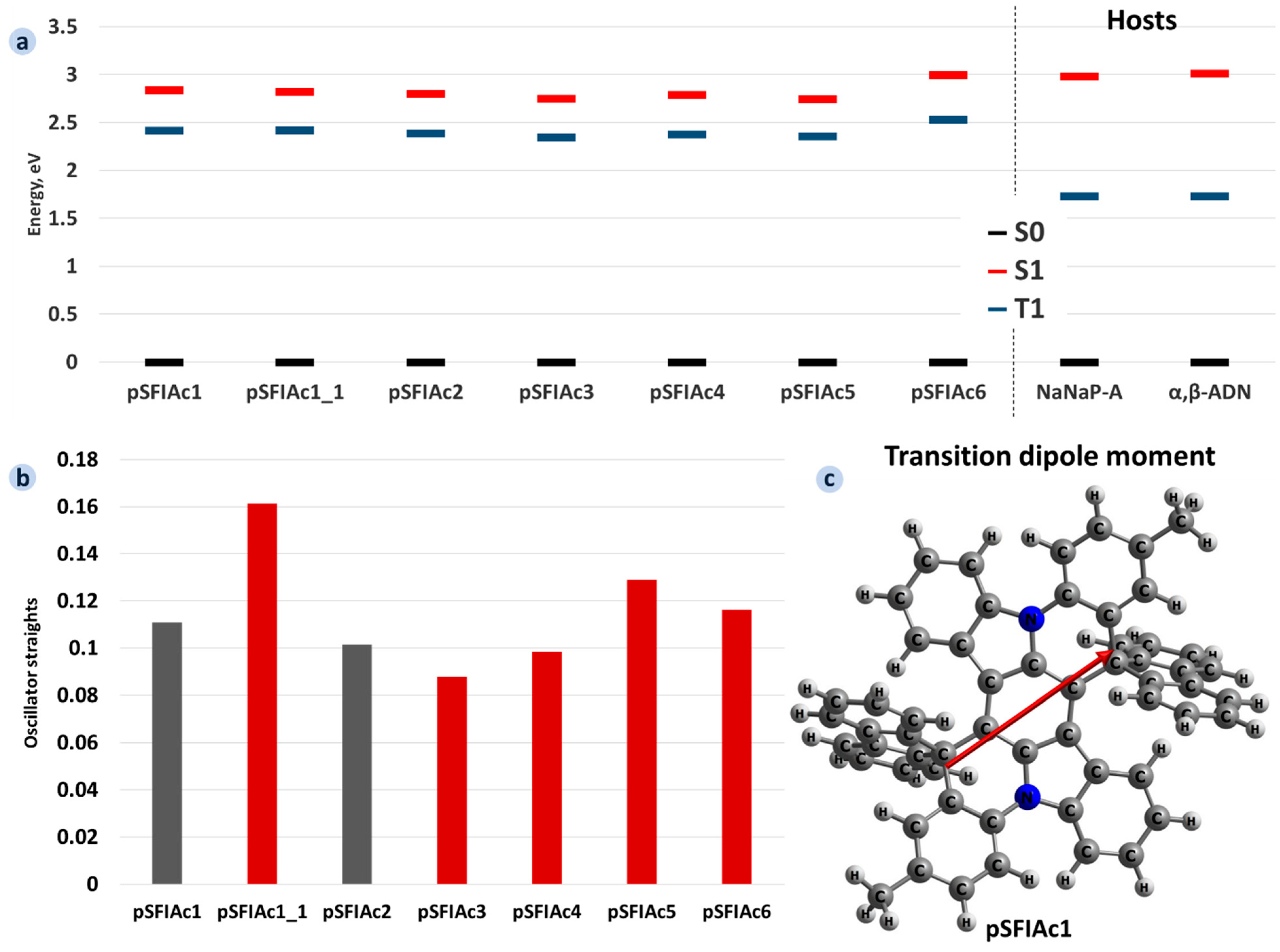
2.1. DFT Calculations for Single Molecules
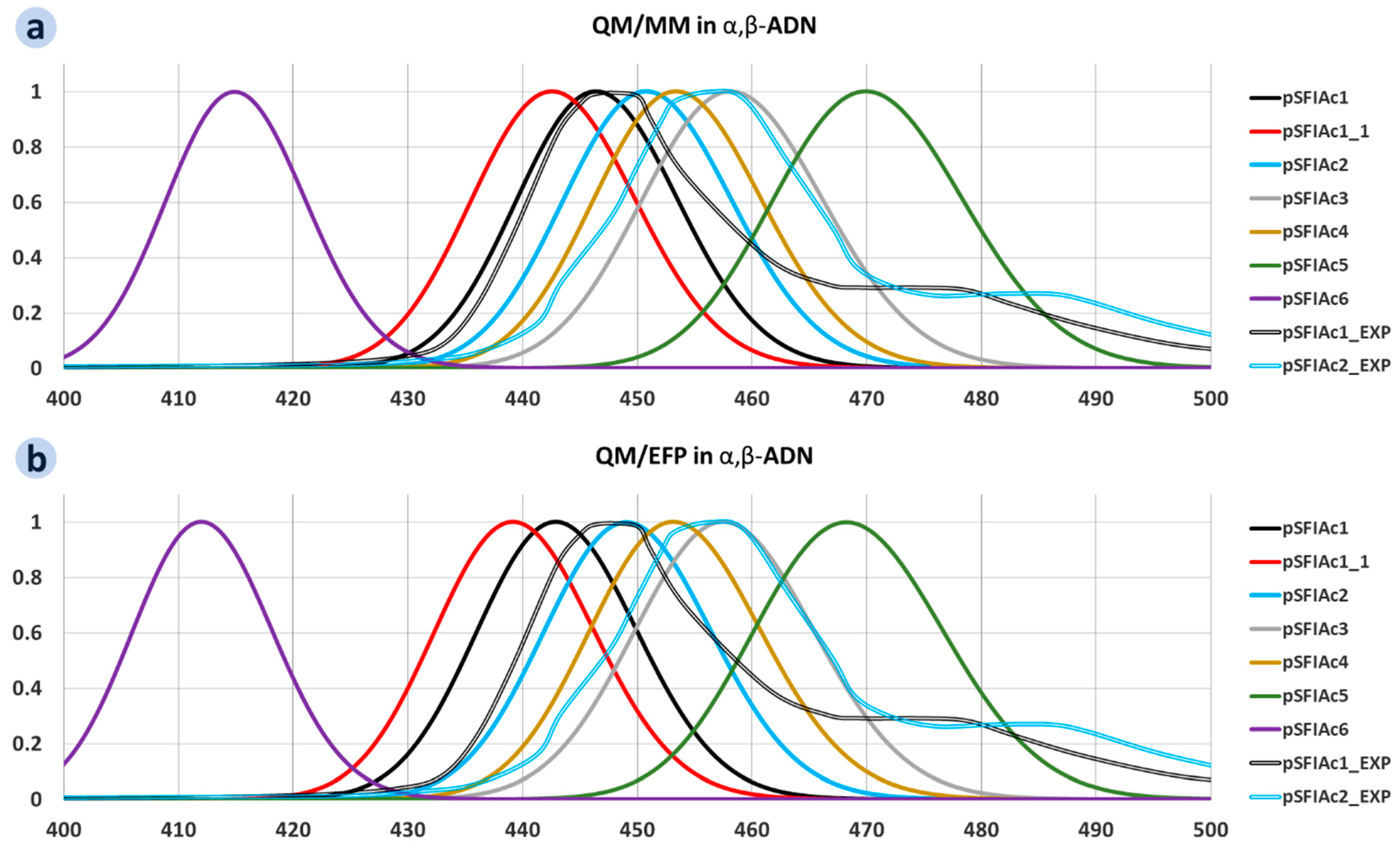
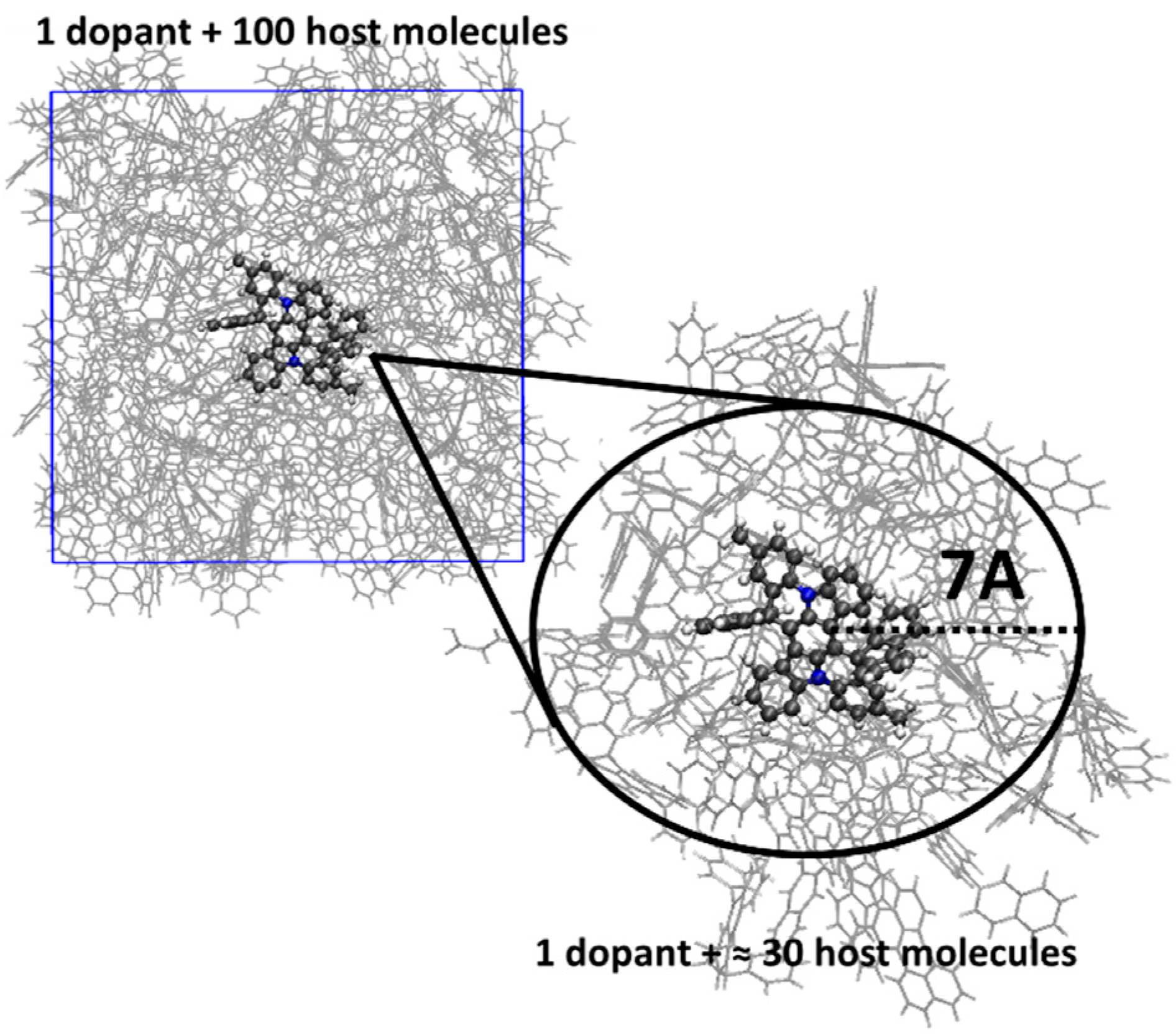
2.2. Multiscale Modelling of Molecules Embedded in Hosts
3. Methods
3.1. Quantum Mechanical (QM) Calculations of Isolated Molecules
3.2. Molecular Dynamics Simulations
3.3. Quantum Mechanics/Molecular Mechanics (QM/MM) Calculations
3.4. Quantum Mechanics/Effective Fragment Potential (QM/EFP) Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.-L.; Ran, Q.; Wang, Q.; Liu, Y.; Hänisch, C.; Reineke, S.; Fan, J.; Liao, L.-S. High-Efficiency Red Organic Light-Emitting Diodes with External Quantum Efficiency Close to 30% Based on a Novel Thermally Activated Delayed Fluorescence Emitter. Adv. Mater. 2019, 31, 1902368. [Google Scholar] [CrossRef]
- Chan, C.-Y.; Tanaka, M.; Lee, Y.-T.; Wong, Y.-W.; Nakanotani, H.; Hatakeyama, T.; Adachi, C. Stable pure-blue hyperfluorescence organic light-emitting diodes with high-efficiency and narrow emission. Nat. Photonics 2021, 15, 203–207. [Google Scholar] [CrossRef]
- Jeon, S.O.; Lee, K.H.; Kim, J.S.; Ihn, S.-G.; Chung, Y.S.; Kim, J.W.; Lee, H.; Kim, S.; Choi, H.; Lee, J.Y. High-efficiency, long-lifetime deep-blue organic light-emitting diodes. Nat. Photonics 2021, 15, 208–215. [Google Scholar] [CrossRef]
- Zhang, D.; Duan, L. TADF sensitization targets deep-blue. Nat. Photonics 2021, 15, 173–174. [Google Scholar] [CrossRef]
- Rayappa Naveen, K.; Prabhu CP, K.; Braveenth, R.; Hyuk Kwon, J. Molecular Design Strategy for Orange Red Thermally Activated Delayed Fluorescence Emitters in Organic Light-Emitting Diodes (OLEDs). Chem. A Eur. J. 2022, 28, e202103532. [Google Scholar] [CrossRef]
- Song, W.; Lee, J.Y. Degradation Mechanism and Lifetime Improvement Strategy for Blue Phosphorescent Organic Light-Emitting Diodes. Adv. Opt. Mater. 2017, 5, 1600901. [Google Scholar] [CrossRef]
