

Emerging Signatures of Hematological Malignancies from Gene Expression and Transcription Factor-Gene Regulations
 , , ,
, , ,  , ,
, ,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Hierarchical Clustering Based on Gene Expression
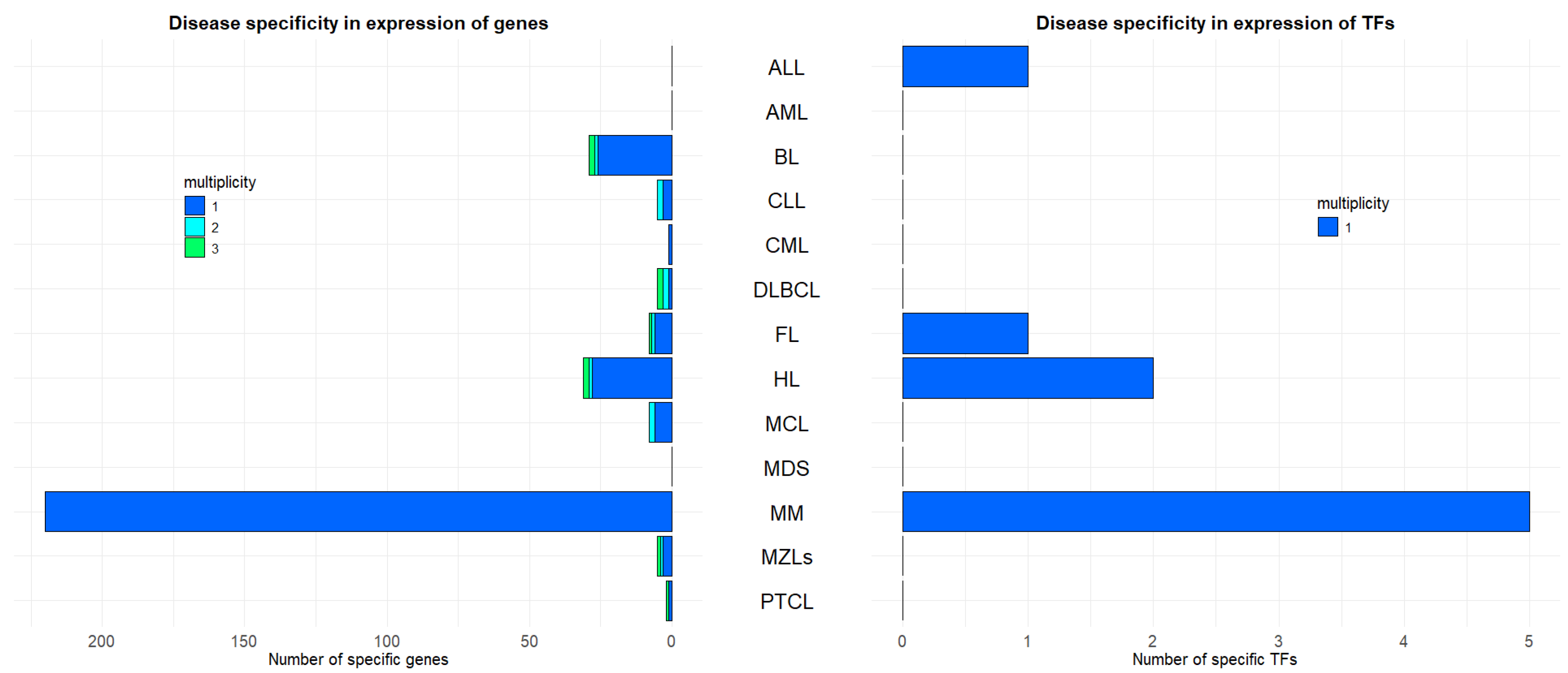
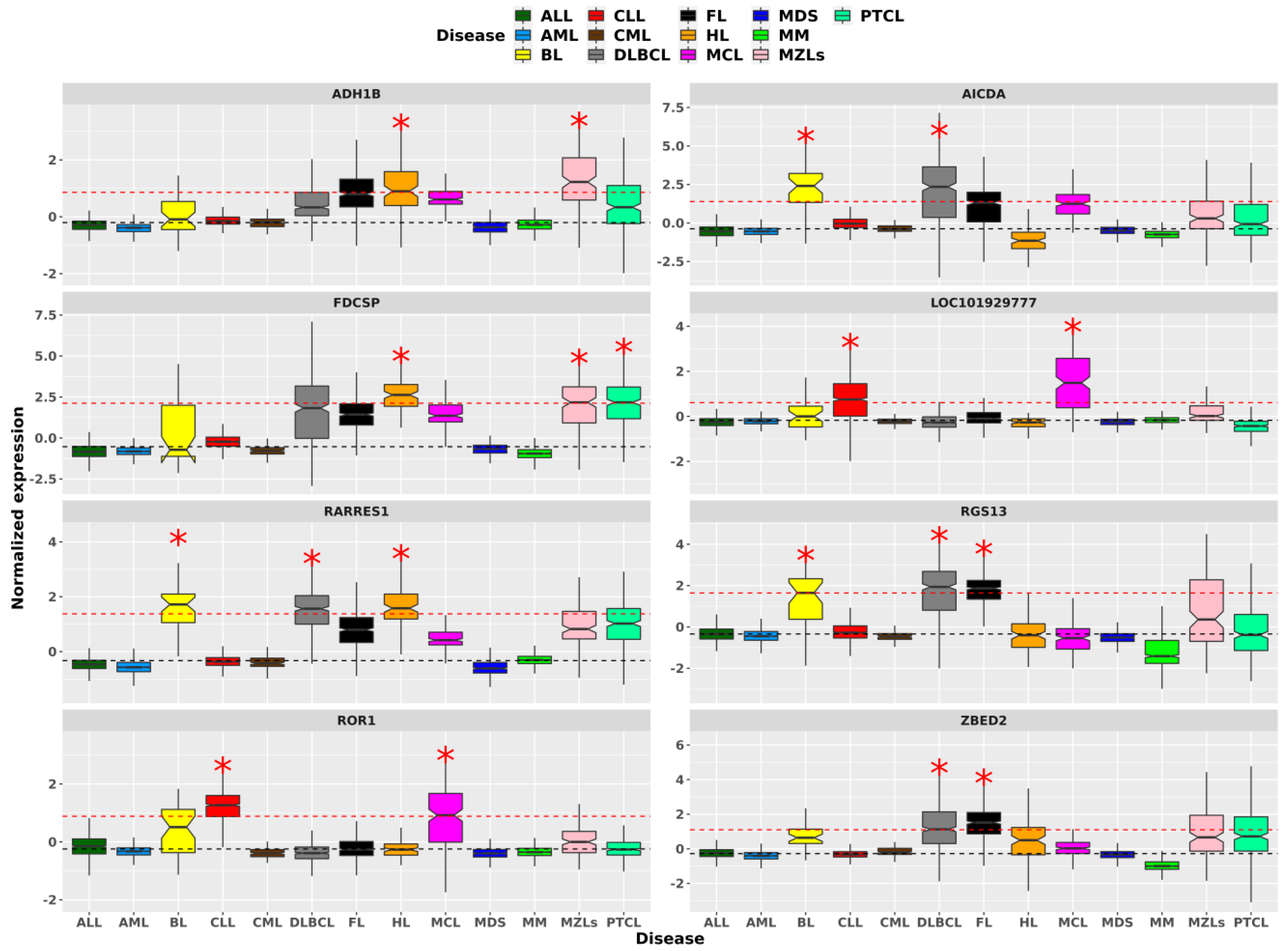
2.2. Hematological Malignancies Indicate Different Over-Expressed Genes and TFs
2.3. Highlighting HMs Similarities and Specificities over Biological Pathways
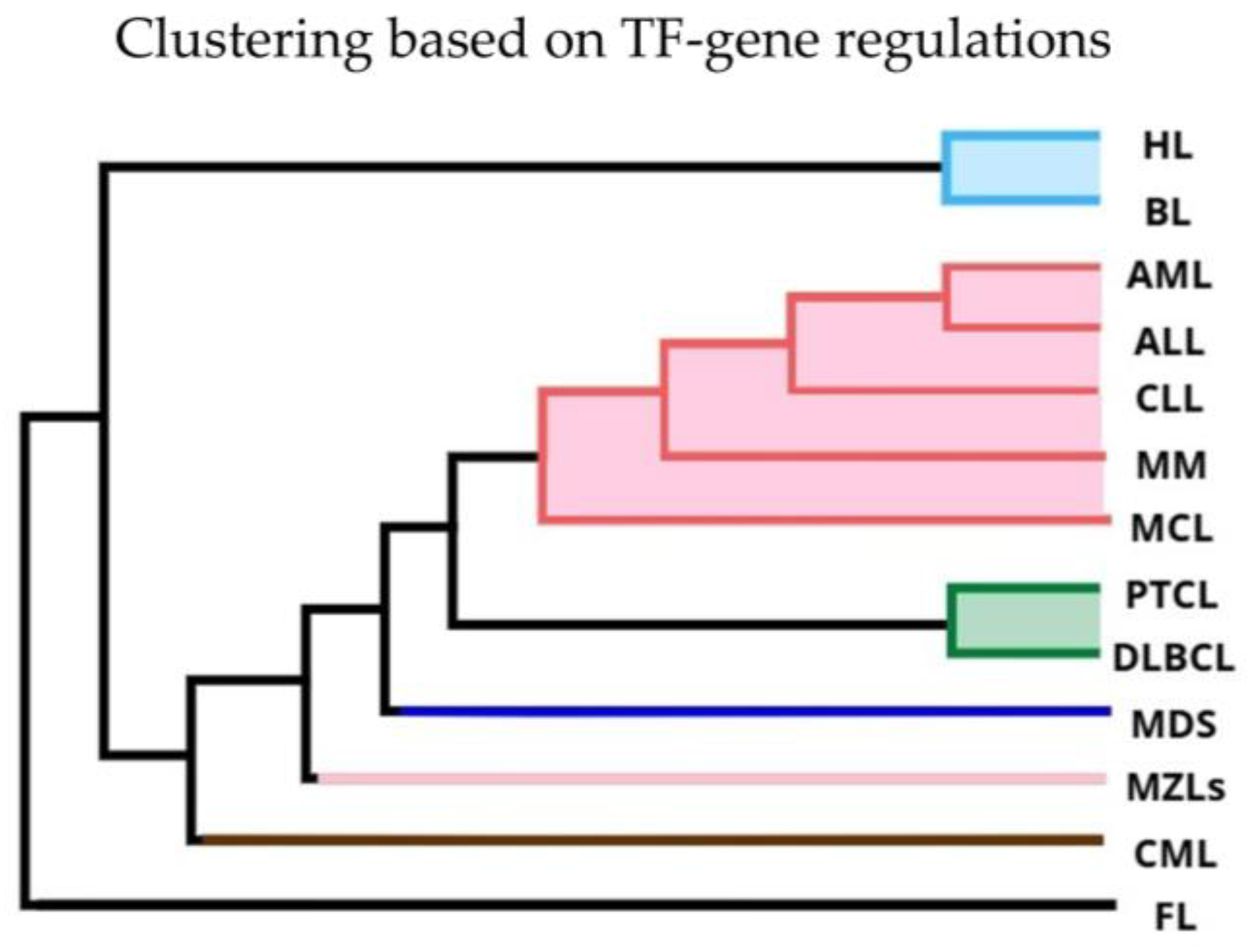
2.4. Hierarchical Clustering on TF-Gene Regulations
2.5. TF-Gene Regulations Highlight Fundamental Biological Functions Across HMs
3. Discussion
3.1. Hierarchical Clustering Based on Gene Expression and TF Shows Divergent Clustering in Leukemias and Lymphomas
3.2. Gene and Transcription Factor Expression Highlight Unique and Overlapping Traits Among HMs
3.3. Different HMs Commonly Emphasize the Same Biological Pathways
3.4. Distinct Regulatory Patterns Emerge Across HMs
4. Conclusions
5. Materials and Methods
5.1. Dataset Pre-Processing and Batch Correction
5.2. Analyses at Gene Expression Levels
5.3. Analyses on TF-Gene Regulations
5.4. Intersection-Based Signatures for Clustering and Targets Prioritization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, N.; Wu, J.; Wang, Q.; Liang, Y.; Li, X.; Chen, G.; Ma, L.; Liu, X.; Zhou, F. Global Burden of Hematologic Malignancies and Evolution Patterns over the Past 30 Years. Blood Cancer J. 2023, 13, 82. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kang, K.; Krahn, J.M.; Croutwater, N.; Lee, K.; Umbach, D.M.; Li, L. A Comprehensive Genomic Pan-Cancer Classification Using The Cancer Genome Atlas Gene Expression Data. BMC Genom. 2017, 18, 508. [Google Scholar] [CrossRef] [PubMed]
- do Valle, Í.F.; Menichetti, G.; Simonetti, G.; Bruno, S.; Zironi, I.; Durso, D.F.; Mombach, J.C.M.; Martinelli, G.; Castellani, G.; Remondini, D. Network Integration of Multi-Tumour Omics Data Suggests Novel Targeting Strategies. Nat. Commun. 2018, 9, 4514. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef]
- Meng, F.; Zhang, K.; Yang, C.; Zhang, K.; Xu, Q.; Ren, R.; Zhou, Y.; Sun, Y.; Peng, Y.; Li, Y.; et al. Prognostic Pathways Guide Drug Indications in Pan-Cancers. Front. Oncol. 2022, 12, 849552. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Lemus, E.; Martínez-García, M. Pathway-Based Drug-Repurposing Schemes in Cancer: The Role of Translational Bioinformatics. Front. Oncol. 2021, 10, 605680. [Google Scholar] [CrossRef]
