
Assays of CFTR Function In Vitro, Ex Vivo and In Vivo

 , and
, and
Abstract
:1. Introduction
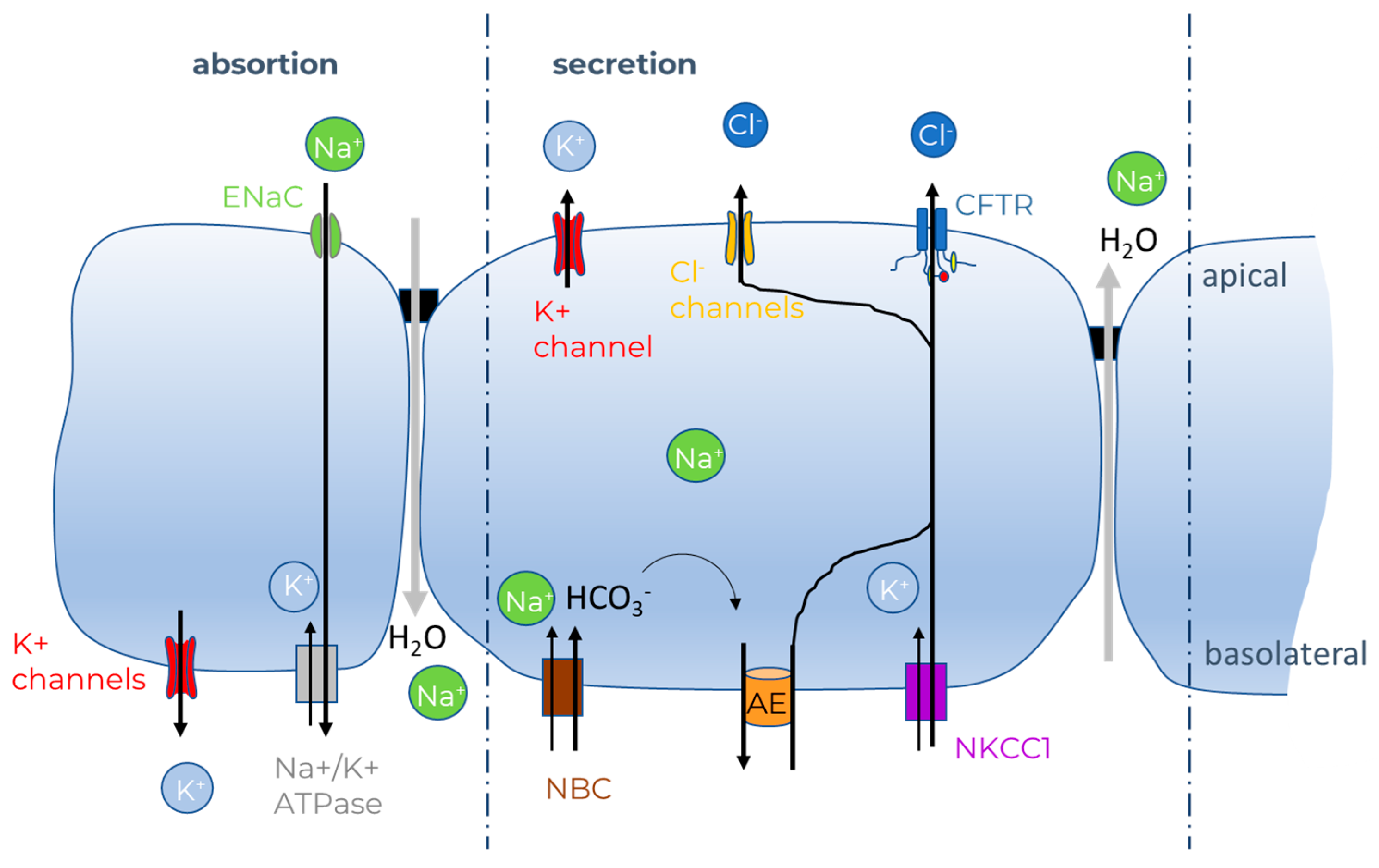
2. CFTR Bioassays
2.1. In Vitro CFTR Bioassays
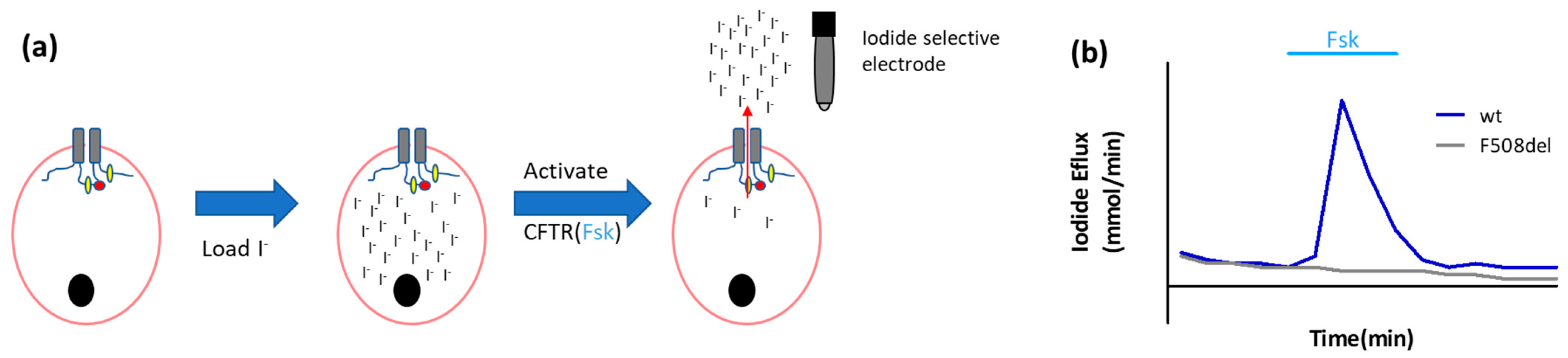
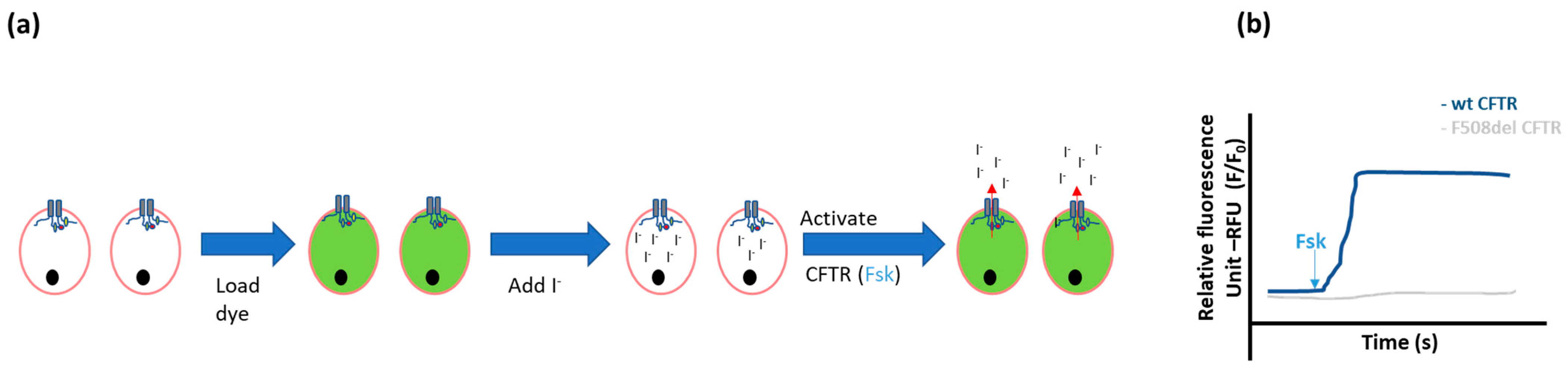
