

Association between ZASP/LDB3 Pro26Ser and Inclusion Body Myopathy



 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
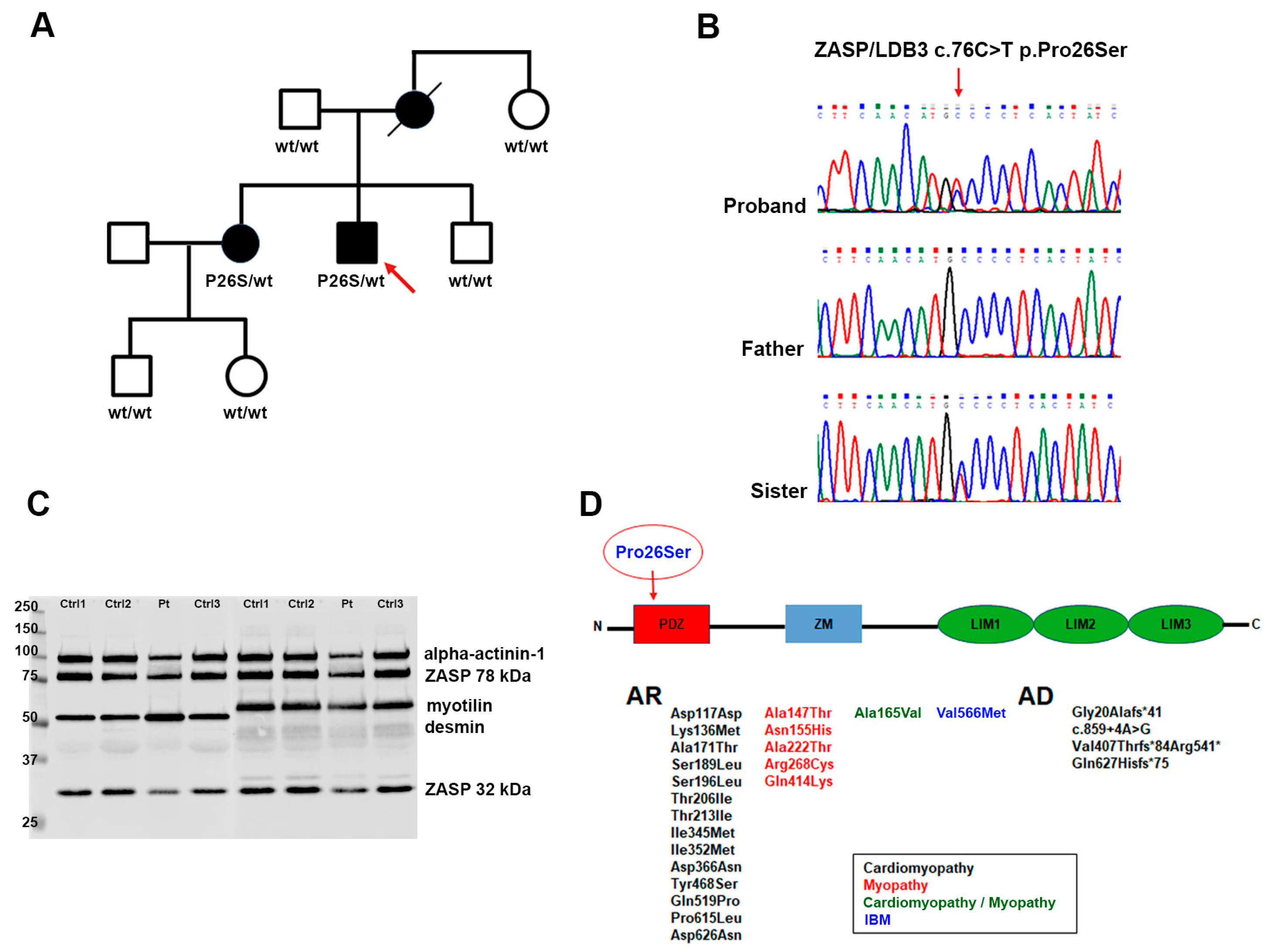
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Muscle Biopsy
3.1.1. Light Microscopy
3.1.2. Electron Microscopy
3.1.3. Western Blot
3.2. Genetic Testing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Callan, A.; Capkun, G.; Vasanthaprasad, V.; Freitas, R.; Needham, M.A. Systematic Review and Meta-Analysis of Prevalence Studies of Sporadic Inclusion Body Myositis. J. Neuromuscul. Dis. 2017, 4, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Nagy, S.; Khan, A.; Machado, P.M.; Houlden, H. Inclusion body myositis: From genetics to clinical trials. J. Neurol. 2023, 270, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Dimachkie, M.M.; Barohn, R.J. Inclusion body myositis. Semin. Neurol. 2012, 32, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A. Inclusion body myositis: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2019, 15, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Gang, Q.; Bettencourt, C.; Machado, P.; Hanna, M.G.; Houlden, H. Sporadic inclusion body myositis: The genetic contributions to the pathogenesis. Orphanet J. Rare Dis. 2014, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Broccolini, A.; Mirabella, M. Hereditary inclusion-body myopathies. Biochim. Biophys. Acta 2015, 1852, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Lilleker, J.B.; Naddaf, E.; Saris, C.G.J.; Schmidt, J.; de Visser, M.; Weihl, C.C.; 272nd ENMC workshop participants. 272nd ENMC international workshop: 10 Years of progress—Revision of the ENMC 2013 diagnostic criteria for inclusion body myositis and clinical trial readiness. 16–18 June 2023, Hoofddorp, The Netherlands. Neuromuscul. Disord. 2024, 37, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Klaavuniemi, T.; Kelloniemi, A.; Ylänne, J. The ZASP-like motif in actinin-associated LIM protein is required for interaction with the alpha-actinin rod and for targeting to the muscle Z-line. J. Biol. Chem. 2004, 279, 26402–26410. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; Blech-Hermoni, Y.; Subedi, K.; Mpamugo, J.; Obeng-Nyarko, C.; Ohman, R.; Molloy, I.; Kates, M.; Hale, J.; Stauffer, S.; et al. Myopathy associated LDB3 mutation causes Z-disc disassembly and protein aggregation through PKCα and TSC2-mTOR downregulation. Commun. Biol. 2021, 4, 355. [Google Scholar] [CrossRef]