- Sonawane, A.R.; Platig, J.; Fagny, M.; Chen, C.Y.; Paulson, J.N.; Lopes-Ramos, C.M.; DeMeo, D.L.; Quackenbush, J.; Glass, K.; Kuijjer, M.L. Understanding Tissue-Specific Gene Regulation. Cell Rep. 2017, 21, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Ben Guebila, M.; Lopes-Ramos, C.M.; Weighill, D.; Sonawane, A.R.; Burkholz, R.; Shamsaei, B.; Platig, J.; Glass, K.; Kuijjer, M.L.; Quackenbush, J. GRAND: A Database of Gene Regulatory Network Models across Human Conditions. Nucleic Acids Res. 2022, 50, D610–D621. [Google Scholar] [CrossRef] [PubMed]
- Bushweller, J.H. Targeting Transcription Factors in Cancer—From Undruggable to Reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Weth, F.R.; Hoggarth, G.B.; Weth, A.F.; Paterson, E.; White, M.P.J.; Tan, S.T.; Peng, L.; Gray, C. Unlocking Hidden Potential: Advancements, Approaches, and Obstacles in Repurposing Drugs for Cancer Therapy. Br. J. Cancer 2023, 130, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Glass, K.; Huttenhower, C.; Quackenbush, J.; Yuan, G.C. Passing Messages between Biological Networks to Refine Predicted Interactions. PLoS ONE 2013, 8, e64832. [Google Scholar] [CrossRef]
- Schlauch, D.; Paulson, J.N.; Young, A.; Glass, K.; Quackenbush, J. Estimating Gene Regulatory Networks with PandaR. Bioinformatics 2017, 33, 2232–2234. [Google Scholar] [CrossRef]
- Pertesi, M.; Went, M.; Hansson, M.; Hemminki, K.; Houlston, R.S.; Nilsson, B. Genetic Predisposition for Multiple Myeloma. Leukemia 2020, 34, 697–708. [Google Scholar] [CrossRef]
- Lind, J.; Aksoy, O.; Prchal-Murphy, M.; Fan, F.; Fulciniti, M.; Stoiber, D.; Bakiri, L.; Wagner, E.F.; Zwickl-Traxler, E.; Sattler, M.; et al. Dual Therapeutic Targeting of MYC and JUNB Transcriptional Programs for Enhanced Anti-Myeloma Activity. Blood Cancer J. 2024, 14, 138. [Google Scholar] [CrossRef] [PubMed]
- Neuse, C.J.; Lomas, O.C.; Schliemann, C.; Shen, Y.J.; Manier, S.; Bustoros, M.; Ghobrial, I.M. Genome Instability in Multiple Myeloma. Leukemia 2020, 34, 2887–2897. [Google Scholar] [CrossRef]
- Peng, Y.; Li, F.; Zhang, P.; Wang, X.; Shen, Y.; Feng, Y.; Jia, Y.; Zhang, R.; Hu, J.; He, A. IGF-1 Promotes Multiple Myeloma Progression through PI3K/Akt-Mediated Epithelial-Mesenchymal Transition. Life Sci. 2020, 249, 117503. [Google Scholar] [CrossRef] [PubMed]
- Pölönen, P.; Mehtonen, J.; Lin, J.; Liuksiala, T.; Häyrynen, S.; Teppo, S.; Mäkinen, A.; Kumar, A.; Malani, D.; Pohjolainen, V.; et al. HEMap: An Interactive Online Resource for Characterizing Molecular Phenotypes across Hematologic Malignancies. Cancer Res. 2019, 79, 2466–2479. [Google Scholar] [CrossRef] [PubMed]