- Tankelevičiūtė, E.; Samuel, I.D.W.; Zysman-Colman, E. The Blue Problem: OLED Stability and Degradation Mechanisms. J. Phys. Chem. Lett. 2024, 15, 1034–1047. [Google Scholar] [CrossRef] [PubMed]
- Kondakov, D.Y. Triplet–triplet annihilation in highly efficient fluorescent organic light-emitting diodes: Current state and future outlook. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2015, 373, 20140321. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Miyamae, T.; Ohata, H.; Tsutsui, T. Direct observations of the charge behavior of a high-efficiency blue organic light-emitting diode under operating conditions using electric-field-induced doubly resonant sum-frequency-generation vibrational spectroscopy. Org. Electron. 2019, 74, 118–125. [Google Scholar] [CrossRef]
- Bezvikonnyi, O.; Bucinskas, A.; Arsenyan, P.; Petrenko, A.; Wei, Z.-Y.; Lee, J.-H.; Volyniuk, D.; Rashid, E.U.; Chiu, T.-L.; Grazulevicius, J.V. Enhancement of Blue Doping-Free and Hyperfluorescent Organic Light Emitting Diode Performance through Triplet–Triplet Annihilation in the Derivatives of Anthracene and Carbazole. ACS Appl. Electron. Mater. 2024, 6, 4489–4503. [Google Scholar] [CrossRef]
- Kim, J.U.; Park, I.S.; Chan, C.-Y.; Tanaka, M.; Tsuchiya, Y.; Nakanotani, H.; Adachi, C. Nanosecond-time-scale delayed fluorescence molecule for deep-blue OLEDs with small efficiency rolloff. Nat. Commun. 2020, 11, 1765. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Cao, F.; Cheng, C.; Tsuboi, T.; Zhu, Y.; Deng, C.; Zheng, X.; Wang, D.; Liu, Z.; Zhang, Q. High Fluorescence Rate of Thermally Activated Delayed Fluorescence Emitters for Efficient and Stable Blue OLEDs. ACS Appl. Mater. Interfaces 2020, 12, 31706–31715. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Shiren, K.; Nakajima, K.; Nomura, S.; Nakatsuka, S.; Kinoshita, K.; Ni, J.; Ono, Y.; Ikuta, T. Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO-LUMO Separation by the Multiple Resonance Effect. Adv. Mater. 2016, 28, 2777–2781. [Google Scholar] [CrossRef]