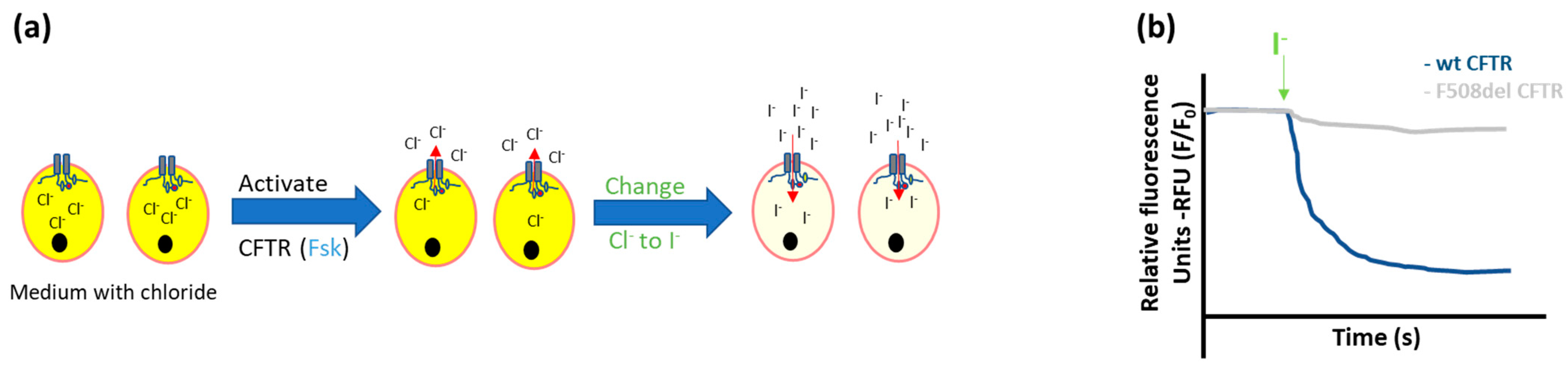
2.1.1. Ion Fluxes
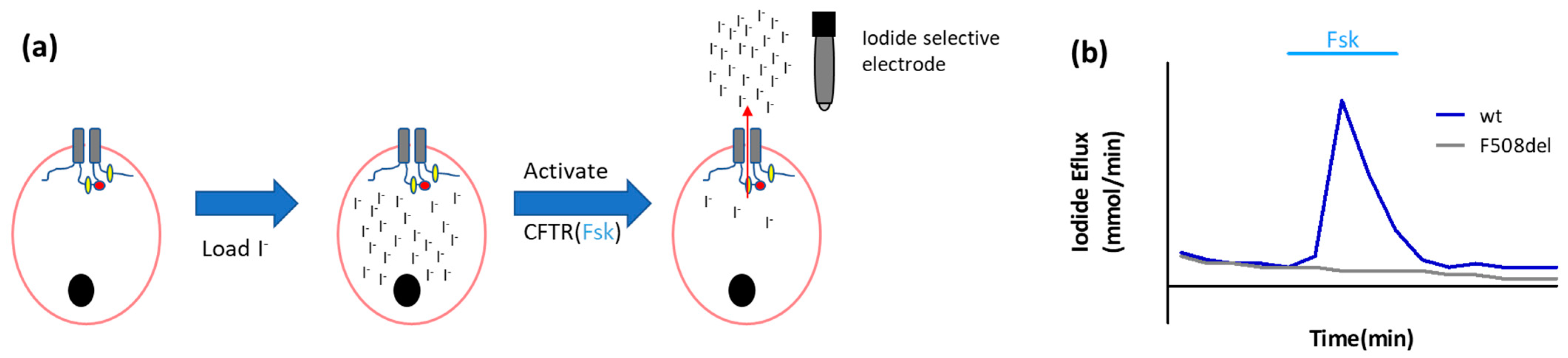
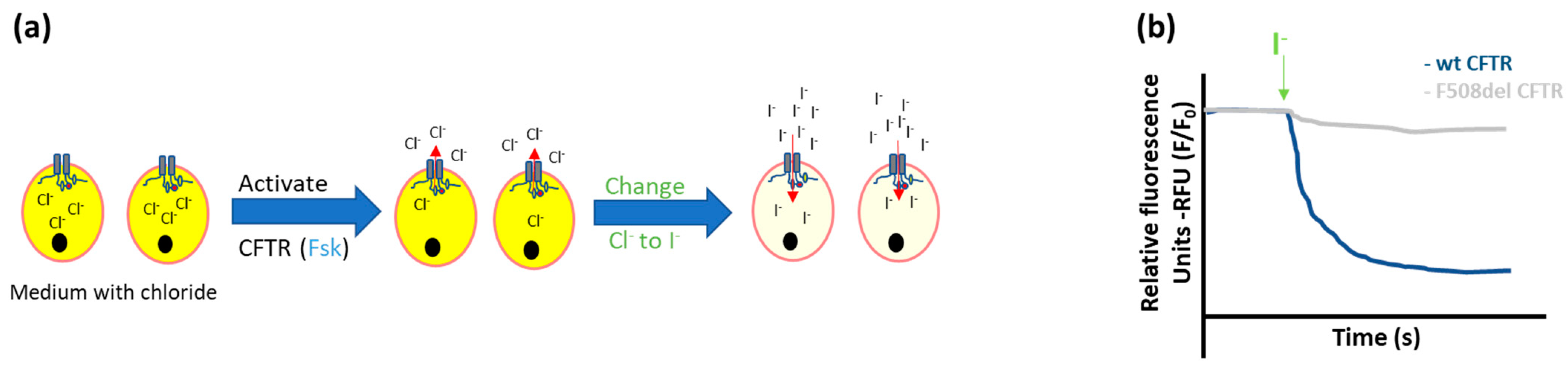
- Iodide efflux
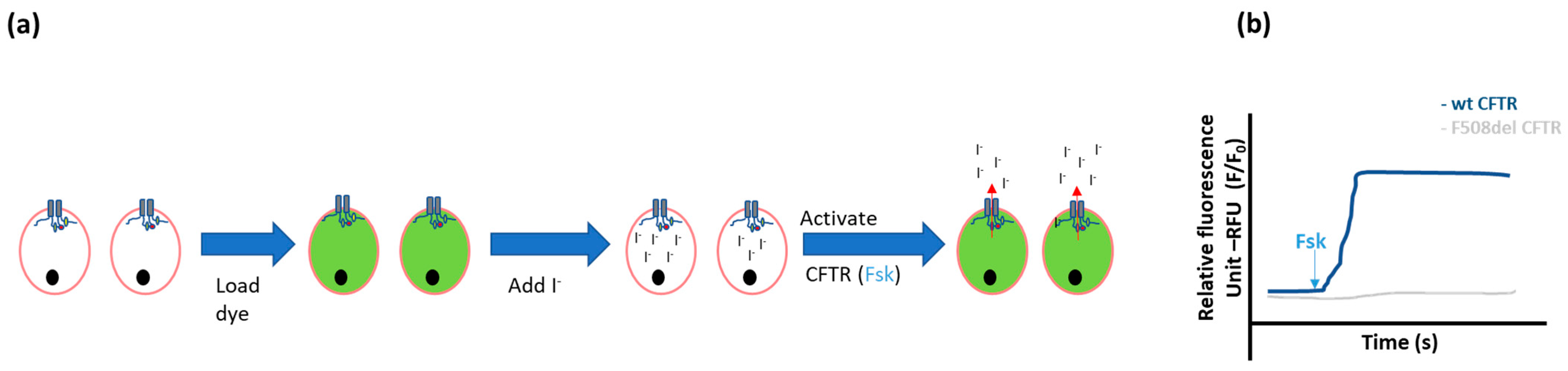
- Halide-sensitive fluorescent probes
