
Advanced Glycation End Products: A Sweet Flavor That Embitters Cardiovascular Disease

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
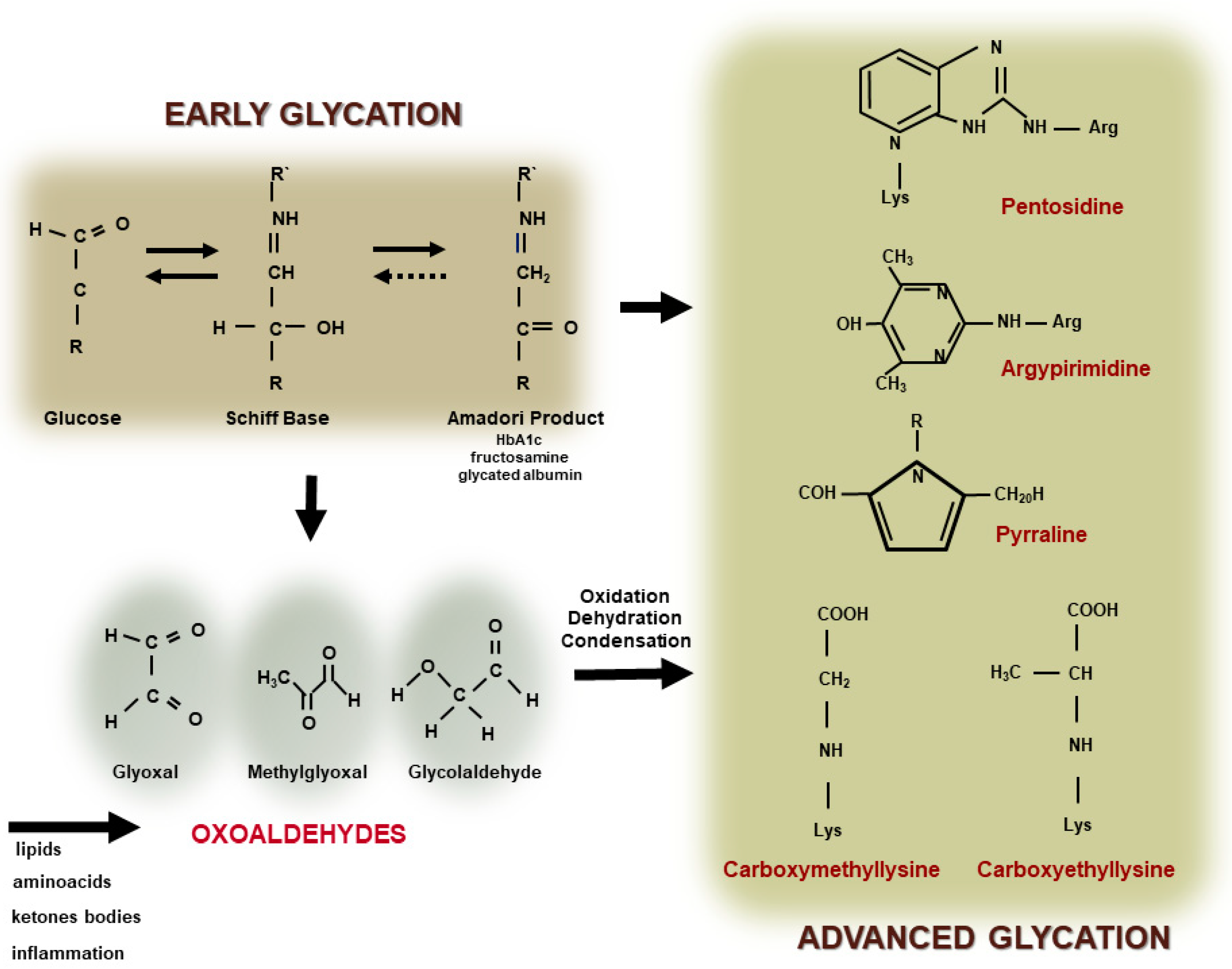
2. Formation of Advanced Glycation End Products
3. AGEs as Indicators of Cardiovascular Burden: Where Are We?
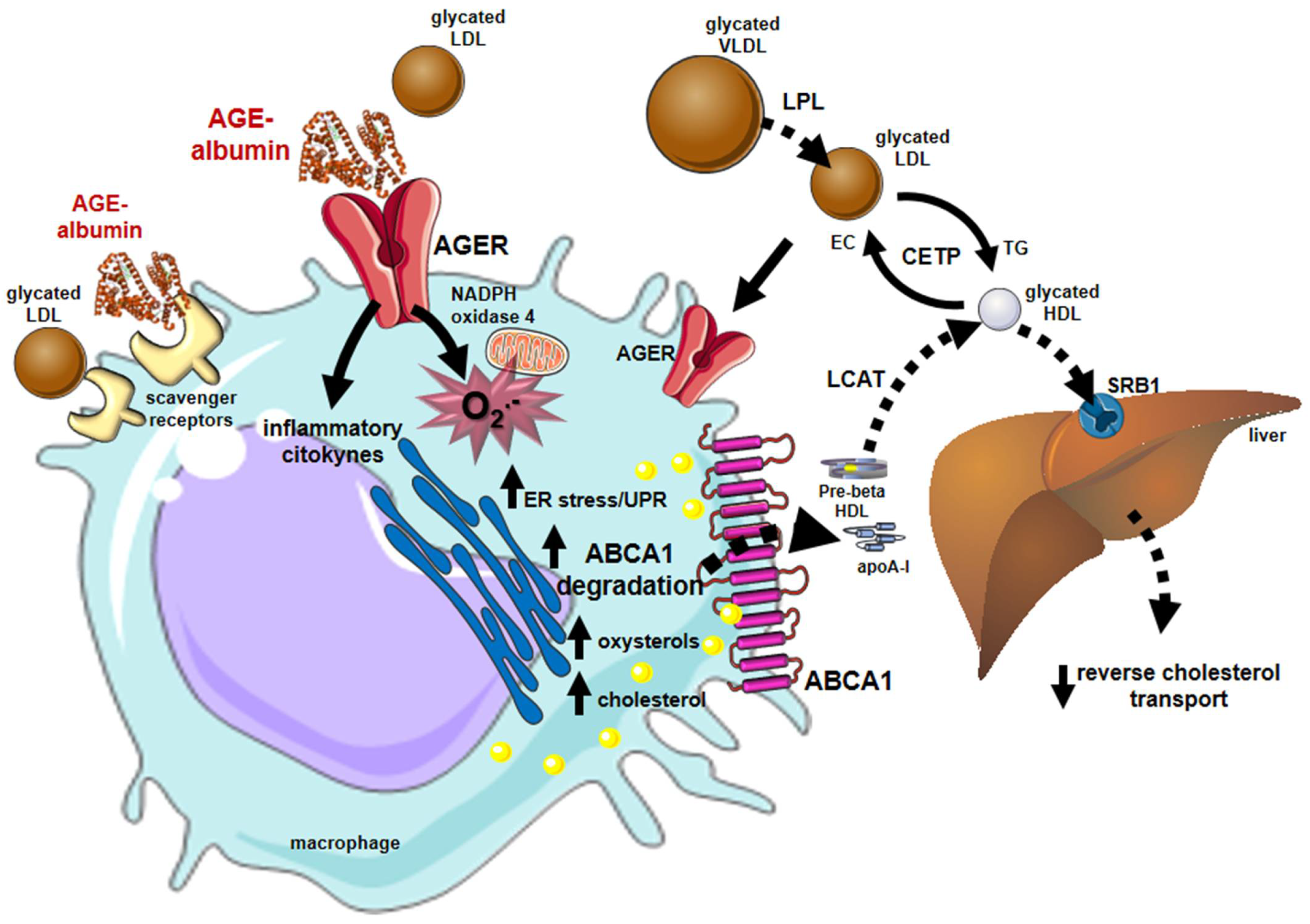
4. Sweet AGEs Driving the Bitter Pathophysiology of CVD
5. AGEs: Where Are We Going?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2021, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Lawes, C.M.M.; Vander Hoorn, S.; Murray, C.J.L.; Ezzati, M. Global and Regional Mortality from Ischaemic Heart Disease and Stroke Attributable to Higher-than-Optimum Blood Glucose Concentration: Comparative Risk Assessment. Lancet 2006, 368, 1651–1659. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.K.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes Mellitus, Fasting Blood Glucose Concentration, and Risk of Vascular Disease: A Collaborative Meta-Analysis of 102 Prospective Studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef]
- Collins, A.J.; Foley, R.N.; Gilbertson, D.T.; Chen, S.-C. United States Renal Data System Public Health Surveillance of Chronic Kidney Disease and End-Stage Renal Disease. Kidney Int. Suppl. 2015, 5, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Moxey, P.W.; Gogalniceanu, P.; Hinchliffe, R.J.; Loftus, I.M.; Jones, K.J.; Thompson, M.M.; Holt, P.J. Lower Extremity Amputations—A Review of Global Variability in Incidence. Diabet. Med. 2011, 28, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Congdon, N.G.; Friedman, D.S.; Lietman, T. Important Causes of Visual Impairment in the World Today. JAMA 2003, 290, 2057–2060. [Google Scholar] [CrossRef]
- Lachin, J.M.; Orchard, T.J.; Nathan, D.M.; DCCT/EDIC Research Group. Update on Cardiovascular Outcomes at 30 Years of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study. Diabetes Care 2014, 37, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.-Y.C.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B.; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive Diabetes Treatment and Cardiovascular Disease in Patients with Type 1 Diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [CrossRef]
- Gaede, P.; Lund-Andersen, H.; Parving, H.-H.; Pedersen, O. Effect of a Multifactorial Intervention on Mortality in Type 2 Diabetes. N. Engl. J. Med. 2008, 358, 580–591. [Google Scholar] [CrossRef]
- Ceriello, A.; Gallo, M.; Armentano, V.; Perriello, G.; Gentile, S.; De Micheli, A.; Associazione Medici Diabetologi. Personalizing Treatment in Type 2 Diabetes: A Self-Monitoring of Blood Glucose Inclusive Innovative Approach. Diabetes Technol. Ther. 2012, 14, 373–378. [Google Scholar] [CrossRef]
- Patton, A.R.; Hill, E.G. Inactivation of Nutrients by Heating With Glucose. Science 1948, 107, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, S.; Blumenfeld, O.; Ranney, H.M. Studies of an Unusual Hemoglobin in Patients with Diabetes Mellitus. Biochem. Biophys. Res. Commun. 1969, 36, 838–843. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and Molecular Cell Biology of Diabetic Complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef] [PubMed]
- Mora-Fernández, C.; Domínguez-Pimentel, V.; de Fuentes, M.M.; Górriz, J.L.; Martínez-Castelao, A.; Navarro-González, J.F. Diabetic Kidney Disease: From Physiology to Therapeutics. J. Physiol. 2014, 592, 3997–4012. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Rabbani, N. Progress in Uremic Toxin Research: Highlights and Hotspots of Protein Glycation in End-Stage Renal Disease. Semin. Dial. 2009, 22, 400–404. [Google Scholar] [CrossRef]
- Dubois, C.; Litke, R.; Rianha, S.; Paul-Constant, C.; Lo Guidice, J.M.; Taront, S.; Tessier, F.J.; Boulanger, E.; Fradin, C. Exposure of Caenorhabditis elegans to Dietary Nε-Carboxymethyllysine Emphasizes Endocytosis as a New Route for Intestinal Absorption of Advanced Glycation End Products. Nutrients 2021, 13, 4398. [Google Scholar] [CrossRef]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef]