- Benyoucef, A.; Haigh, J.J.; Brand, M. Unveiling the Complexity of Transcription Factor Networks in Hematopoietic Stem Cells: Implications for Cell Therapy and Hematological Malignancies. Front. Oncol. 2023, 13, 1151343. [Google Scholar] [CrossRef]
- Zhuang, S.; Yang, Z.; Cui, Z.; Zhang, Y.; Che, F. Epigenetic Alterations and Advancement of Lymphoma Treatment. Ann. Hematol. 2023, 103, 1435–1454. [Google Scholar] [CrossRef] [PubMed]
- Calero-Nieto, F.J.; Ng, F.S.; Wilson, N.K.; Hannah, R.; Moignard, V.; Leal-Cervantes, A.I.; Jimenez-Madrid, I.; Diamanti, E.; Wernisch, L.; Göttgens, B. Key Regulators Control Distinct Transcriptional Programmes in Blood Progenitor and Mast Cells. EMBO J. 2014, 33, 1212–1226. [Google Scholar] [CrossRef]
- Van De Donk, N.W.C.J.; Moreau, P.; Plesner, T.; Palumbo, A.; Gay, F.; Laubach, J.P.; Malavasi, F.; Avet-Loiseau, H.; Mateos, M.V.; Sonneveld, P.; et al. Clinical Efficacy and Management of Monoclonal Antibodies Targeting CD38 and SLAMF7 in Multiple Myeloma. Blood 2016, 127, 681–695. [Google Scholar] [CrossRef]
- Ryu, J.; Koh, Y.; Park, H.; Kim, D.Y.; Kim, D.C.; Byun, J.M.; Lee, H.J.; Yoon, S.S. Highly Expressed Integrin-A8 Induces Epithelial to Mesenchymal Transition-Like Features in Multiple Myeloma with Early Relapse. Mol. Cells 2016, 39, 898–908. [Google Scholar] [CrossRef]
- Xu, C.; Gao, M.; Zhang, J.; Fu, Y. IL5RA as an Immunogenic Cell Death-Related Predictor in Progression and Therapeutic Response of Multiple Myeloma. Sci. Rep. 2023, 13, 8528. [Google Scholar] [CrossRef]
- Zhang, X.-D.; Baladandayuthapani, V.; Lin, H.Y.; Barlogie, B.; Usmani, S.Z.; Yang, L.; Orlowski, R.Z. Identification Of Tight Junction Protein (TJP)-1 As a Modulator and Biomarker Of Proteasome Inhibitor Sensitivity In Multiple Myeloma. Blood 2013, 122, 123. [Google Scholar] [CrossRef]
- Rodriguez-Otero, P.; van de Donk, N.W.C.J.; Pillarisetti, K.; Cornax, I.; Vishwamitra, D.; Gray, K.; Hilder, B.; Tolbert, J.; Renaud, T.; Masterson, T.; et al. GPRC5D as a Novel Target for the Treatment of Multiple Myeloma: A Narrative Review. Blood Cancer J. 2024, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D. The Role of the Wnt-Signaling Antagonist DKK1 in the Development of Osteolytic Lesions in Multiple Myeloma. N. Engl. J. Med. 2003, 349, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- van Andel, H.; Kocemba, K.A.; Spaargaren, M.; Pals, S.T. Aberrant Wnt Signaling in Multiple Myeloma: Molecular Mechanisms and Targeting Options. Leukemia 2019, 33, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Niens, M.; Visser, L.; Nolte, I.M.; Van Der Steege, G.; Diepstra, A.; Cordano, P.; Jarrett, R.F.; Te Meerman, G.J.; Poppema, S.; Van Den Berg, A. Serum Chemokine Levels in Hodgkin Lymphoma Patients: Highly Increased Levels of CCL17 and CCL22. Br. J. Haematol. 2008, 140, 527–536. [Google Scholar] [CrossRef]