- Cai, X.; Su, S.-J. Marching Toward Highly Efficient, Pure-Blue, and Stable Thermally Activated Delayed Fluorescent Organic Light-Emitting Diodes. Adv. Funct. Mater. 2018, 28, 1802558. [Google Scholar] [CrossRef]
- Meng, G.; Zhang, D.; Wei, J.; Zhang, Y.; Huang, T.; Liu, Z.; Yin, C.; Hong, X.; Wang, X.; Zeng, X.; et al. Highly efficient and stable deep-blue OLEDs based on narrowband emitters featuring an orthogonal spiro-configured indolo[3,2,1-de]acridine structure. Chem. Sci. 2022, 13, 5622–5630. [Google Scholar] [CrossRef]
- Chen, N.; Yan, B. Recent Theoretical and Experimental Progress in Circularly Polarized Luminescence of Small Organic Molecules. Molecules 2018, 23, 3376. [Google Scholar] [CrossRef]
- Sosorev, A.Y.; Nuraliev, M.K.; Feldman, E.V.; Maslennikov, D.R.; Borshchev, O.V.; Skorotetcky, M.S.; Surin, N.M.; Kazantsev, M.S.; Ponomarenko, S.A.; Paraschuk, D.Y. Impact of terminal substituents on the electronic, vibrational and optical properties of thiophene–phenylene co-oligomers. Phys. Chem. Chem. Phys. 2019, 21, 11578–11588. [Google Scholar] [CrossRef]
- Freidzon, A.; Dubinets, N.; Bagaturyants, A. Theoretical Study of Charge-Transfer Exciplexes in Organic Photovoltaics. J. Phys. Chem. A 2022, 126, 2111–2118. [Google Scholar] [CrossRef]
- Ma, M.; Wang, Y.; Sun, C. Synchronous or stepwise Mechanism? A theoretical study on the Excited-State double proton transfer properties of shikonin and acetylshikosin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2024, 308, 123689. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, A.; Dubinets, N.; Koshkin, A.; Rykova, E. Study of the behavior of the 4-DASPI dye in samples of silicate hydrogel by fluorescence spectroscopy and quantum chemistry. J. Mol. Liq. 2024, 393, 123651. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, Y.; Sun, C. External electric field induced emission behavior for ESIPT-based 2-(benzo[d]thiazol-2-yl)-4-(pyren-1-yl)phenol towards near-infrared region. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2025, 325, 125045. [Google Scholar] [CrossRef] [PubMed]