- Cai, W.; Gao, Q.D.; Zhu, L.; Peppa, M.; He, C.; Vlassara, H. Oxidative stress-inducing carbonyl compounds from common foods: Novel mediators of cellular dysfunction. Mol. Med. 2002, 8, 337–346. [Google Scholar] [CrossRef]
- Maessen, D.E.; Hanssen, N.M.; Scheijen, J.L.; van der Kallen, C.J.; van Greevenbroek, M.M.; Stehouwer, C.D.; Schalkwijk, C.G. Post-Glucose Load Plasma α-Dicarbonyl Concentrations Are Increased in Individuals With Impaired Glucose Metabolism and Type 2 Diabetes: The CODAM Study. Diabetes Care 2015, 38, 913–920. [Google Scholar] [CrossRef]
- Watanabe, M.; Kawai, Y.; Kitayama, M.; Akao, H.; Motoyama, A.; Wakasa, M.; Saito, R.; Aoki, H.; Fujibayashi, K.; Tsuchiya, T.; et al. Diurnal glycemic fluctuation is associated with severity of coronary artery disease in prediabetic patients: Possible role of nitrotyrosine and glyceraldehyde-derived advanced glycation end products. J. Cardiol. 2017, 69, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.R.; Lachin, J.; DCCT/EDIC Research Group; Monnier, V.M. Skin Advanced Glycation End Products Glucosepane and Methylglyoxal Hydroimidazolone Are Independently Associated with Long-Term Microvascular Complication Progression of Type 1 Diabetes. Diabetes 2015, 64, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.U.; Thorpe, S.R.; Baynes, J.W. Identification of N Epsilon-Carboxymethyllysine as a Degradation Product of Fructoselysine in Glycated Protein. J. Biol. Chem. 1986, 261, 4889–4894. [Google Scholar] [CrossRef]
- Vlassara, H.; Palace, M.R. Diabetes and Advanced Glycation Endproducts. J. Intern. Med. 2002, 251, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Baidoshvili, A.; Niessen, H.W.M.; Stooker, W.; Huybregts, R.A.J.M.; Hack, C.E.; Rauwerda, J.A.; Meijer, C.J.L.M.; Eijsman, L.; van Hinsbergh, V.W.M.; Schalkwijk, C.G. N(Omega)-(Carboxymethyl)Lysine Depositions in Human Aortic Heart Valves: Similarities with Atherosclerotic Blood Vessels. Atherosclerosis 2004, 174, 287–292. [Google Scholar] [CrossRef]
- Nerlich, A.G.; Schleicher, E.D. N(Epsilon)-(Carboxymethyl)Lysine in Atherosclerotic Vascular Lesions as a Marker for Local Oxidative Stress. Atherosclerosis 1999, 144, 41–47. [Google Scholar] [CrossRef]
- Thomas, C.J.; Cleland, T.P.; Sroga, G.E.; Vashishth, D. Accumulation of Carboxymethyl-Lysine (CML) in Human Cortical Bone. Bone 2018, 110, 128–133. [Google Scholar] [CrossRef]
- Charlton, A.; Garzarella, J.; Jandeleit-Dahm, K.A.M.; Jha, J.C. Oxidative Stress and Inflammation in Renal and Cardiovascular Complications of Diabetes. Biology 2020, 10, 18. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The Role of Advanced Glycation End-Products in the Development of Coronary Artery Disease in Patients with and without Diabetes Mellitus: A Review. Mol. Med. 2018, 24, 59. [Google Scholar] [CrossRef]
- Wautier, M.-P.; Guillausseau, P.-J.; Wautier, J.-L. Activation of the Receptor for Advanced Glycation End Products and Consequences on Health. Diabetes Metab. Syndr. 2017, 11, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Glycation & the RAGE Axis: Targeting Signal Transduction through DIAPH1. Expert Rev. Proteom. 2017, 14, 147–156. [Google Scholar] [CrossRef]
- MacLean, M.; Derk, J.; Ruiz, H.H.; Juranek, J.K.; Ramasamy, R.; Schmidt, A.M. The Receptor for Advanced Glycation End Products (RAGE) and DIAPH1: Implications for Vascular and Neuroinflammatory Dysfunction in Disorders of the Central Nervous System. Neurochem. Int. 2019, 126, 154–164. [Google Scholar] [CrossRef]
- Van Zoelen, M.A.D.; Yang, H.; Florquin, S.; Meijers, J.C.M.; Akira, S.; Arnold, B.; Nawroth, P.P.; Bierhaus, A.; Tracey, K.J.; van der Poll, T. Role of Toll-like Receptors 2 and 4, and the Receptor for Advanced Glycation End Products in High-Mobility Group Box 1-Induced Inflammation in Vivo. Shock 2009, 31, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, W.; Han, J.; Khan, Z.A.; Fang, Q.; Jin, Y.; Chen, X.; Zhang, Y.; Wang, M.; Qian, J.; et al. MD2 Activation by Direct AGE Interaction Drives Inflammatory Diabetic Cardi.......iom.m.myopathy. Nat. Commun. 2020, 11, 2148. [Google Scholar] [CrossRef]