- Skinnider, B.F.; Kapp, U.; Mak, T.W. Interleukin 13: A Growth Factor in Hodgkin Lymphoma. Int. Arch. Allergy Immunol. 2001, 126, 267–276. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Marshall, A.; Chavez, E.A.; Miyata-Takata, T.; Ben-Neriah, S.; Unrau, D.; Telenius, A.; et al. Single-Cell Profiling Reveals the Importance of CXCL13/ CXCR5 Axis Biology in Lymphocyte-Rich Classic Hodgkin Lymphoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2105822118. [Google Scholar] [CrossRef]
- Iijima, K.; Yamada, H.; Miharu, M.; Imadome, K.I.; Miyagawa, Y.; Akimoto, S.; Kobayashi, K.; Okita, H.; Nakazawa, A.; Fujiwara, S.; et al. ZNF385B Is Characteristically Expressed in Germinal Center B Cells and Involved in B-Cell Apoptosis. Eur. J. Immunol. 2012, 42, 3405–3415. [Google Scholar] [CrossRef]
- El-Mallawany, N.K.; Day, N.; Ayello, J.; Van De Ven, C.; Conlon, K.; Fermin, D.; Basrur, V.; Elenitoba-Johnson, K.; Lim, M.; Cairo, M.S. Differential Proteomic Analysis of Endemic and Sporadic Epstein-Barr Virus-Positive and Negative Burkitt Lymphoma. Eur. J. Cancer 2015, 51, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Pehlivan, M.; Sercan, Z.; Sercan, H.O. SFRP1 Promoter Methylation Is Associated with Persistent Philadelphia Chromosome in Chronic Myeloid Leukemia. Leuk. Res. 2009, 33, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, Q.; Li, C.; Chen, Z.; Deng, M.; Weng, H.; Zhu, X. Analysis of Endoplasmic Reticulum Stress-Related Gene Signature for the Prognosis and Pattern in Diffuse Large B Cell Lymphoma. Sci. Rep. 2023, 13, 13894. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ma, L.; Wei, H.; Nie, F.; Ning, J.; Jiang, T. MiR-1256 Inhibits Cell Proliferation and Cell Cycle Progression in Papillary Thyroid Cancer by Targeting 5-Hydroxy Tryptamine Receptor 3A. Hum. Cell 2020, 33, 630–640. [Google Scholar] [CrossRef]
- Zohren, F.; Bruns, I.; Pechtel, S.; Schroeder, T.; Fenk, R.; Czibere, A.; Maschmeyer, G.; Kofahl-Krause, D.; Niederle, N.; Heil, G.; et al. Prognostic Value of Circulating Bcl-2/IgH Levels in Patients with Follicular Lymphoma Receiving First-Line Immunochemotherapy. Blood 2015, 126, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Mozos, A.; Royo, C.; Hartmann, E.; De Jong, D.; Baró, C.; Valera, A.; Fu, K.; Weisenburger, D.D.; Delabie, J.; Chuang, S.S.; et al. SOX11 Expression Is Highly Specific for Mantle Cell Lymphoma and Identifies the Cyclin D1-Negative Subtype. Haematologica 2009, 94, 1555. [Google Scholar] [CrossRef] [PubMed]