- Faulkner, G.; Pallavicini, A.; Formentin, E.; Comelli, A.; Ievolella, C.; Trevisan, S.; Bortoletto, G.; Scannapieco, P.; Salamon, M.; Mouly, V.; et al. ZASP: A new Z-band alternatively spliced PDZ-motif protein. J. Cell Biol. 1999, 146, 465–475. [Google Scholar] [CrossRef]
- van Ham, M.; Hendriks, W. PDZ domains-glue and guide. Mol. Biol. Rep. 2003, 30, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ruiz, J.; Bajraktari, I.; Ohman, R.; Banerjee, S.; Gribble, K.; Kaufman, J.D.; Wingfield, P.T.; Griggs, R.C.; Fischbeck, K.H.; et al. Z-disc-associated, alternatively spliced, PDZ motif-containing protein (ZASP) mutations in the actin-binding domain cause disruption of skeletal muscle actin filaments in myofibrillar myopathy. J. Biol. Chem. 2014, 289, 13615–13626. [Google Scholar] [CrossRef] [PubMed]
- Vatta, M.; Mohapatra, B.; Jimenez, S.; Sanchez, X.; Faulkner, G.; Perles, Z.; Sinagra, G.; Lin, J.H.; Vu, T.M.; Zhou, Q.; et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J. Am. Coll. Cardiol. 2003, 42, 2014–2027. [Google Scholar] [CrossRef]
- Theis, J.L.; Bos, J.M.; Bartleson, V.B.; Will, M.L.; Binder, J.; Vatta, M.; Towbin, J.A.; Gersh, B.J.; Ommen, S.R.; Ackerman, M.J. Echocardiographic-determined septal morphology in Z-disc hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 351, 896–902. [Google Scholar] [CrossRef]
- Cai, H.; Yabe, I.; Sato, K.; Kano, T.; Nakamura, M.; Hozen, H.; Sasaki, H. Clinical, pathological, and genetic mutation analysis of sporadic inclusion body myositis in Japanese people. J. Neurol. 2012, 259, 1913–1922. [Google Scholar] [CrossRef]
- Selcen, D.; Engel, A.G. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann. Neurol. 2005, 57, 269–276. [Google Scholar] [CrossRef]
- Griggs, R.; Vihola, A.; Hackman, P.; Talvinen, K.; Haravuori, H.; Faulkner, G.; Eymard, B.; Richard, I.; Selcen, D.; Engel, A.; et al. Zaspopathy in a large classic late-onset distal myopathy family. Brain 2007, 130, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Wadmore, K.; Azad, A.J.; Gehmlich, K. The Role of Z-disc Proteins in Myopathy and Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 3058. [Google Scholar] [CrossRef]