- Cai, W.; Torreggiani, M.; Zhu, L.; Chen, X.; He, J.C.; Striker, G.E.; Vlassara, H. AGER1 Regulates Endothelial Cell NADPH Oxidase-Dependent Oxidant Stress via PKC-Delta: Implications for Vascular Disease. Am. J. Physiol. Cell Physiol. 2010, 298, C624–C634. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Lu, C.; Vlassara, H. Advanced Glycation End Product (AGE) Receptor 1 Suppresses Cell Oxidant Stress and Activation Signaling via EGF Receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 13801–13806. [Google Scholar] [CrossRef]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.; Zhu, L.; Vlassara, H. Advanced Glycation Endproduct (AGE) Receptor 1 Is a Negative Regulator of the Inflammatory Response to AGE in Mesangial Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Cai, W.; Goodman, S.; Pyzik, R.; Yong, A.; Chen, X.; Zhu, L.; Neade, T.; Beeri, M.; Silverman, J.M.; et al. Protection against Loss of Innate Defenses in Adulthood by Low Advanced Glycation End Products (AGE) Intake: Role of the Antiinflammatory AGE Receptor-1. J. Clin. Endocrinol. Metab. 2009, 94, 4483–4491. [Google Scholar] [CrossRef]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH Oxidase by AGE Links Oxidant Stress to Altered Gene Expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Torreggiani, M.; Liu, H.; Wu, J.; Zheng, F.; Cai, W.; Striker, G.; Vlassara, H. Advanced Glycation End Product Receptor-1 Transgenic Mice Are Resistant to Inflammation, Oxidative Stress, and Post-Injury Intimal Hyperplasia. Am. J. Pathol. 2009, 175, 1722–1732. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Adachi, H.; Osuga, J.; Ohashi, K.; Yahagi, N.; Sekiya, M.; Okazaki, H.; Tomita, S.; Iizuka, Y.; Shimano, H.; et al. FEEL-1 and FEEL-2 Are Endocytic Receptors for Advanced Glycation End Products. J. Biol. Chem. 2003, 278, 12613–12617. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. The Enzymatic Defence against Glycation in Health, Disease and Therapeutics: A Symposium to Examine the Concept. Biochem. Soc. Trans. 2003, 31, 1341–1342. [Google Scholar] [CrossRef]
- Yumnam, S.; Subedi, L.; Kim, S.Y. Glyoxalase System in the Progression of Skin Aging and Skin Malignancies. Int. J. Mol. Sci. 2021, 22, 310. [Google Scholar] [CrossRef] [PubMed]
- Selvin, E.; Rawlings, A.M.; Lutsey, P.L.; Maruthur, N.; Pankow, J.S.; Steffes, M.; Coresh, J. Fructosamine and Glycated Albumin and the Risk of Cardiovascular Outcomes and Death. Circulation 2015, 132, 269–277. [Google Scholar] [CrossRef]
- Blaauw, J.; Smit, A.J.; van Pampus, M.G.; van Doormaal, J.J.; Aarnoudse, J.G.; Rakhorst, G.; Graaff, R. Skin Autofluorescence, a Marker of Advanced Glycation End Products and Oxidative Stress, Is Increased in Recently Preeclamptic Women. Am. J. Obstet. Gynecol. 2006, 195, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.-H.; Huang, P.-H.; Huang, S.-S.; Wu, T.-C.; Chen, J.-W.; Lin, S.-J. Plasma Levels of Soluble Receptor for Advanced Glycation End Products Are Associated with Endothelial Function and Predict Cardiovascular Events in Nondiabetic Patients. Coron. Artery Dis. 2009, 20, 267–273. [Google Scholar] [CrossRef]
- Baumann, M.; Richart, T.; Sollinger, D.; Pelisek, J.; Roos, M.; Kouznetsova, T.; Eckstein, H.-H.; Heemann, U.; Staessen, J.A. Association between Carotid Diameter and the Advanced Glycation End Product N-Epsilon-Carboxymethyllysine (CML). Cardiovasc. Diabetol. 2009, 8, 45. [Google Scholar] [CrossRef]
- Semba, R.D.; Bandinelli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Plasma Carboxymethyl-Lysine, an Advanced Glycation End Product, and All-Cause and Cardiovascular Disease Mortality in Older Community-Dwelling Adults. J. Am. Geriatr. Soc. 2009, 57, 1874–1880. [Google Scholar] [CrossRef]
- Odani, H.; Iijima, K.; Nakata, M.; Miyata, S.; Kusunoki, H.; Yasuda, Y.; Hiki, Y.; Irie, S.; Maeda, K.; Fujimoto, D. Identification of N(Omega)-Carboxymethylarginine, a New Advanced Glycation Endproduct in Serum Proteins of Diabetic Patients: Possibility of a New Marker of Aging and Diabetes. Biochem. Biophys. Res. Commun. 2001, 285, 1232–1236. [Google Scholar] [CrossRef]