- Harder, L.; Eschenburg, G.; Zech, A.; Kriebitzsch, N.; Otto, B.; Streichert, T.; Behlich, A.S.; Dierck, K.; Klingler, B.; Hansen, A.; et al. Aberrant ZNF423 Impedes B Cell Differentiation and Is Linked to Adverse Outcome of ETV6-RUNX1 Negative B Precursor Acute Lymphoblastic Leukemia. J. Exp. Med. 2013, 210, 2289. [Google Scholar] [CrossRef]
- Lollies, A.; Hartmann, S.; Schneider, M.; Bracht, T.; Weiß, A.L.; Arnolds, J.; Klein-Hitpass, L.; Sitek, B.; Hansmann, M.L.; Küppers, R.; et al. An Oncogenic Axis of STAT-Mediated BATF3 Upregulation Causing MYC Activity in Classical Hodgkin Lymphoma and Anaplastic Large Cell Lymphoma. Leukemia 2018, 32, 92–101. [Google Scholar] [CrossRef]
- Lee, N.; Kim, S.M.; Lee, Y.; Jeong, D.; Yun, J.; Ryu, S.; Yoon, S.S.; Ahn, Y.O.; Hwang, S.M.; Lee, D.S. Prognostic Value of Integrated Cytogenetic, Somatic Variation, and Copy Number Variation Analyses in Korean Patients with Newly Diagnosed Multiple Myeloma. PLoS ONE 2021, 16, e0246322. [Google Scholar] [CrossRef] [PubMed]
- Teater, M.; Dominguez, P.M.; Redmond, D.; Chen, Z.; Ennishi, D.; Scott, D.W.; Cimmino, L.; Ghione, P.; Chaudhuri, J.; Gascoyne, R.D.; et al. AICDA Drives Epigenetic Heterogeneity and Accelerates Germinal Center-Derived Lymphomagenesis. Nat. Commun. 2018, 9, 222. [Google Scholar] [CrossRef]
- Cui, B.; Ghia, E.M.; Chen, L.; Rassenti, L.Z.; DeBoever, C.; Widhopf, G.F.; Yu, J.; Neuberg, D.S.; Wierda, W.G.; Rai, K.R.; et al. High-Level ROR1 Associates with Accelerated Disease Progression in Chronic Lymphocytic Leukemia. Blood 2016, 128, 2931–2940. [Google Scholar] [CrossRef] [PubMed]
- Soltani, M.; Zhao, Y.; Xia, Z.; Ganjalikhani Hakemi, M.; Bazhin, A.V. The Importance of Cellular Metabolic Pathways in Pathogenesis and Selective Treatments of Hematological Malignancies. Front. Oncol. 2021, 11, 767026. [Google Scholar] [CrossRef] [PubMed]
- Yusenko, M.V.; Trentmann, A.; Casolari, D.A.; Abdel Ghani, L.; Lenz, M.; Horn, M.; Dörner, W.; Klempnauer, S.; Mootz, H.D.; Arteaga, M.F.; et al. C/EBPβ Is a MYB- and P300-Cooperating pro-Leukemogenic Factor and Promising Drug Target in Acute Myeloid Leukemia. Oncogene 2021, 40, 4746–4758. [Google Scholar] [CrossRef] [PubMed]
- Burk, O.; Mink, S.; Ringwald, M.; Klempnauer, K.H. Synergistic Activation of the Chicken Mim-1 Gene by V-myb and C/EBP Transcription Factors. EMBO J. 1993, 12, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Pellegrino, E.; Mattioli, M.; Agnelli, L.; Lombardi, L.; Boccalatte, F.; Costa, G.; Ruggeri, B.A.; Cheng, M.; Chiarle, R.; et al. Functional Validation of the Anaplastic Lymphoma Kinase Signature Identifies CEBPB and Bcl2A1 as Critical Target Genes. J. Clin. Investig. 2006, 116, 3171–3182. [Google Scholar] [CrossRef]