- Dubinets, N.O.; Freidzon, A.Y.; Bagaturyants, A.A. Use of effective fragment potentials for simulation of excited states in an inhomogeneous environment. Int. J. Quantum Chem. 2020, 120, e26071. [Google Scholar] [CrossRef]
- Stasyuk, O.A.; Szatylowicz, H.; Krygowski, T.M.; Fonseca Guerra, C. How amino and nitro substituents direct electrophilic aromatic substitution in benzene: An explanation with Kohn–Sham molecular orbital theory and Voronoi deformation density analysis. Phys. Chem. Chem. Phys. 2016, 18, 11624–11633. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Gao, J. Hybrid Quantum and Molecular Mechanical Simulations: An Alternative Avenue to Solvent Effects in Organic Chemistry. Acc. Chem. Res. 1996, 29, 298–305. [Google Scholar] [CrossRef]
- Gordon, M.S.; Slipchenko, L.; Li, H.; Jensen, J.H. Chapter 10 The Effective Fragment Potential: A General Method for Predicting Intermolecular Interactions. In Annual Reports in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2007; Volume 3, pp. 177–193. [Google Scholar]
- Slipchenko, L.V.; Gurunathan, P.K. Effective Fragment Potential Method: Past, Present, and Future. In Fragmentation: Toward Accurate Calculations on Complex Molecular Systems; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Zhang, G.; Musgrave, C.B. Comparison of DFT Methods for Molecular Orbital Eigenvalue Calculations. J. Phys. Chem. A 2007, 111, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Hernández, D.D.; Tarakeshwar, P.; Gust, D.; Moore, T.A.; Moore, A.L.; Mujica, V. Simple and accurate correlation of experimental redox potentials and DFT-calculated HOMO/LUMO energies of polycyclic aromatic hydrocarbons. J. Mol. Model. 2013, 19, 2845–2848. [Google Scholar] [CrossRef]