- Halide-sensitive (HS) yellow fluorescent protein (YFP) quenching
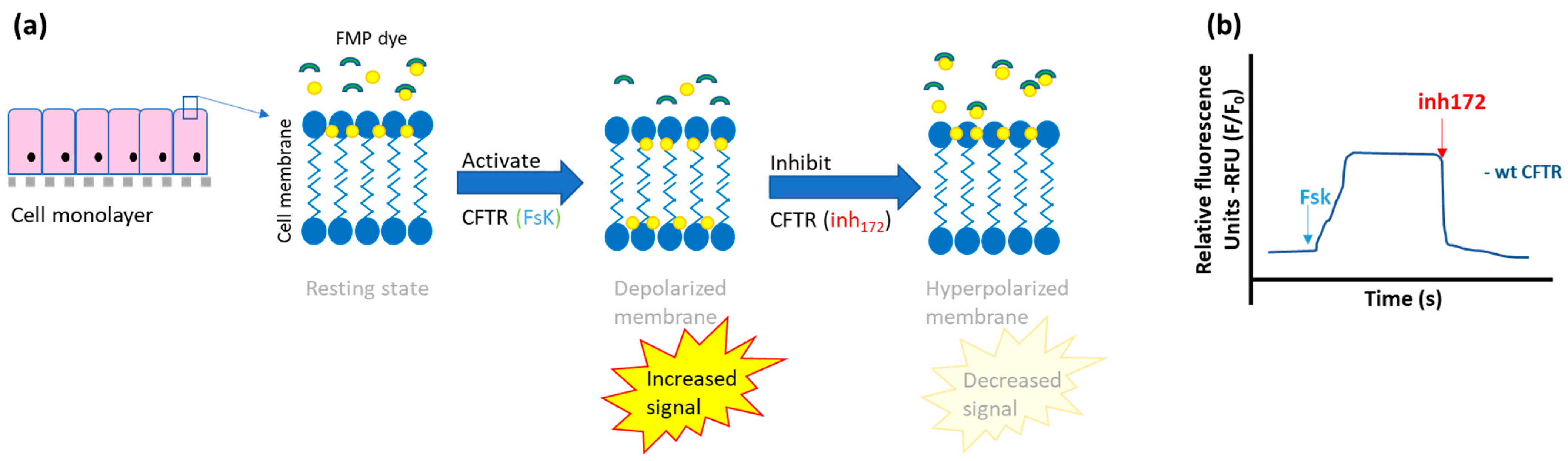
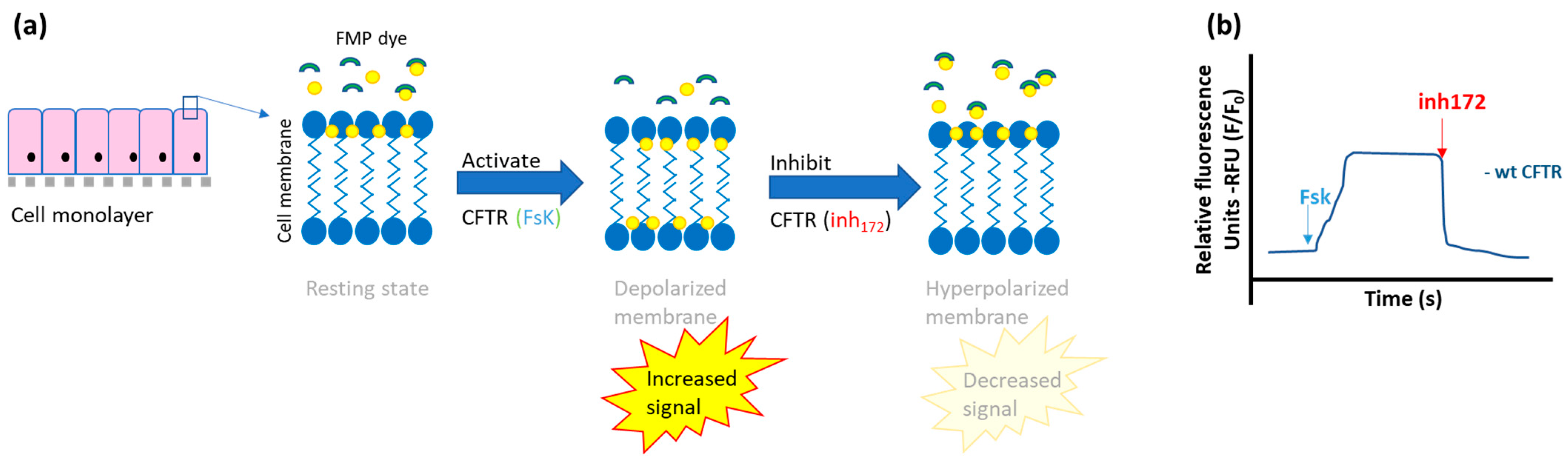
- Fluorescent-based membrane potential assay
2.1.2. Electrophysiological Methods
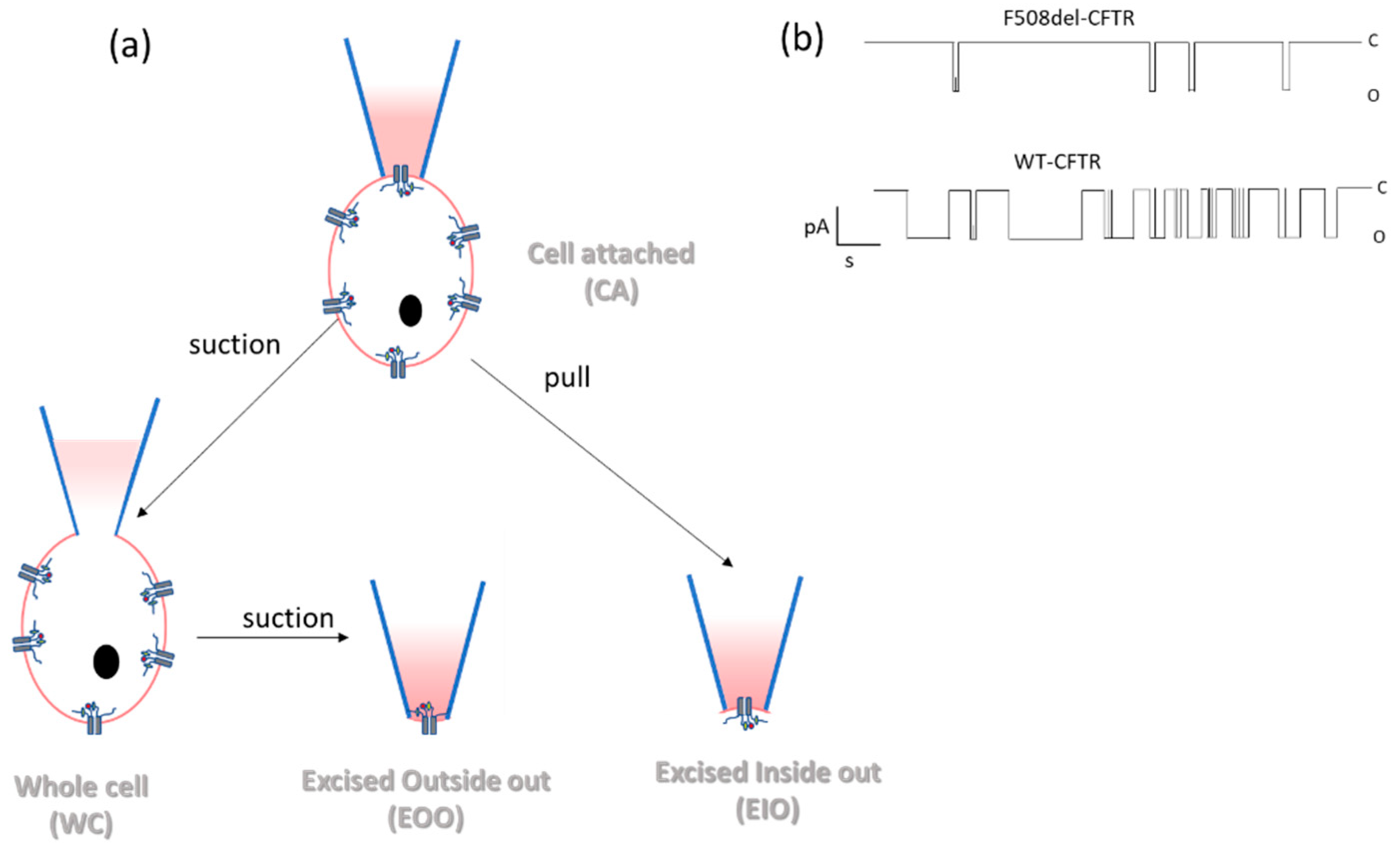