- Cassandrini, D.; Merlini, L.; Pilla, F.; Cenni, V.; Santi, S.; Faldini, C.; Santorelli, F.M.; Sabatelli, P. Protein aggregates and autophagy involvement in a family with a mutation in Z-band alternatively spliced PDZ-motif protein. Neuromuscul. Disord. 2021, 31, 44–51. [Google Scholar] [CrossRef]
- Zhou, Q.; Ruiz-Lozano, P.; Martone, M.E.; Chen, J. Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to alpha-actinin-2 and protein kinase C. J. Biol. Chem. 1999, 274, 19807–19813. [Google Scholar] [CrossRef]
- Ripolone, M.; Velardo, D.; Mondello, S.; Zanotti, S.; Magri, F.; Minuti, E.; Cazzaniga, S.; Fortunato, F.; Ciscato, P.; Tiberio, F.; et al. Muscle histological changes in a large cohort of patients affected with Becker muscular dystrophy. Acta Neuropathol. Commun. 2022, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, V.; Sewry, C.A. Muscle Biopsy, A Practical Approach, 3rd ed.; Saunders Elsevier: London, UK, 2007; pp. 28–39. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piga, D.; Zanotti, S.; Ripolone, M.; Napoli, L.; Ciscato, P.; Gibertini, S.; Maggi, L.; Fortunato, F.; Rigamonti, A.; Ronchi, D.; et al. Association between ZASP/LDB3 Pro26Ser and Inclusion Body Myopathy. Int. J. Mol. Sci. 2024, 25, 6547. https://doi.org/10.3390/ijms25126547
Piga D, Zanotti S, Ripolone M, Napoli L, Ciscato P, Gibertini S, Maggi L, Fortunato F, Rigamonti A, Ronchi D, et al. Association between ZASP/LDB3 Pro26Ser and Inclusion Body Myopathy. International Journal of Molecular Sciences. 2024; 25(12):6547. https://doi.org/10.3390/ijms25126547
Chicago/Turabian StylePiga, Daniela, Simona Zanotti, Michela Ripolone, Laura Napoli, Patrizia Ciscato, Sara Gibertini, Lorenzo Maggi, Francesco Fortunato, Andrea Rigamonti, Dario Ronchi, and et al. 2024. "Association between ZASP/LDB3 Pro26Ser and Inclusion Body Myopathy" International Journal of Molecular Sciences 25, no. 12: 6547. https://doi.org/10.3390/ijms25126547
APA StylePiga, D., Zanotti, S., Ripolone, M., Napoli, L., Ciscato, P., Gibertini, S., Maggi, L., Fortunato, F., Rigamonti, A., Ronchi, D., Comi, G. P., Corti, S., & Sciacco, M. (2024). Association between ZASP/LDB3 Pro26Ser and Inclusion Body Myopathy. International Journal of Molecular Sciences, 25(12), 6547. https://doi.org/10.3390/ijms25126547