- Sampathkumar, R.; Balasubramanyam, M.; Rema, M.; Premanand, C.; Mohan, V. A Novel Advanced Glycation Index and Its Association with Diabetes and Microangiopathy. Metabolism 2005, 54, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Guerin-Dubourg, A.; Cournot, M.; Planesse, C.; Debussche, X.; Meilhac, O.; Rondeau, P.; Bourdon, E. Association between Fluorescent Advanced Glycation End-Products and Vascular Complications in Type 2 Diabetic Patients. Biomed. Res. Int. 2017, 2017, 7989180. [Google Scholar] [CrossRef]
- Kratochvilová, M.; Zakiyanov, O.; Kalousová, M.; Kříha, V.; Zima, T.; Tesař, V. Associations of Serum Levels of Advanced Glycation End Products with Nutrition Markers and Anemia in Patients with Chronic Kidney Disease. Ren Fail. 2011, 33, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, K.; Nejima, J.; Takano, T.; Ohta, M.; Hashimoto, H. Increased Serum Concentrations of Advanced Glycation End Products: A Marker of Coronary Artery Disease Activity in Type 2 Diabetic Patients. Heart 2001, 85, 87–91. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Ares, S.; Cardelo, M.P.; Gutiérrez-Mariscal, F.M.; Torres-Peña, J.D.; García-Rios, A.; Katsiki, N.; Malagón, M.M.; López-Miranda, J.; Pérez-Martínez, P.; Yubero-Serrano, E.M. Endothelial Dysfunction and Advanced Glycation End Products in Patients with Newly Diagnosed Versus Established Diabetes: From the CORDIOPREV Study. Nutrients 2020, 12, 238. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tsujimoto, T.; Yasuda, K.; Chujo, D.; Ohsugi, M.; Tanabe, A.; Ueki, K.; Kajio, H. Poorly Controlled Type 2 Diabetes with No Progression of Diabetes-Related Complications and Low Levels of Advanced Glycation End Products: A Case Report. Medicine 2019, 98, e16573. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, Y.; Daida, H.; Morimoto, T.; Kasai, T.; Miyauchi, K.; Yamagishi, S.; Takeuchi, M.; Hiro, T.; Kimura, T.; Nakagawa, Y.; et al. Relationship between Advanced Glycation End Products and Plaque Progression in Patients with Acute Coronary Syndrome: The JAPAN-ACS Sub-Study. Cardiovasc. Diabetol. 2013, 12, 5. [Google Scholar] [CrossRef]
- Di Pino, A.; Mangiafico, S.; Urbano, F.; Scicali, R.; Scandura, S.; D’Agate, V.; Piro, S.; Tamburino, C.; Purrello, F.; Rabuazzo, A.M. HbA1c Identifies Subjects With Prediabetes and Subclinical Left Ventricular Diastolic Dysfunction. J. Clin. Endocrinol. Metab. 2017, 102, 3756–3764. [Google Scholar] [CrossRef] [PubMed]
- Montaner, J.; Mendioroz, M.; Delgado, P.; García-Berrocoso, T.; Giralt, D.; Merino, C.; Ribó, M.; Rosell, A.; Penalba, A.; Fernández-Cadenas, I.; et al. Differentiating Ischemic from Hemorrhagic Stroke Using Plasma Biomarkers: The S100B/RAGE Pathway. J. Proteom. 2012, 75, 4758–4765. [Google Scholar] [CrossRef]
- Simm, A.; Philipp, C.; Friedrich, I.; Scheubel, R.J.; Hofmann, H.-S.; Meibodi, K.H.; Sablotzki, A.; Silber, R.-E.; Börgermann, J. Intraoperative SRAGE Kinetics. A New Age-Related Outcome Predictor of Cardiac Surgery. Z. Gerontol. Geriatr. 2014, 47, 666–672. [Google Scholar] [CrossRef]
- Di Pino, A.; Urbano, F.; Scicali, R.; Di Mauro, S.; Filippello, A.; Scamporrino, A.; Piro, S.; Purrello, F.; Rabuazzo, A.M. 1 h Postload Glycemia Is Associated with Low Endogenous Secretory Receptor for Advanced Glycation End Product Levels and Early Markers of Cardiovascular Disease. Cells 2019, 8, 910. [Google Scholar] [CrossRef] [PubMed]
- Karnosová, P.; Mateřánková, M.; Seidlerová, J.; Mayer, O.; Filipovský, J.; Karnos, V. Soluble RAGEs and Cardiovascular Risk Factors in Adult Offspring of Patients with Premature Coronary Heart Disease. Blood Press 2020, 29, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Devangelio, E.; Santilli, F.; Formoso, G.; Ferroni, P.; Bucciarelli, L.; Michetti, N.; Clissa, C.; Ciabattoni, G.; Consoli, A.; Davì, G. Soluble RAGE in Type 2 Diabetes: Association with Oxidative Stress. Free Radic. Biol. Med. 2007, 43, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N.; Matsuhisa, M.; Kaneto, H.; Matsuoka, T.; Sakamoto, K.; Yasuda, T.; Umayahara, Y.; Kosugi, K.; Yamasaki, Y. Serum Endogenous Secretory RAGE Level Is an Independent Risk Factor for the Progression of Carotid Atherosclerosis in Type 1 Diabetes. Atherosclerosis 2009, 204, 288–292. [Google Scholar] [CrossRef]
