
Flow Cytometric Analysis and Sorting of Murine Enteric Nervous System Cells: An Optimized Protocol
 , and
, and
Abstract
1. Introduction
2. Results
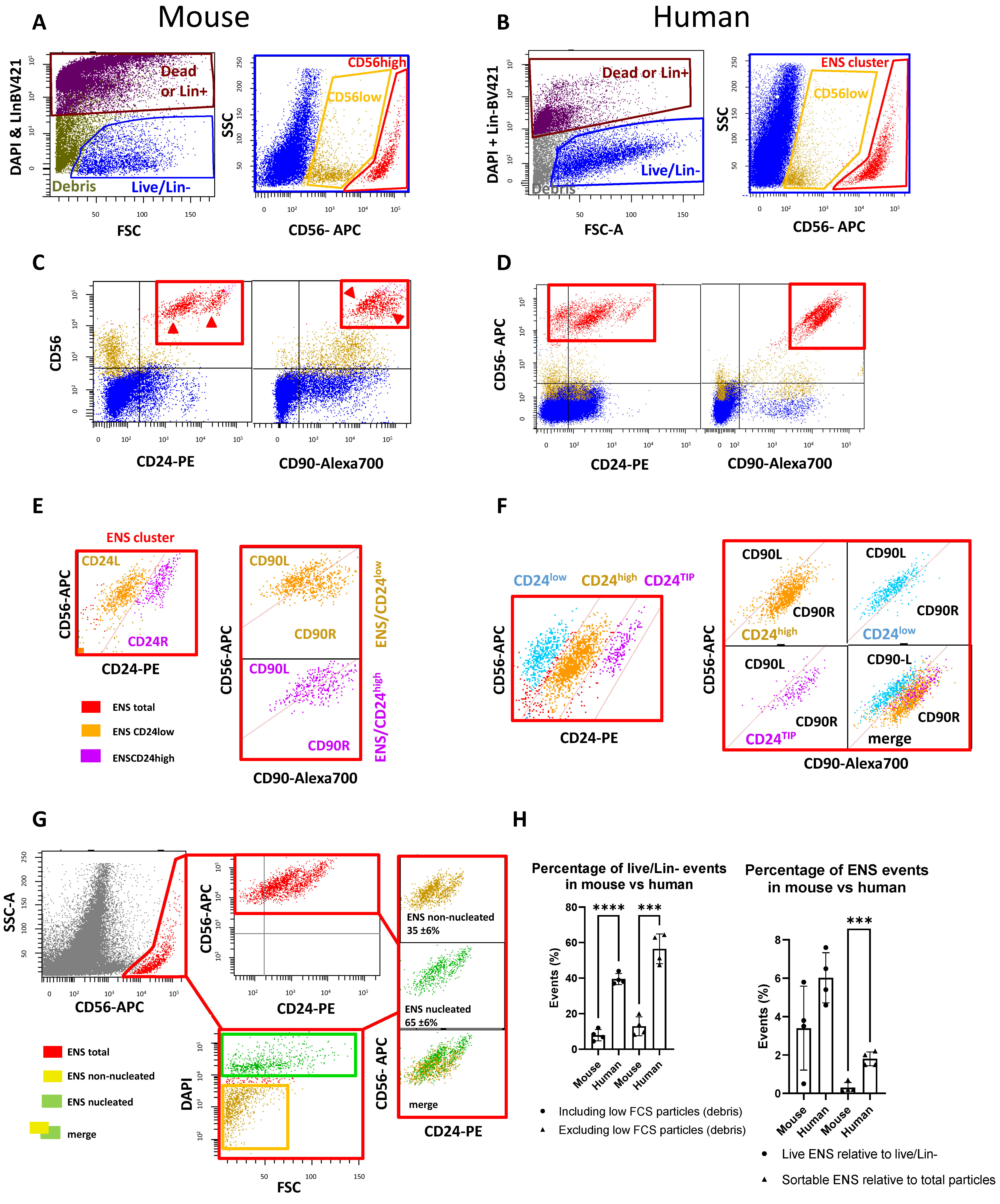
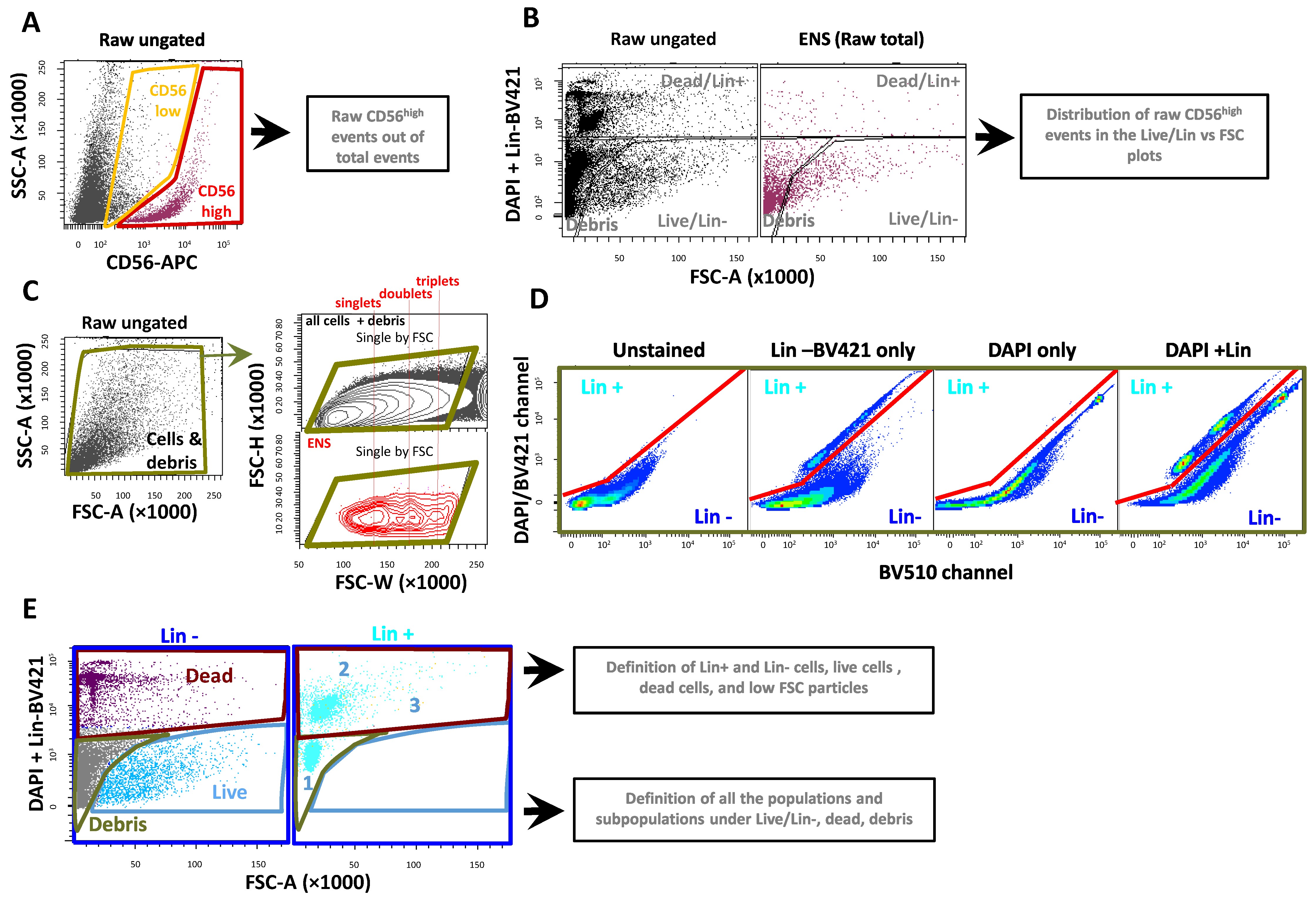
2.1. Mouse ENS Cells Are Successfully Isolated with the Human Protocol, but with Low Viability
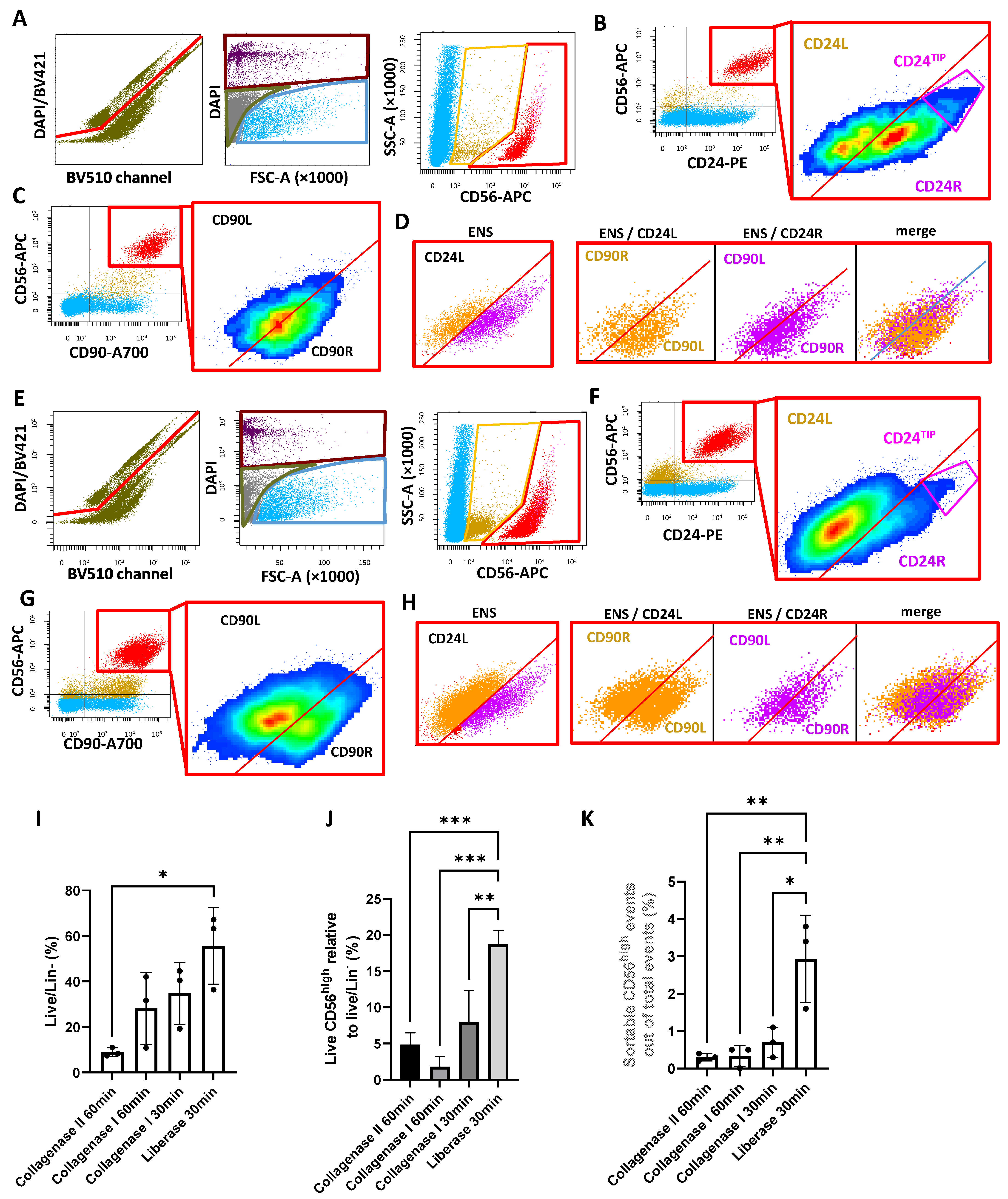
2.2. New Dissociation Protocols for Isolation of Murine ENS Cells
2.2.1. New Dissociation Protocols and Gating Strategy
2.2.2. The Liberase-Based Protocol Increases Overall Viability and ENS Recovery from Mouse Colons
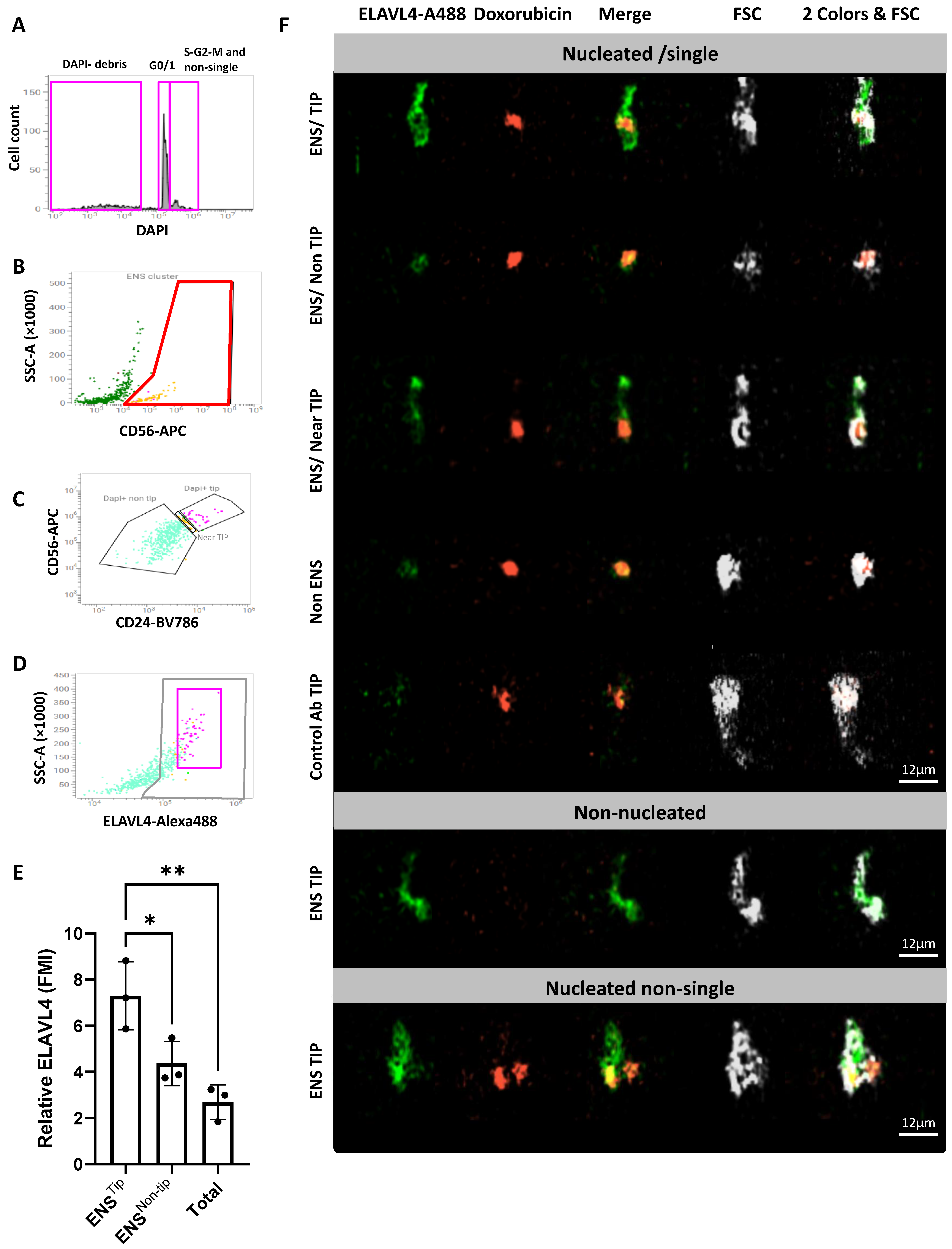
2.3. Validation and Subdivision of the ENS Cluster
2.4. CD56/CD24TIP Is Enriched in Neurons
3. Discussion
4. Materials and Methods
4.1. Animals and Intestinal Isolation
4.2. Dissociation of Mouse Colon Tissue
4.3. Extracellular and Intracellular Staining
4.4. Fluorescence-Activated Cell Sorting
4.5. Gene Expression Analysis
4.6. Cell Culture
4.7. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baghdadi, M.B.; Kim, T.-H. The multiple roles of enteric glial cells in intestinal homeostasis and regeneration. Semin. Cell Dev. Biol. 2023, 150–151, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.J.; Hu, H. Enteric nervous system: Sensory transduction, neural circuits and gastrointestinal motility. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Gershon, M.D. Enteric nervous system development: What could possibly go wrong? Nat. Rev. Neurosci. 2018, 19, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Avetisyan, M.; Schill, E.M.; Heuckeroth, R.O. Building a second brain in the bowel. J. Clin. Investig. 2015, 125, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.M.; Bon-Frauches, A.C.; Keszthelyi, D.; Melotte, V.; Boesmans, W. The enteric nervous system in gastrointestinal disease etiology. Cell Mol. Life Sci. 2021, 78, 4713–4733. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.D.; López, S.H.; Sengupta, R.; Shenoy, A.; Schneider, S.; Wright, C.M.; Feldman, M.; Furth, E.; Valdivieso, F.; Lemke, A.; et al. Robust, 3-Dimensional Visualization of Human Colon Enteric Nervous System without Tissue Sectioning. Gastroenterology 2020, 158, 2221–2235.e5. [Google Scholar] [CrossRef] [PubMed]
- Windster, J.D.; Sacchetti, A.; Schaaf, G.J.; Bindels, E.M.; Hofstra, R.M.; Wijnen, R.M.; Sloots, C.E.; Alves, M.M. A combinatorial panel for flow cytometry-based isolation of enteric nervous system cells from human intestine. EMBO Rep. 2023, 24, e55789. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.; Wu, Y.; Liu, Y.; Wang, N.; Su, M.; Qin, D.; Zhong, W.; Zhao, X.; Zhu, Y.; He, Q.; et al. Establishment and identification of an animal model of Hirschsprung disease in suckling mice. Pediatr. Res. 2023, 94, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- McQuade, R.M.; Singleton, L.M.; Wu, H.; Lee, S.; Constable, R.; Di Natale, M.; Ringuet, M.T.; Berger, J.P.; Kauhausen, J.; Parish, C.L.; et al. The association of enteric neuropathy with gut phenotypes in acute and progressive models of Parkinson’s disease. Sci. Rep. 2021, 11, 7934. [Google Scholar] [CrossRef] [PubMed]
- Baydi, Z.; Limami, Y.; Khalki, L.; Zaid, N.; Naya, A.; Mtairag, E.M.; Oudghiri, M.; Zaid, Y. An Update of Research Animal Models of Inflammatory Bowel Disease. Sci. World J. 2021, 2021, 7479540. [Google Scholar] [CrossRef] [PubMed]
- Drokhlyansky, E.; Smillie, C.S.; Van Wittenberghe, N.; Ericsson, M.; Griffin, G.K.; Eraslan, G.; Dionne, D.; Cuoco, M.S.; Goder-Reiser, M.N.; Sharova, T.; et al. The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 2020, 182, 1606–1622.e23. [Google Scholar] [CrossRef] [PubMed]
- Llorente, C. Isolation of Myenteric and Submucosal Plexus from Mouse Gastrointestinal Tract and Subsequent Co-Culture with Small Intestinal Organoids. Cells 2024, 13, 815. [Google Scholar] [CrossRef] [PubMed]
- Schonkeren, S.L.; Küthe, T.T.; Idris, M.; Bon-Frauches, A.C.; Boesmans, W.; Melotte, V. The gut brain in a dish: Murine primary enteric nervous system cell cultures. Neurogastroenterol. Motil. 2022, 34, e14215. [Google Scholar] [CrossRef] [PubMed]