- Valeur, B.; Berberan-Santos, M.N. Molecular Fluorescence: Principles and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Yersin, H.; Czerwieniec, R.; Shafikov, M.Z.; Suleymanova, A.F. TADF Material Design: Photophysical Background and Case Studies Focusing on Cu(I) and Ag(I) Complexesa. In Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence; Wiley-VCH: Weinheim, Germany, 2018; Chapter 1; pp. 1–60. [Google Scholar] [CrossRef]
- Sanderson, S.; Philippa, B.; Vamvounis, G.; Burn, P.L.; White, R.D. Elucidating the effects of guest-host energy level alignment on charge transport in phosphorescent OLEDs. Appl. Phys. Lett. 2019, 115, 263301. [Google Scholar] [CrossRef]
- Cheng, C.; Zhu, Y.; Tsuboi, T.; Deng, C.; Lou, W.; Liu, T.; Wang, D.; Zhang, Q. Extremely stable deep-blue organic light-emitting diodes employing diindolophenazine-based fluorophore with narrow-band emission and a shallow LUMO level. Chem. Eng. J. 2023, 474, 145691. [Google Scholar] [CrossRef]
- Dubinets, N.O.; Dominskiy, D.I.; Tukachev, N.V.; Sosorev, A.Y. Reliability of DFT and multiscale modelling for prediction of emission properties of blue TADF luminophores. Opt. Mater. 2024, 157, 116244. [Google Scholar] [CrossRef]
- Odinokov, A.V.; Dubinets, N.O.; Bagaturyants, A.A. pyEFP: Automatic decomposition of the complex molecular systems into rigid polarizable fragments. J. Comput. Chem. 2018, 39, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Quantum Density Oscillations in an Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, A1697–A1705. [Google Scholar] [CrossRef]
- Matsuzawa, N.N.; Ishitani, A.; Dixon, D.A.; Uda, T. Time-Dependent Density Functional Theory Calculations of Photoabsorption Spectra in the Vacuum Ultraviolet Region. J. Phys. Chem. A 2001, 105, 4953–4962. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ziesche, P.; Eschrig, H. Electronic Structure of Solids ’91; Akademie Verlag: Berlin, Germany, 1991. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef]