- Thomas, M.C.; Söderlund, J.; Lehto, M.; Mäkinen, V.-P.; Moran, J.L.; Cooper, M.E.; Forsblom, C.; Groop, P.-H.; FinnDiane Study Group. Soluble Receptor for AGE (RAGE) Is a Novel Independent Predictor of All-Cause and Cardiovascular Mortality in Type 1 Diabetes. Diabetologia 2011, 54, 2669–2677. [Google Scholar] [CrossRef]
- Thomas, M.C.; Woodward, M.; Neal, B.; Li, Q.; Pickering, R.; Marre, M.; Williams, B.; Perkovic, V.; Cooper, M.E.; Zoungas, S.; et al. Relationship between Levels of Advanced Glycation End Products and Their Soluble Receptor and Adverse Outcomes in Adults with Type 2 Diabetes. Diabetes Care 2015, 38, 1891–1897. [Google Scholar] [CrossRef]
- Du, R.; Zhang, R.Y.; Lu, L.; Shen, Y.; Pu, L.J.; Zhu, Z.B.; Zhang, Q.; Hu, J.; Yang, Z.K.; Ding, F.H.; et al. Increased Glycated Albumin and Decreased EsRAGE Levels in Serum Are Related to Negative Coronary Artery Remodeling in Patients with Type 2 Diabetes: An Intravascular Ultrasound Study. Cardiovasc. Diabetol. 2018, 17, 149. [Google Scholar] [CrossRef]
- Yoon, K.H.; Steinberg, H.; Teng, R.; Golm, G.T.; Lee, M.; O’Neill, E.A.; Kaufman, K.D.; Goldstein, B.J. Efficacy and Safety of Initial Combination Therapy with Sitagliptin and Pioglitazone in Patients with Type 2 Diabetes: A 54-Week Study. Diabetes Obes. Metab. 2012, 14, 745–752. [Google Scholar] [CrossRef]
- Paradela-Dobarro, B.; Agra, R.M.; Álvarez, L.; Varela-Román, A.; García-Acuña, J.M.; González-Juanatey, J.R.; Álvarez, E.; García-Seara, F.J. The Different Roles for the Advanced Glycation End Products Axis in Heart Failure and Acute Coronary Syndrome Settings. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 1050–1060. [Google Scholar] [CrossRef]
- Nakamura, K.; Yamagishi, S.; Adachi, H.; Kurita-Nakamura, Y.; Matsui, T.; Yoshida, T.; Sato, A.; Imaizumi, T. Elevation of Soluble Form of Receptor for Advanced Glycation End Products (SRAGE) in Diabetic Subjects with Coronary Artery Disease. Diabetes Metab. Res. Rev. 2007, 23, 368–371. [Google Scholar] [CrossRef]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.; Hitman, G.; Neil, A.; Livingstone, S.; Charlton-Menys, V.; Bao, W.; Demicco, D.A.; Preston, G.M.; et al. Total Soluble and Endogenous Secretory Receptor for Advanced Glycation End Products as Predictive Biomarkers of Coronary Heart Disease Risk in Patients with Type 2 Diabetes: An Analysis from the CARDS Trial. Diabetes 2011, 60, 2379–2385. [Google Scholar] [CrossRef]
- Ebert, H.; Lacruz, M.E.; Kluttig, A.; Simm, A.; Greiser, K.H.; Tiller, D.; Kartschmit, N.; Mikolajczyk, R. Association between Advanced Glycation End Products, Their Soluble Receptor, and Mortality in the General Population: Results from the CARLA Study. Exp. Gerontol. 2020, 131, 110815. [Google Scholar] [CrossRef] [PubMed]
- Reichert, S.; Triebert, U.; Santos, A.N.; Hofmann, B.; Schaller, H.-G.; Schlitt, A.; Schulz, S. Soluble Form of Receptor for Advanced Glycation End Products and Incidence of New Cardiovascular Events among Patients with Cardiovascular Disease. Atherosclerosis 2017, 266, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Castagnini, M.; Del Turco, S.; Epistolato, M.C.; Righini, P.; Sangiorgi, G.M.; De Caterina, R.; Tanganelli, P. High Plasma Levels of the Soluble Receptor for Advanced Glycation Endproducts in Patients with Symptomatic Carotid Atherosclerosis. Eur. J. Clin. Investig. 2009, 39, 1065–1072. [Google Scholar] [CrossRef]
- Tan, A.L.Y.; Sourris, K.C.; Harcourt, B.E.; Thallas-Bonke, V.; Penfold, S.; Andrikopoulos, S.; Thomas, M.C.; O’Brien, R.C.; Bierhaus, A.; Cooper, M.E.; et al. Disparate Effects on Renal and Oxidative Parameters Following RAGE Deletion, AGE Accumulation Inhibition, or Dietary AGE Control in Experimental Diabetic Nephropathy. Am. J. Physiol. Renal Physiol. 2010, 298, F763–F770. [Google Scholar] [CrossRef] [PubMed]
- Machado-Lima, A.; López-Díez, R.; Iborra, R.T.; de Souza Pinto, R.; Daffu, G.; Shen, X.; Nakandakare, E.R.; Machado, U.F.; Corrêa-Giannella, M.L.C.; Schmidt, A.M.; et al. RAGE Mediates Cholesterol Efflux Impairment in Macrophages Caused by Human Advanced Glycated Albumin. Int. J. Mol. Sci. 2020, 21, 7265. [Google Scholar] [CrossRef]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of Accelerated Diabetic Atherosclerosis by the Soluble Receptor for Advanced Glycation Endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef]