- Patch clamp technique
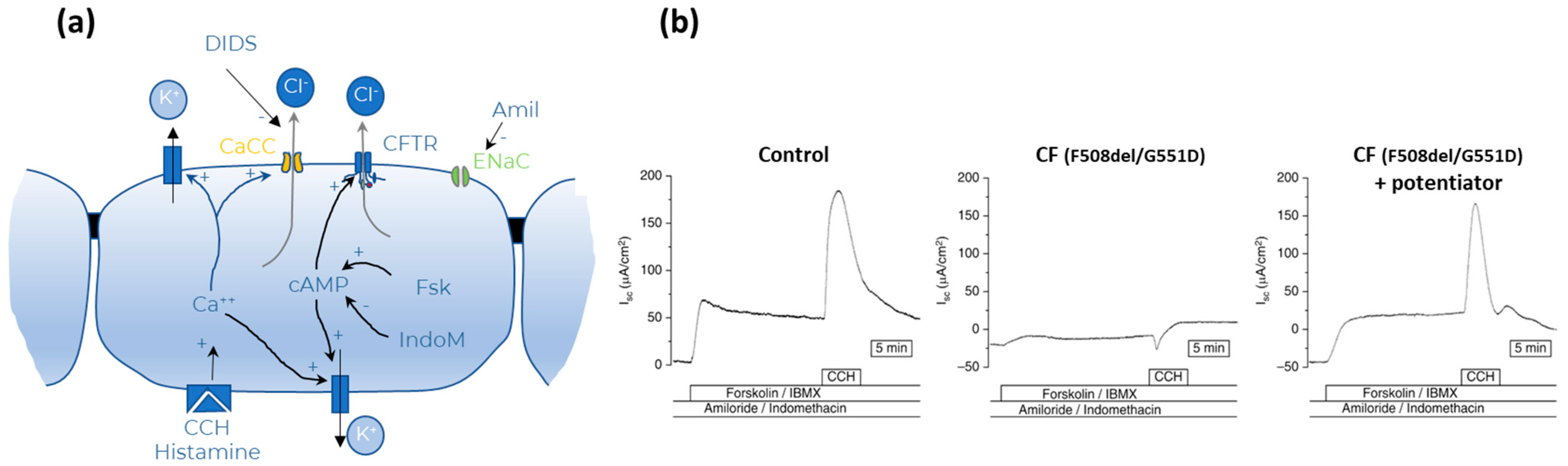
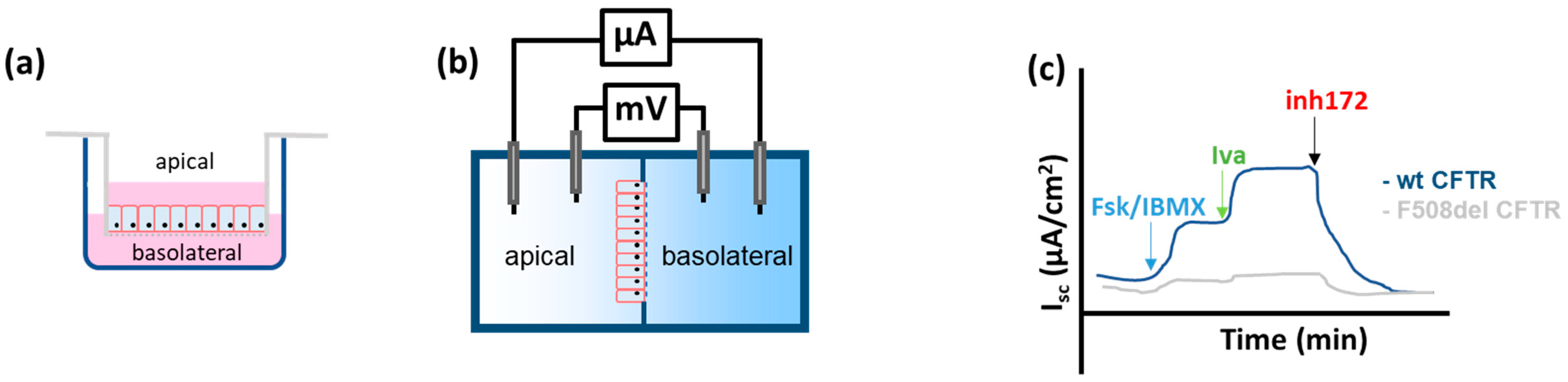
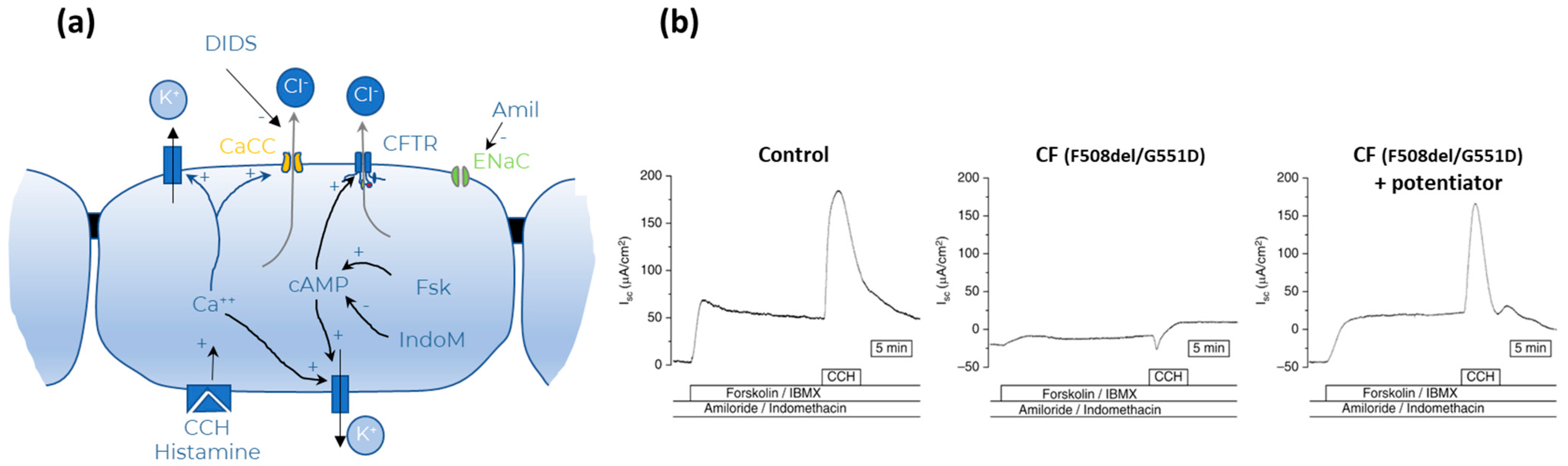
- Transepithelial short circuit current (Isc) measurements—Ussing Chamber