- Klempnauer, K.H. C/EBPβ Sustains the Oncogenic Program of AML Cells by Cooperating with MYB and Co-Activator P300 in a Transcriptional Module. Exp. Hematol. 2022, 108, 8–15. [Google Scholar] [CrossRef]
- Matherne, M.G.; Phillips, E.S.; Embrey, S.J.; Burke, C.M.; Machado, H.L. Emerging Functions of C/EBPβ in Breast Cancer. Front. Oncol. 2023, 13, 1111522. [Google Scholar] [CrossRef]
- Hatanaka, A.; Nakada, S.; Matsumoto, G.; Satoh, K.; Aketa, I.; Watanabe, A.; Hirakawa, T.; Tsujita, T.; Waku, T.; Kobayashi, A. The Transcription Factor NRF1 (NFE2L1) Activates Aggrephagy by Inducing P62 and GABARAPL1 after Proteasome Inhibition to Maintain Proteostasis. Sci. Rep. 2023, 13, 14405. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Zhang, K.; Hong, Z.; Liu, Z.; Liu, Z.; Li, G.; Xu, Y.; Pi, J.; Fu, J.; et al. Understanding the Transcription Factor NFE2L1/NRF1 from the Perspective of Hallmarks of Cancer. Antioxidants 2024, 13, 758. [Google Scholar] [CrossRef]
- Seçilmiş, D.; Hillerton, T.; Morgan, D.; Tjärnberg, A.; Nelander, S.; Nordling, T.E.M.; Sonnhammer, E.L.L. Uncovering Cancer Gene Regulation by Accurate Regulatory Network Inference from Uninformative Data. NPJ Syst. Biol. Appl. 2020, 6, 37. [Google Scholar] [CrossRef]
- Nakamura-García, A.K.; Espinal-Enríquez, J. The Network Structure of Hematopoietic Cancers. Sci. Rep. 2023, 13, 19837. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Li, Y.; Ma, L.; Xu, Q.; Tan, F.; Chen, G. GRNdb: Decoding the Gene Regulatory Networks in Diverse Human and Mouse Conditions. Nucleic Acids Res. 2021, 49, D97–D103. [Google Scholar] [CrossRef] [PubMed]
- Unger Avila, P.; Padvitski, T.; Leote, A.C.; Chen, H.; Saez-Rodriguez, J.; Kann, M.; Beyer, A. Gene Regulatory Networks in Disease and Ageing. Nat. Rev. Nephrol. 2024, 20, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Affymetrix, Inc. GeneChip Expression Analysis: Data Analysis Fundamentals; Affymatrix: Santa Clara, CA, USA, 2004. [Google Scholar]
- McCall, M.N.; Murakami, P.N.; Lukk, M.; Huber, W.; Irizarry, R.A. Assessing Affymetrix GeneChip Microarray Quality. BMC Bioinform. 2011, 12, 137. [Google Scholar] [CrossRef]