- Eckel, R.H.; Bornfeldt, K.E.; Goldberg, I.J. Cardiovascu.ul.l.l.lar Disease in Diabetes, beyond Glucose. Cell Metab. 2021, 33, 1519–1545. [Google Scholar] [CrossRef]
- Miyata, T.; Hori, O.; Zhang, J.; Yan, S.D.; Ferran, L.; Iida, Y.; Schmidt, A.M. The Receptor for Advanced Glycation End Products (RAGE) Is a Central Mediator of the Interaction of AGE-Beta2microglobulin with Human Mononuclear Phagocytes via an Oxidant-Sensitive Pathway. Implications for the Pathogenesis of Dialysis-Related Amyloidosis. J. Clin. Investig. 1996, 98, 1088–1094. [Google Scholar] [CrossRef]
- Inoue, K.; Kawahara, K.; Biswas, K.K.; Ando, K.; Mitsudo, K.; Nobuyoshi, M.; Maruyama, I. HMGB1 Expression by Activated Vascular Smooth Muscle Cells in Advanced Human Atherosclerosis Plaques. Cardiovasc. Pathol. 2007, 16, 136–143. [Google Scholar] [CrossRef]
- Yozgatli, K.; Lefrandt, J.D.; Noordzij, M.J.; Oomen, P.H.N.; Brouwer, T.; Jager, J.; Castro Cabezas, M.; Smit, A.J. Accumulation of Advanced Glycation End Products Is Associated with Macrovascular Events and Glycaemic Control with Microvascular Complications in Type 2 Diabetes Mellitus. Diabet. Med. 2018, 35, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.; Betriu, À.; Yeramian, A.; Fernández, E.; Purroy, F.; Sánchez-de-la-Torre, M.; Pamplona, R.; Miquel, E.; Kerkeni, M.; Hernández, C.; et al. Skin Autofluorescence Measurement in Subclinical Atheromatous Disease: Results from the ILERVAS Project. J. Atheroscler. Thromb. 2019, 26, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Jujić, A.; Östling, G.; Persson, M.; Engström, G.; Nilsson, P.M.; Melander, O.; Magnusson, M. Skin Autofluorescence as a Measure of Advanced Glycation End Product Levels Is Associated with Carotid Atherosclerotic Plaque Burden in an Elderly Population. Diab. Vasc. Dis. Res. 2019, 16, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Sun, W.; Gao, X.; Sell, D.R.; Cleary, P.A.; Lachin, J.M.; Genuth, S.; DCCT/EDIC Research Group. Skin Collagen Advanced Glycation Endproducts (AGEs) and the Long-Term Progression of Sub-Clinical Cardiovascular Disease in Type 1 Diabetes. Cardiovasc. Diabetol. 2015, 14, 118. [Google Scholar] [CrossRef]
- Passarelli, M.; Catanozi, S.; Nakandakare, E.R.; Rocha, J.C.; Morton, R.E.; Shimabukuro, A.F.; Quintão, E.C. Plasma Lipoproteins from Patients with Poorly Controlled Diabetes Mellitus and “in Vitro” Glycation of Lipoproteins Enhance the Transfer Rate of Cholesteryl Ester from HDL to Apo-B-Containing Lipoproteins. Diabetologia 1997, 40, 1085–1093. [Google Scholar] [CrossRef][Green Version]
- Matsuki, K.; Tamasawa, N.; Yamashita, M.; Tanabe, J.; Murakami, H.; Matsui, J.; Imaizumi, T.; Satoh, K.; Suda, T. Metformin Restores Impaired HDL-Mediated Cholesterol Efflux Due to Glycation. Atherosclerosis 2009, 206, 434–438. [Google Scholar] [CrossRef]
- De Souza Pinto, R.; Castilho, G.; Paim, B.A.; Machado-Lima, A.; Inada, N.M.; Nakandakare, E.R.; Vercesi, A.E.; Passarelli, M. Inhibition of Macrophage Oxidative Stress Prevents the Reduction of ABCA-1 Transporter Induced by Advanced Glycated Albumin. Lipids 2012, 47, 443–450. [Google Scholar] [CrossRef]
- Okuda, L.S.; Castilho, G.; Rocco, D.D.F.M.; Nakandakare, E.R.; Catanozi, S.; Passarelli, M. Advanced Glycated Albumin Impairs HDL Anti-Inflammatory Activity and Primes Macrophages for Inflammatory Response That Reduces Reverse Cholesterol Transport. Biochim. Biophys. Acta 2012, 1821, 1485–1492. [Google Scholar] [CrossRef]
- Castilho, G.; Okuda, L.S.; Pinto, R.S.; Iborra, R.T.; Nakandakare, E.R.; Santos, C.X.; Laurindo, F.R.; Passarelli, M. ER Stress Is Associated with Reduced ABCA-1 Protein Levels in Macrophages Treated with Advanced Glycated Albumin—Reversal by a Chemical Chaperone. Int. J. Biochem. Cell Biol. 2012, 44, 1078–1086. [Google Scholar] [CrossRef]
- Iborra, R.T.; Machado-Lima, A.; Castilho, G.; Nunes, V.S.; Abdalla, D.S.P.; Nakandakare, E.R.; Passarelli, M. Advanced Glycation in Macrophages Induces Intracellular Accumulation of 7-Ketocholesterol and Total Sterols by Decreasing the Expression of ABCA-1 and ABCG-1. Lipids Health Dis. 2011, 10, 172. [Google Scholar] [CrossRef]