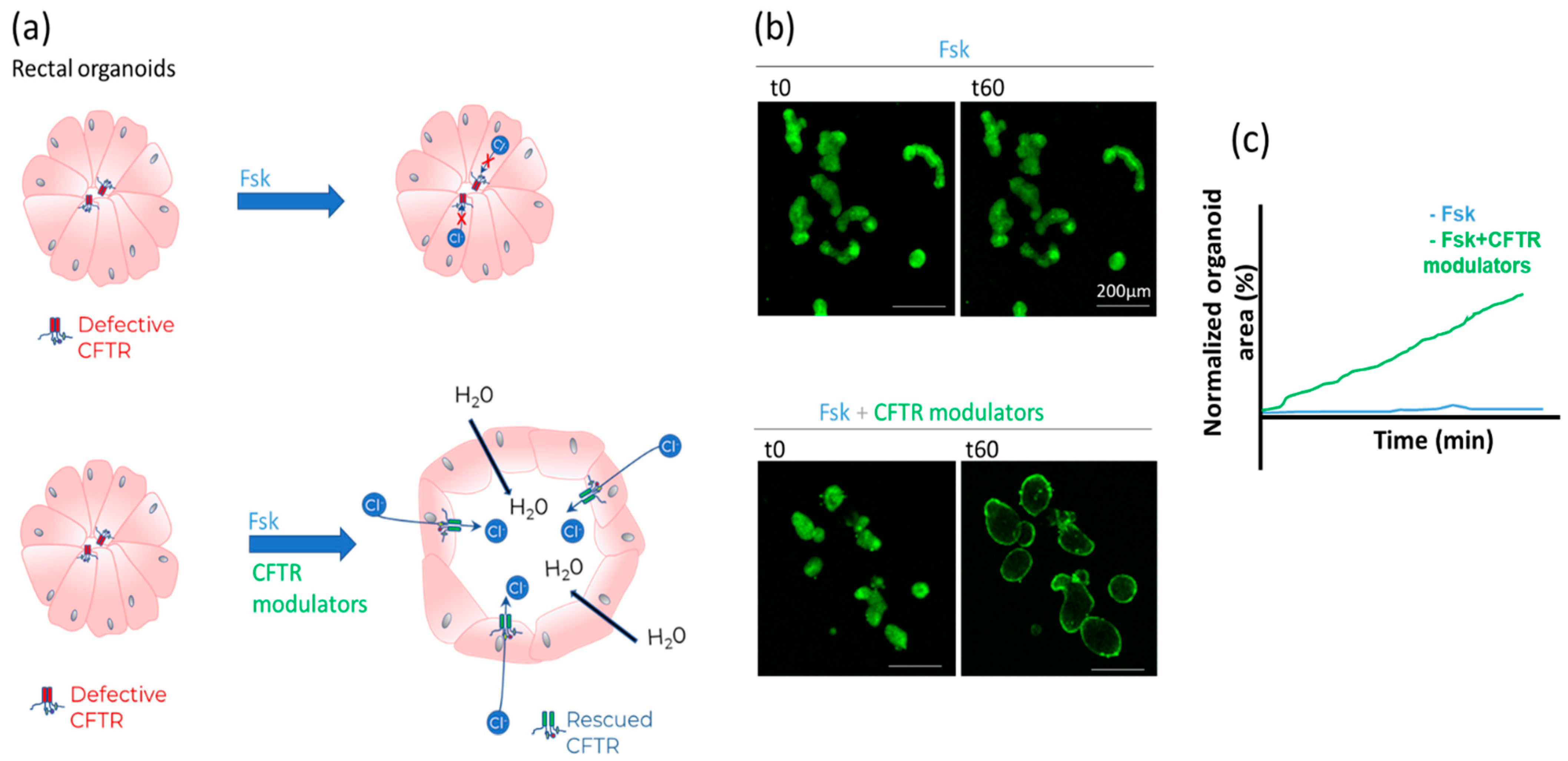
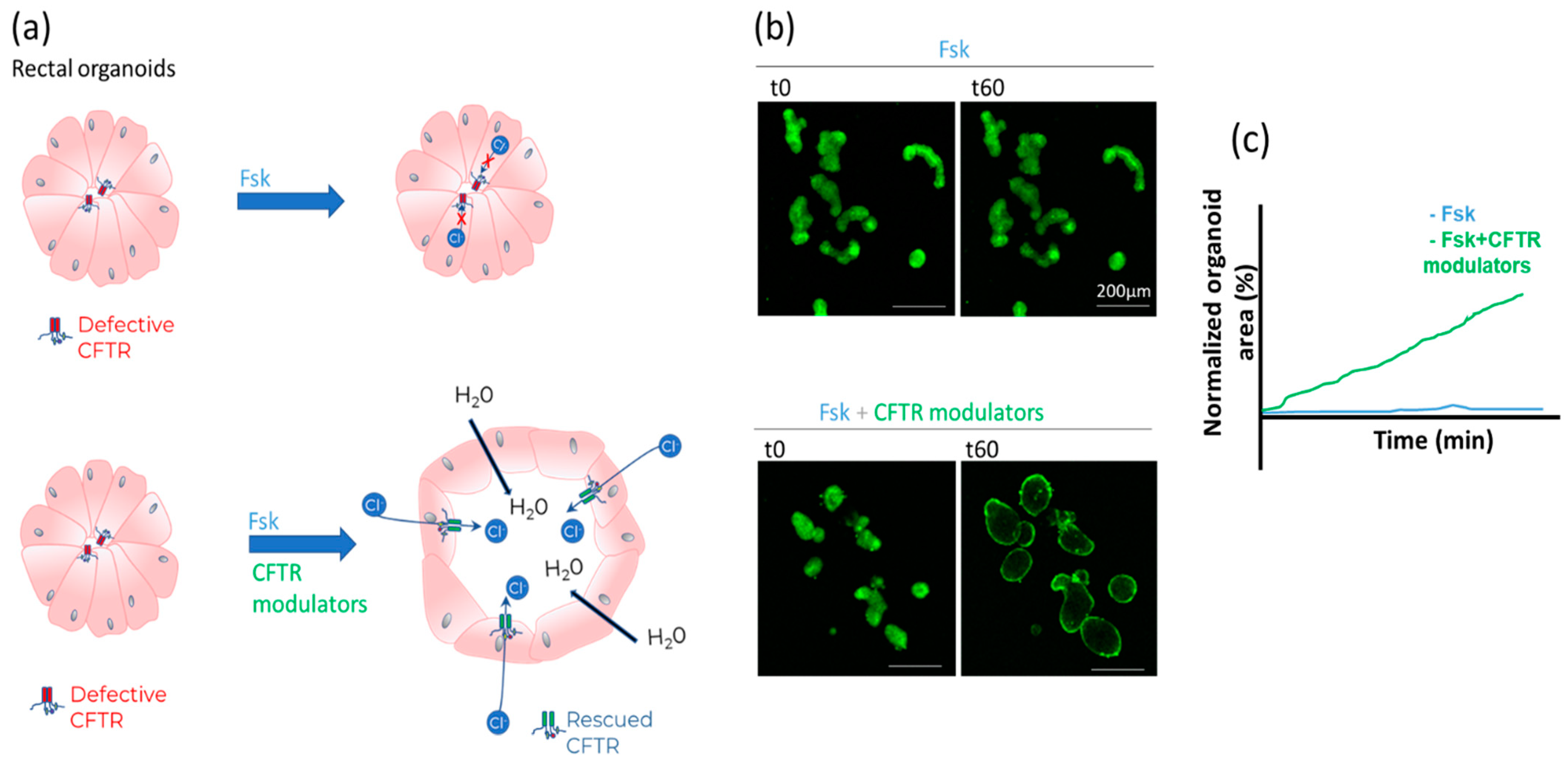
- Forskolin-induced swelling (FIS) assay in organoids
2.2. Ex Vivo CFTR Bioassays
Intestinal Current Measurements (ICM)
2.3. In Vivo CFTR Bioassays
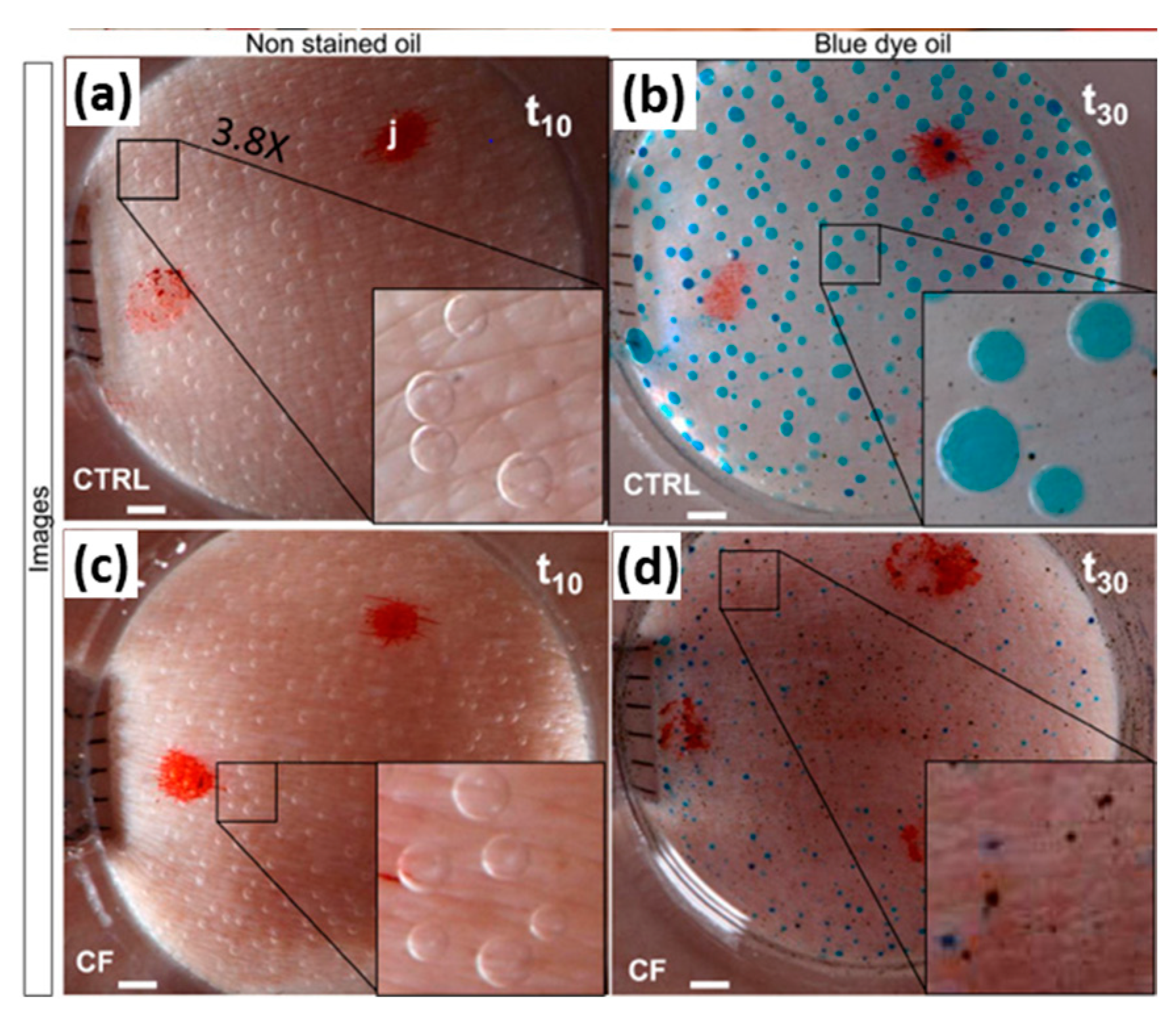