- Morarach, K.; Mikhailova, A.; Knoflach, V.; Memic, F.; Kumar, R.; Li, W.; Ernfors, P.; Marklund, U. Diversification of molecularly defined myenteric neuron classes revealed by single-cell RNA sequencing. Nat. Neurosci. 2021, 24, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Windster, J.D.; Kakiailatu, N.J.M.; Kuil, L.E.; Antanaviciute, A.; Sacchetti, A.; MacKenzie, K.C.; Peulen-Zink, J.; Kan, T.W.; Bindels, E.; de Pater, E.; et al. Human Enteric Glia Diversity in Health and Disease: New Avenues for the Treatment of Hirschsprung Disease. Gastroenterology 2024, 168, 965–979.e12. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Manufacturer | Cat. No. | Final Concentration | Dissociation Medium 1 |
|---|---|---|---|---|
| DMEM/F12 | Gibco (Waltham, MA, USA) | 11320-074 | N/A | A & B & C |
| HEPES (1M) | Thermo Fisher Scientific (Waltham, MA, USA) | 15630106 | 10 mM | A & B & C |
| DNase I | Sigma-Aldrich (St. Louis, MO, USA) | 11284932001 | 200 µg/mL | A & B & C |
| Dispase | Gibco (Waltham, MA, USA) | 17105-041 | 0.25 mg/mL | A |
| Dispase | Gibco (Waltham, MA, USA) | 17105-041 | 1 mg/mL | B |
| FBS | Capricorn Scientific (Ebsdorfergrund, Germany) | FBS-12A | 5% | B |
| Collagenase II | Gibco (Waltham, MA, USA) | 17101-017 | 3mg/mL | A |
| Collagenase I | Gibco (Waltham, MA, USA) | 17101-015 | 1 mg/mL | B |
| Liberase | Roche (Basel, Switzerland) | 5401119001 | 0.5 mg/mL | C |
| Fluorophore | Lasers | BP Filter (nm) | LP Filter (nm) | |