- Iborra, R.T.; Machado-Lima, A.; Okuda, L.S.; Pinto, P.R.; Nakandakare, E.R.; Machado, U.F.; Correa-Giannella, M.L.; Pickford, R.; Woods, T.; Brimble, M.A.; et al. AGE-Albumin Enhances ABCA1 Degradation by Ubiquitin-Proteasome and Lysosomal Pathways in Macrophages. J. Diabetes Complicat. 2018, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Machado-Lima, A.; Iborra, R.T.; Pinto, R.S.; Sartori, C.H.; Oliveira, E.R.; Nakandakare, E.R.; Stefano, J.T.; Giannella-Neto, D.; Corrêa-Giannella, M.L.C.; Passarelli, M. Advanced Glycated Albumin Isolated from Poorly Controlled Type 1 Diabetes Mellitus Patients Alters Macrophage Gene Expression Impairing ABCA-1-Mediated Reverse Cholesterol Transport. Diabetes Metab. Res. Rev. 2013, 29, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Machado-Lima, A.; Iborra, R.T.; Pinto, R.S.; Castilho, G.; Sartori, C.H.; Oliveira, E.R.; Okuda, L.S.; Nakandakare, E.R.; Giannella-Neto, D.; Machado, U.F.; et al. In Type 2 Diabetes Mellitus Glycated Albumin Alters Macrophage Gene Expression Impairing ABCA1-Mediated Cholesterol Efflux. J. Cell. Physiol. 2015, 230, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Myoishi, M.; Hao, H.; Minamino, T.; Watanabe, K.; Nishihira, K.; Hatakeyama, K.; Asada, Y.; Okada, K.; Ishibashi-Ueda, H.; Gabbiani, G.; et al. Increased Endoplasmic Reticulum Stress in Atherosclerotic Plaques Associated with Acute Coronary Syndrome. Circulation 2007, 116, 1226–1233. [Google Scholar] [CrossRef]
- Wang, Z.; Bao, Z.; Ding, Y.; Xu, S.; Du, R.; Yan, J.; Li, L.; Sun, Z.; Shao, C.; Gu, W. Nε-Carboxymethyl-Lysine-Induced PI3K/Akt Signaling Inhibition Promotes Foam Cell Apoptosis and Atherosclerosis Progression. Biomed. Pharmacother. 2019, 115, 108880. [Google Scholar] [CrossRef]
- Paradela-Dobarro, B.; Bravo, S.B.; Rozados-Luís, A.; González-Peteiro, M.; Varela-Román, A.; González-Juanatey, J.R.; García-Seara, J.; Alvarez, E. Inflammatory Effects of in Vivo Glycated Albumin from Cardiovascular Patients. Biomed. Pharmacother. 2019, 113, 108763. [Google Scholar] [CrossRef]
- Minanni, C.A.; Machado-Lima, A.; Iborra, R.T.; Okuda, L.S.; de Souza Pinto, R.; de Fátima Mello Santana, M.; de Araújo Lira, A.L.; Nakandakare, E.R.; Côrrea-Giannella, M.L.C.; Passarelli, M. Persistent Effect of Advanced Glycated Albumin Driving Inflammation and Disturbances in Cholesterol Efflux in Macrophages. Nutrients 2021, 13, 3633. [Google Scholar] [CrossRef]
- Santana, M.F.M.; Lira, A.L.A.; Pinto, R.S.; Minanni, C.A.; Silva, A.R.M.; Sawada, M.I.B.A.C.; Nakandakare, E.R.; Correa-Giannella, M.L.C.; Queiroz, M.S.; Ronsein, G.E.; et al. Enrichment of Apolipoprotein A-IV and Apolipoprotein D in the HDL Proteome Is Associated with HDL Functions in Diabetic Kidney Disease without Dialysis. Lipids Health Dis. 2020, 19, 205. [Google Scholar] [CrossRef]
- De Araújo Lira, A.L.; de Fátima Mello Santana, M.; de Souza Pinto, R.; Minanni, C.A.; Iborra, R.T.; de Lima, A.M.S.; Correa-Giannella, M.L.; Passarelli, M.; Queiroz, M.S. Serum albumin modified by carbamoylation impairs macrophage cholesterol efflux in diabetic kidney disease. J. Diabetes Complicat. 2021, 35, 107969. [Google Scholar] [CrossRef]
- Xue, J.; Yuan, Z.; Wu, Y.; Liu, Y.; Zhao, Y.; Zhang, W.; Tian, Y.; Liu, W.; Liu, Y.; Kishimoto, C. High Glucose Promotes Intracellular Lipid Accumulation in Vascular Smooth Muscle Cells by Impairing Cholesterol Influx and Efflux Balance. Cardiovasc. Res. 2010, 86, 141–150. [Google Scholar] [CrossRef]
- Daffu, G.; Shen, X.; Senatus, L.; Thiagarajan, D.; Abedini, A.; Hurtado Del Pozo, C.; Rosario, R.; Song, F.; Friedman, R.A.; Ramasamy, R.; et al. RAGE Suppresses ABCG1-Mediated Macrophage Cholesterol Efflux in Diabetes. Diabetes 2015, 64, 4046–4060. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Raghavan, S.; Shanmugam, G.; Shanmugam, N. Ligation of RAGE with Ligand S100B Attenuates ABCA1 Expression in Monocytes. Metabolism 2013, 62, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, Y.-R.; Li, P.-C.; Feng, B. Atorvastatin Blocks Advanced Glycation End Products Induced Reduction in Macrophage Cholesterol Efflux Mediated With ATP-Binding Cassette Transporters G 1. Circ. J. 2019, 83, 1954–1964. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ishida, T.; Yasuda, T.; Kojima, Y.; Honjo, T.; Yamamoto, Y.; Yamamoto, H.; Ishibashi, S.; Hirata, K.; Hayashi, Y. RAGE Mediates Oxidized LDL-Induced pro-Inflammatory Eff.f.fects and Atherosclerosis in Non-Diabetic LDL Receptor-Deficient Mice. Cardiovasc. Res. 2009, 82, 371–381. [Google Scholar] [CrossRef]