- York, D.M.; Karplus, M. A Smooth Solvation Potential Based on the Conductor-Like Screening Model. J. Phys. Chem. A 1999, 103, 11060–11079. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Vreven, T.; Morokuma, K. Hybrid Methods: ONIOM(QM:MM) and QM/MM. In Annual Reports in Computational Chemistry; Spellmeyer, D.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 2, pp. 35–51. [Google Scholar]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L., III; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Dubinets, N.; Slipchenko, L.V. Effective Fragment Potential Method for H-Bonding: How To Obtain Parameters for Nonrigid Fragments. J. Phys. Chem. A 2017, 121, 5301–5312. [Google Scholar] [CrossRef]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubinets, N.O.; Sosorev, A.Y. Fluorescence Properties of Novel Multiresonant Indolocarbazole Derivatives for Deep-Blue OLEDs from Multiscale Computer Modelling. Molecules 2025, 30, 255. https://doi.org/10.3390/molecules30020255
Dubinets NO, Sosorev AY. Fluorescence Properties of Novel Multiresonant Indolocarbazole Derivatives for Deep-Blue OLEDs from Multiscale Computer Modelling. Molecules. 2025; 30(2):255. https://doi.org/10.3390/molecules30020255
Chicago/Turabian StyleDubinets, Nikita O., and Andrey Yu. Sosorev. 2025. "Fluorescence Properties of Novel Multiresonant Indolocarbazole Derivatives for Deep-Blue OLEDs from Multiscale Computer Modelling" Molecules 30, no. 2: 255. https://doi.org/10.3390/molecules30020255
APA StyleDubinets, N. O., & Sosorev, A. Y. (2025). Fluorescence Properties of Novel Multiresonant Indolocarbazole Derivatives for Deep-Blue OLEDs from Multiscale Computer Modelling. Molecules, 30(2), 255. https://doi.org/10.3390/molecules30020255