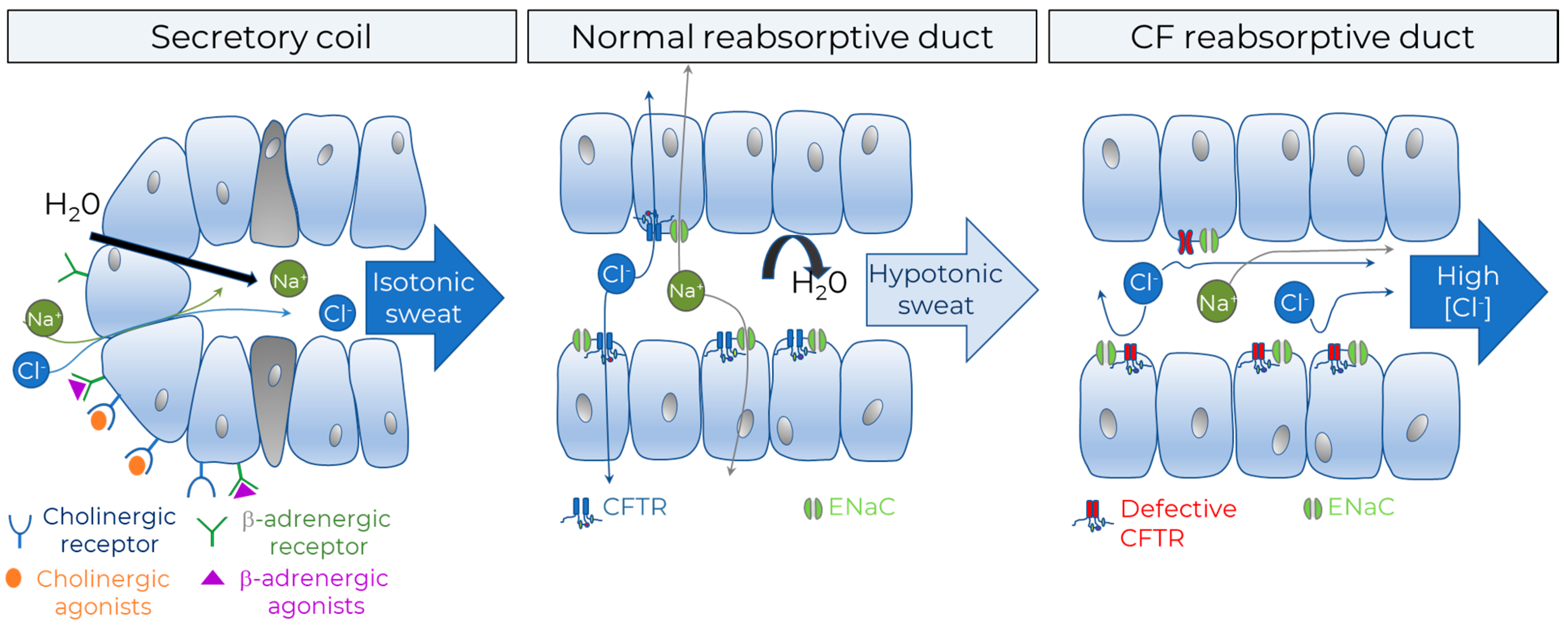
2.3.1. Sweat Chloride Concentration (SCC)
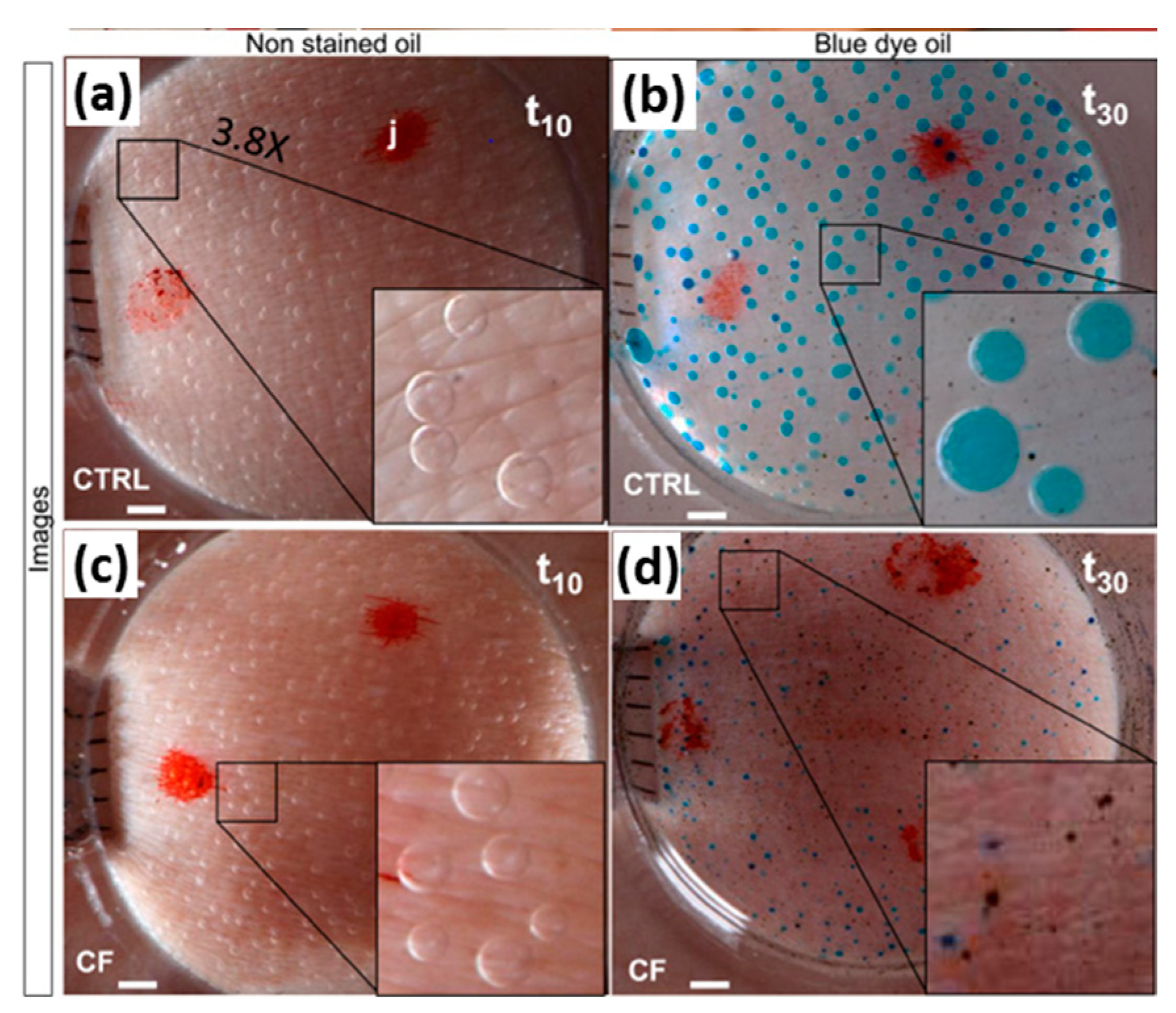
2.3.2. β-Adrenergic Sweat Test
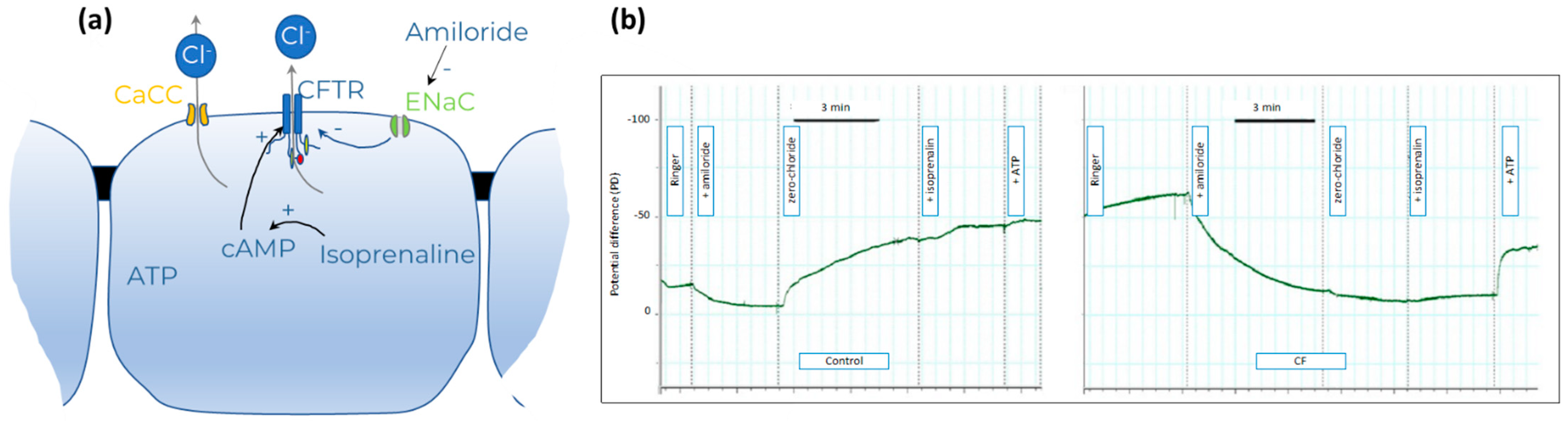
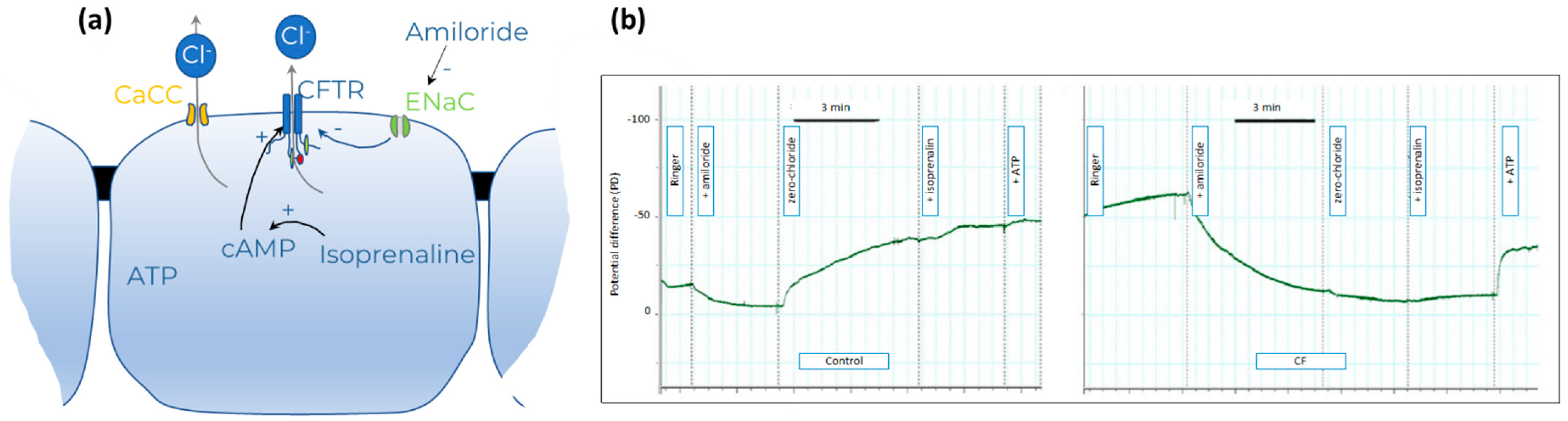
2.3.3. Nasal Potential Difference (NPD)
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Who Is TRIKAFTA® For?|TRIKAFTA® (Elexacaftor/Tezacaftor/Ivacaftor and Ivacaftor) n.d. Available online: https://www.trikafta.com/who-trikafta-is-for (accessed on 6 January 2022).