- Gomes, D.J.; Velosa, A.P.; Okuda, L.S.; Fusco, F.B.; da Silva, K.S.; Pinto, P.R.; Nakandakare, E.R.; Correa-Giannella, M.L.; Woods, T.; Brimble, M.A.; et al. Glycated Albumin Induces Lipid Infiltration in Mice Aorta Independently of DM and RAS Local Modulation by Inducing Lipid Peroxidation and Inflammation. J. Diabetes Complicat. 2016, 30, 1614–1621. [Google Scholar] [CrossRef]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M.; et al. Advanced Glycation End Products-Induced Insulin Resistance Involves Repression of Skeletal Muscle GLUT4 Expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, K.S.; Pinto, P.R.; Fabre, N.T.; Gomes, D.J.; Thieme, K.; Okuda, L.S.; Iborra, R.T.; Freitas, V.G.; Shimizu, M.H.M.; Teodoro, W.R.; et al. N-Acetylcysteine Counteracts Adipose Tissue Macrophage Infiltration and Insulin Resistance Elicited by Advanced Glycated Albumin in Healthy Rats. Front. Physiol. 2017, 8, 723. [Google Scholar] [CrossRef]
- Uribarri, J.; del Castillo, M.D.; de la Maza, M.P.; Filip, R.; Gugliucci, A.; Luevano-Contreras, C.; Macías-Cervantes, M.H.; Markowicz Bastos, D.H.; Medrano, A.; Menini, T.; et al. Dietary Advanced Glycation End Products and Their Role in Health and Disease. Adv. Nutr. 2015, 6, 461–473. [Google Scholar] [CrossRef]
- Gill, V.; Kumar, V.; Singh, K.; Kumar, A.; Kim, J.-J. Advanced Glycation End Products (AGEs) May Be a Striking Link Between Modern Diet and Health. Biomolecules 2019, 9, 888. [Google Scholar] [CrossRef]
- Kosmopoulos, M.; Drekolias, D.; Zavras, P.D.; Piperi, C.; Papavassiliou, A.G. Impact of Advanced Glycation End Products (AGEs) Signaling in Coronary Artery Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 611–619. [Google Scholar] [CrossRef]
- Corica, D.; Aversa, T.; Ruggeri, R.M.; Cristani, M.; Alibrandi, A.; Pepe, G.; De Luca, F.; Wasniewska, M. Could AGE/RAGE-Related Oxidative Homeostasis Dysregulation Enhance Susceptibility to Pathogenesis of Cardio-Metabolic Complications in Childhood Obesity? Front. Endocrinol. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed]
- Cavero-Redondo, I.; Soriano-Cano, A.; Álvarez-Bueno, C.; Cunha, P.G.; Martínez-Hortelano, J.A.; Garrido-Miguel, M.; Berlanga-Macías, C.; Martínez-Vizcaíno, V. Skin Autofluorescence-Indicated Advanced Glycation End Products as Predictors of Cardiovascular and All-Cause Mortality in High-Risk Subjects: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2018, 7, e009833. [Google Scholar] [CrossRef] [PubMed]
- Da Moura Semedo, C.; Webb, M.; Waller, H.; Khunti, K.; Davies, M. Skin Autofluorescence, a Non-Invasive Marker of Advanced Glycation End Products: Clinical Relevance and Limitations. Postgrad. Med. J. 2017, 93, 289–294. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, R.S.; Minanni, C.A.; de Araújo Lira, A.L.; Passarelli, M. Advanced Glycation End Products: A Sweet Flavor That Embitters Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 2404. https://doi.org/10.3390/ijms23052404
Pinto RS, Minanni CA, de Araújo Lira AL, Passarelli M. Advanced Glycation End Products: A Sweet Flavor That Embitters Cardiovascular Disease. International Journal of Molecular Sciences. 2022; 23(5):2404. https://doi.org/10.3390/ijms23052404
Chicago/Turabian StylePinto, Raphael S., Carlos A. Minanni, Aécio Lopes de Araújo Lira, and Marisa Passarelli. 2022. "Advanced Glycation End Products: A Sweet Flavor That Embitters Cardiovascular Disease" International Journal of Molecular Sciences 23, no. 5: 2404. https://doi.org/10.3390/ijms23052404
APA StylePinto, R. S., Minanni, C. A., de Araújo Lira, A. L., & Passarelli, M. (2022). Advanced Glycation End Products: A Sweet Flavor That Embitters Cardiovascular Disease. International Journal of Molecular Sciences, 23(5), 2404. https://doi.org/10.3390/ijms23052404