| Hoechst/DAPI/BV421 | 405 nm | 450/40 | - | |
| FITC/A488/Cycle Green | 488 nm | 530/30 | 502 | |
| PE | 561 nm | 582/15 | - | |
| APC | 633 nm | 660/20 | - | |
| Alexa 700 | 633 nm | 730/45 | 690 | |
| BV605 | 405 nm | 610/20 | 570 | |
| BV786 | 405 nm | 780/60 | 750 | |
| BV510 | 405 nm | 530/30 | 502 | |
| Primary Antibody | Reactivity 1 | Fluorochrome | Supplier; Cat.# | Dilution |
| CD56 | H | APC | Biolegend (San Diego, CA, USA); 362504 | 1:40 |
| CD90 | H | Alexa 700 | Sony Biotechnology (San Jose, CA, USA); 2240600 | 1:40 |
| CD24 | H | PE | BD (Franklin Lakes, NJ, USA); 555428 | 1:20 |
| CD31 | H | BV421 | Biolegend (San Diego, CA, USA); 564089 | 1:0 |
| CD45 | H | BV421 | Biolegend (San Diego, CA, USA); 304031 | 1:40 |
| CD24 | M | PE | Biolegend (San Diego, CA, USA); 101807 | 4 µg/mL |
| CD24 | M | BV786 | BD (Franklin Lakes, NJ, USA); 744470 | 4 µg/mL |
| CD24 | M | BV605 | Biolegend (San Diego, CA, USA); 101827 | 4 µg/mL |
| CD56 | M | APC | R&D systems (Minneapolis, MN, USA); FAB2408A-100UG | 4 µg/mL |
| CD90 | M | Alexa 700 | Biolegend (San Diego, CA, USA); 105320 | 4 µg/mL |
| CD45 | M | BV421 | Biolegend (San Diego, CA, USA); 103133 | 4 µg/mL |
| CD31 | M | BV421 | Biolegend (San Diego, CA, USA); 102423 | 4 µg/mL |
| TER119 | M | BV421 | BD (Franklin Lakes, NJ, USA); 563998 | 4 µg/mL |
| TUBB3 | M/H | Alexa 555 | BD (Franklin Lakes, NJ, USA); 560339 | 1 µg/mL |