- McCall, M.N.; Bolstad, B.M.; Irizarry, R.A. Frozen Robust Multiarray Analysis (FRMA). Biostatistics 2010, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Freytag, S.; Gagnon-Bartsch, J.; Speed, T.P.; Bahlo, M. Systematic Noise Degrades Gene Co-Expression Signals but Can Be Corrected. BMC Bioinform. 2015, 16, 309. [Google Scholar] [CrossRef] [PubMed]
- Everitt, B.S.; Landau, S.; Leese, M. Cluster Analysis. In Encyclopedia of Pain; Springer: Berlin/Heidelberg, Germany, 1974; pp. 403–404. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining Clusters from a Hierarchical Cluster Tree: The Dynamic Tree Cut Package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast Gene Set Enrichment Analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Francesconi, M.; Remondini, D.; Neretti, N.; Sedivy, J.M.; Cooper, L.N.; Verondini, E.; Milanesi, L.; Castellani, G. Reconstructing Networks of Pathways via Significance Analysis of Their Intersections. BMC Bioinform. 2008, 9, S9. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.L.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2024, 52, D1265–D1275. [Google Scholar] [CrossRef]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hematological Malignancies | Enriched KEGG Pathways |
|---|---|
| ALL | Lysosome (41), DNA replication (21) |
| CLL | Purine metabolism (49), B-cell receptor signaling pathway (37) |
| CML | Endocytosis (58), Insulin signaling pathway (44), Erbb signaling pathway (24), Pentose phosphate pathway (14) |
| MM | Regulation of actin cytoskeleton (74), Fc gamma r mediated phagocytosis (47), T-cell receptor signaling pathway (45), Fc epsilon RI signaling pathway (30), Nod like receptor signaling pathway (28) |
| CLL, MM | Natural killer cell mediated cytotoxicity (22) |
| CML, MM | Primary immunodeficiency (7) |
| DLBCL, MM | Leukocyte transendothelial migration (12) |
| MDS, MM | Hematopoietic cell lineage (17) |
| MM, MZLs | Oxidative phosphorylation (35) |
| ALL, MZLs, PTCL | Ribosome (39) |
| ALL, AML, FL, MDS, MZLs | Cell cycle (21) |
| ALL, AML, MDS, MM, MZLs | Spliceosome (30) |
| ALL, AML, BL, DLBCL, MCL, MDS, MZLs, PTCL | Focal adhesion (22) |
| ALL, DLBCL, FL, HL, MCL, MM, MZLs, PTCL | Chemokine signaling pathway (8) |
| AML, CML, DLBCL, FL, HL, MDS, MM, PTCL | Antigen processing and presentation (5) |
| AML, CLL, CML, FL, HL, MDS, MM, MZLs, PTCL | Graft versus host disease (5) |
| ALL, AML, BL, CML, DLBCL, HL, MCL, MDS, MZLs, PTCL | ECM receptor interaction (12) |
| ALL, AML, BL, DLBCL, FL, HL, MCL, MDS, MZLs, PTCL | Complement and coagulation cascades (12) |
| ALL, AML, DLBCL, FL, HL, MCL, MDS, MM, MZLs, PTCL | Cytokine cytokine receptor interaction (9) |
| AML, CLL, CML, DLBCL, FL, HL, MCL, MDS, MM, MZLs, PTCL | Cell adhesion molecules cams (5), Allograft rejection (4) |