| TUBB3 | M/H | Alexa 488 | Biolegend (San Diego, CA, USA); A488-435L | 1 µg/mL |
| SOX10 | M/H | Unconjugated | ThermoFisher (Waltham, MA, USA); 10422-1-AP | 0.4 µg/mL |
| ELAVL4 | M/H | CL 488 | Proteintech (Rosemont, IL, USA); 67835-1-Ig | 1 µg/mL |
| Control Mouse IgG | - | Unconjugated | Santa Cruz Biotechnology (Dallas, TX, USA); sc-2025 | 1 µg/mL |
| Control Rabbit IgG | - | Unconjugated | Biolegend (San Diego, CA, USA); 910801 | 0.4 µg/mL |
| Control Mouse IgG2a | PE | BD Pharmingen™; 555574 | 1 µg/mL | |
| Secondary Antibody | Reactivity | Supplier; Cat.# | Dilution | |
| Goat anti-rabbit (Fab) | Rabbit IgGs | PE | Jackson ImmunoResearch (West Grove, PA, USA); 111-117-008 | - |
| Donkey anti-mouse (Fab) | Mouse IgGs | Alexa 488 | Jackson ImmunoResearch (West Grove, PA, USA); 715-547-003 | - |
| Nuclear Staining | ||||
| Dye Cycle™ Green | - | - | ThermoFisher (Waltham, MA, USA); V35004 | 1:4000 2 |
| Doxorubicin | - | - | Pharmachemie (Haarlem, The Netherlands), 51.223.805 | 0.4 µg/mL 2 |
| DAPI | - | - | Sigma-Aldrich (St. Louis, MO, USA) | 1 µg/mL 2 |
| Gene | Forward Primer (5→3) | Reverse Primer (5→3) |
|---|---|---|
| Tubb3 | CAGATAGGGGCCAAGTTCTGG | GTTGTCGGGCCTGAATAGGT |
| Elavl4 | TCAGACTCCAGACCAAAACCA | TGATGCGACCGTATTGAGAGA |
| Sox10 | GCAAGACACTAGGCAAGCTC | CCTCTCAGCCTCCTCAATGA |
| Ncam1 | CACCATCTACAACGCCAACA | GGGGTTGGTGCATTCTTGAA |
| β-Actin | CTCCACCAGTCTTAAATGGA | AACATAACAACTCTGCAGTCA |
| Gapdh | ACTTTGGCATTGTGGAAGGG | ACAGTCTTCTGGGTGGCAGTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karkala, F.; de Bosscher, I.; Windster, J.D.; Stroebel, S.; van Zanten, L.; Alves, M.M.; Sacchetti, A. Flow Cytometric Analysis and Sorting of Murine Enteric Nervous System Cells: An Optimized Protocol. Int. J. Mol. Sci. 2025, 26, 4824. https://doi.org/10.3390/ijms26104824
Karkala F, de Bosscher I, Windster JD, Stroebel S, van Zanten L, Alves MM, Sacchetti A. Flow Cytometric Analysis and Sorting of Murine Enteric Nervous System Cells: An Optimized Protocol. International Journal of Molecular Sciences. 2025; 26(10):4824. https://doi.org/10.3390/ijms26104824
Chicago/Turabian StyleKarkala, Faidra, Indy de Bosscher, Jonathan D. Windster, Savio Stroebel, Lars van Zanten, Maria M. Alves, and Andrea Sacchetti. 2025. "Flow Cytometric Analysis and Sorting of Murine Enteric Nervous System Cells: An Optimized Protocol" International Journal of Molecular Sciences 26, no. 10: 4824. https://doi.org/10.3390/ijms26104824
APA StyleKarkala, F., de Bosscher, I., Windster, J. D., Stroebel, S., van Zanten, L., Alves, M. M., & Sacchetti, A. (2025). Flow Cytometric Analysis and Sorting of Murine Enteric Nervous System Cells: An Optimized Protocol. International Journal of Molecular Sciences, 26(10), 4824. https://doi.org/10.3390/ijms26104824