| HMs | Enriched Pathway | TF | Common Driving Genes |
|---|---|---|---|
| ALL, AML, BL, CLL, CML, DLBCL, FL, HL, MCL, MDS, MM, MZLs, PTCL | AMINOACYL TRNA BIOSYNTHESIS (hsa00970) | CEBPB | AARS, CARS, EPRS, FARSA, GARS, HARS and other 10 genes |
| MAPK SIGNALING PATHWAY (hsa04010) | EPAS1 | AKT1, AKT2, BDNF, CACNB4, FGFR1, FGFR4, MAP2K2, MAPT, NTRK2, TGFB1 and other 50 genes | |
| CELL CYCLE (hsa04110) | NFE2L1 | CDKN1A, CHEK1, CHEK2, MDM2, MYC, SMAD2, TP53, WEE1 and other 22 genes | |
| UBIQUITIN MEDIATED PROTEOLYSIS (hsa04120) | NFE2L1 | MDM2, XIAP and other 33 genes | |
| CHEMOKINE SIGNALING PATHWAY (hsa04062) | NFKB2 | AKT2, PIK3CD, PIK3R1, PRKACA, PRKCD and other 12 genes | |
| FOCAL ADHESION (hsa04510) | NR4A3 | COL2A1, ERBB2, FLNA, FLT4, IGF1R, LAMA1, PDGFRA, PDGFRB, PIK3R1, VEGFA and other 50 genes | |
| AML, CLL, HL, MCL, PTCL | ABC TRANSPORTERS (hsa02010) | CEBPB | ABCC6, CFTR, ABCC1, ABCD3, ABCC8, ABCC3,ABCG1, ABCC5, ABCC4, ABCA8, ABCB9, ABCA6, ABCA5, ABCC10, ABCA13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dall’Olio, D.; Magnani, F.; Casadei, F.; Matteuzzi, T.; Curti, N.; Merlotti, A.; Simonetti, G.; Della Porta, M.G.; Remondini, D.; Tarozzi, M.; et al. Emerging Signatures of Hematological Malignancies from Gene Expression and Transcription Factor-Gene Regulations. Int. J. Mol. Sci. 2024, 25, 13588. https://doi.org/10.3390/ijms252413588
Dall’Olio D, Magnani F, Casadei F, Matteuzzi T, Curti N, Merlotti A, Simonetti G, Della Porta MG, Remondini D, Tarozzi M, et al. Emerging Signatures of Hematological Malignancies from Gene Expression and Transcription Factor-Gene Regulations. International Journal of Molecular Sciences. 2024; 25(24):13588. https://doi.org/10.3390/ijms252413588
Chicago/Turabian StyleDall’Olio, Daniele, Federico Magnani, Francesco Casadei, Tommaso Matteuzzi, Nico Curti, Alessandra Merlotti, Giorgia Simonetti, Matteo Giovanni Della Porta, Daniel Remondini, Martina Tarozzi, and et al. 2024. "Emerging Signatures of Hematological Malignancies from Gene Expression and Transcription Factor-Gene Regulations" International Journal of Molecular Sciences 25, no. 24: 13588. https://doi.org/10.3390/ijms252413588
APA StyleDall’Olio, D., Magnani, F., Casadei, F., Matteuzzi, T., Curti, N., Merlotti, A., Simonetti, G., Della Porta, M. G., Remondini, D., Tarozzi, M., & Castellani, G. (2024). Emerging Signatures of Hematological Malignancies from Gene Expression and Transcription Factor-Gene Regulations. International Journal of Molecular Sciences, 25(24), 13588. https://doi.org/10.3390/ijms252413588