- Maule, G.; Ensinck, M.; Bulcaen, M.; Carlon, M.S. Rewriting CFTR to cure cystic fibrosis. Prog. Mol. Biol. Transl. Sci. 2021, 182, 185–224. [Google Scholar] [CrossRef] [PubMed]
- Ensinck, M.; Mottais, A.; Detry, C.; Leal, T.; Carlon, M.S. On the Corner of Models and Cure: Gene Editing in Cystic Fibrosis. Front. Pharmacol. 2021, 12, 662110. [Google Scholar] [CrossRef]
- Cuyx, S.; De Boeck, K. Treating the Underlying Cystic Fibrosis Transmembrane Conductance Regulator Defect in Patients with Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 762–774. [Google Scholar] [CrossRef]
- Cai, Z.; Sohma, Y.; Bompadre, S.G.; Sheppard, D.N.; Hwang, T.-C. Application of High-Resolution Single-Channel Recording to Functional Studies of Cystic Fibrosis Mutants. Methods Mol. Biol. 2011, 741, 419–441. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, K.; Amaral, M. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [Green Version]
- McShane, A.J.; Bajrami, B.; Ramos, A.A.; Diego-Limpin, P.A.; Farrokhi, V.; Coutermarsh, B.A.; Stanton, B.A.; Jensen, T.; Riordan, J.R.; Wetmore, D.; et al. Targeted Proteomic Quantitation of the Absolute Expression and Turnover of Cystic Fibrosis Transmembrane Conductance Regulator in the Apical Plasma Membrane. J. Proteome Res. 2014, 13, 4676–4685. [Google Scholar] [CrossRef] [Green Version]
- Prins, S.; Langron, E.; Hastings, C.; Hill, E.J.; Stefan, A.C.; Griffin, L.D.; Vergani, P. Fluorescence assay for simultaneous quantification of CFTR ion-channel function and plasma membrane proximity. J. Biol. Chem. 2020, 295, 16529–16544. [Google Scholar] [CrossRef] [PubMed]
- Awatade, N.T.; Wong, S.L.; Hewson, C.K.; Fawcett, L.; Kicic, A.; Jaffe, A.; Waters, S.A. Human Primary Epithelial Cell Models: Promising Tools in the Era of Cystic Fibrosis Personalized Medicine. Front. Pharmacol. 2018, 9, 1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarron, A.; Parsons, D.; Donnelley, M. Animal and Cell Culture Models for Cystic Fibrosis: Which Model Is Right for Your Application? Am. J. Pathol. 2021, 191, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Cholon, D.M.; Gentzsch, M. Recent progress in translational cystic fibrosis research using precision medicine strategies. J. Cyst. Fibros. 2018, 17, S52–S60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laselva, O.; Conese, M. Three-Dimensional Airway Spheroids and Organoids for Cystic Fibrosis Research. J. Respir. 2021, 1, 229–247. [Google Scholar] [CrossRef]
- Keegan, D.; Brewington, J. Nasal Epithelial Cell-Based Models for Individualized Study in Cystic Fibrosis. Int. J. Mol. Sci. 2021, 22, 4448. [Google Scholar] [CrossRef]
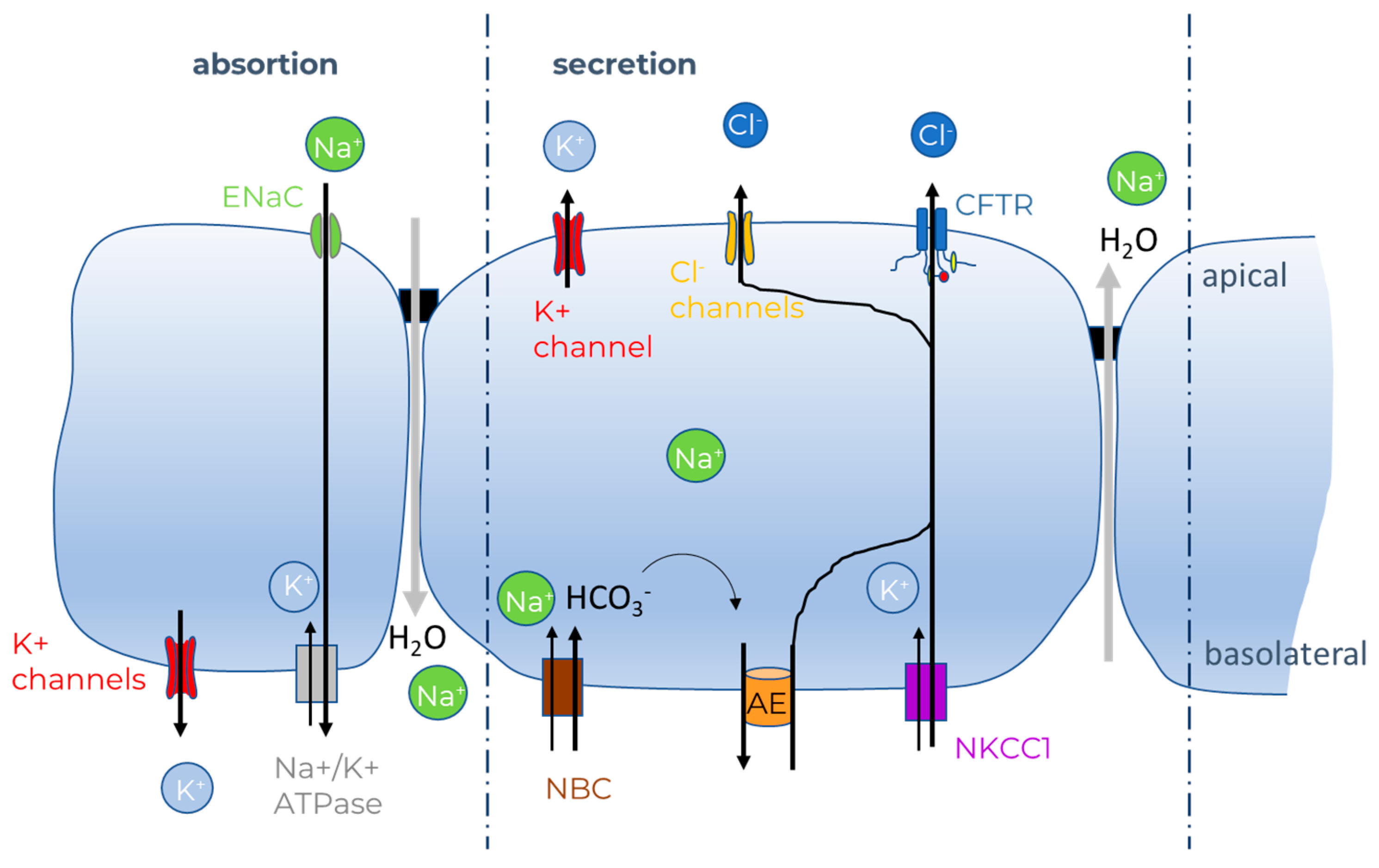
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. [Google Scholar] [CrossRef]
- Moran, O.; Zegarra-Moran, O. On the measurement of the functional properties of the CFTR. J. Cyst. Fibros. 2008, 7, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Norez, C.; Heda, G.D.; Jensen, T.; Kogan, I.; Hughes, L.K.; Auzanneau, C.; Dérand, R.; Bulteau-Pignoux, L.; Li, C.; Ramjeesingh, M.; et al. Determination of CFTR chloride channel activity and pharmacology using radiotracer flux methods. J. Cyst. Fibros. 2004, 3, 119–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaandrager, A.B.; Bajnath, R.; Groot, J.A.; Bot, A.G.; De Jonge, H.R. Ca2+ and cAMP activate different chloride efflux pathways in HT-29.cl19A colonic epithelial cell line. Am. J. Physiol. Liver Physiol. 1991, 261, G958–G965. [Google Scholar] [CrossRef] [PubMed]
- Long, K.J.; Walsh, K.B. Iodide efflux measurements with an iodide-selective electrode: A non-radioactive procedure for monitoring cellular chloride transport. J. Tissue Cult. Methods 1997, 19, 207–212. [Google Scholar] [CrossRef]
- Ramalho, A.S.; Clarke, L.A.; Sousa, M.; Felicio, V.; Barreto, C.; Lopes, C.; Amaral, M.D. Comparative ex vivo, in vitro and in silico analyses of a CFTR splicing mutation: Importance of functional studies to establish disease liability of mutations. J. Cyst. Fibros. 2016, 15, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Vidović, D.; Carlon, M.; Da Cunha, M.F.; Dekkers, J.F.; Hollenhorst, M.I.; Bijvelds, M.J.C.; Ramalho, A.S.; Haute, C.V.D.; Ferrante, M.; Baekelandt, V.; et al. rAAV-CFTRΔR Rescues the Cystic Fibrosis Phenotype in Human Intestinal Organoids and Cystic Fibrosis Mice. Am. J. Respir. Crit. Care Med. 2016, 193, 288–298. [Google Scholar] [CrossRef]
- Mansoura, M.K.; Biwersi, J.; Ashlock, M.A.; Verkman, A. Fluorescent Chloride Indicators to Assess the Efficacy of CFTR cDNA Delivery. Hum. Gene Ther. 1999, 10, 861–875. [Google Scholar] [CrossRef]
- Robinson, E.; MacDonald, K.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.; Galietta, L.J. In Vitro/Ex Vivo Fluorescence Assays of CFTR Chloride Channel Function. In Cystic Fibrosis in the 21st Century; Karger Publishers: Basel, Switzerland, 2006; Volume 34, pp. 93–101. [Google Scholar] [CrossRef]
- Munkonge, F.; Alton, E.W.; Andersson, C.; Davidson, H.; Dragomir, A.; Edelman, A.; Farley, R.; Hjelte, L.; McLachlan, G.; Stern, M.; et al. Measurement of halide efflux from cultured and primary airway epithelial cells using fluorescence indicators. J. Cyst. Fibros. 2004, 3, 171–176. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; De Gregorio, F.; Salvadori, L.; Grassia, R.; Leone, C.; De Rosa, G.; et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014, 10, 2053–2074. [Google Scholar] [CrossRef] [Green Version]
- Abbattiscianni, A.C.; Favia, M.; Mancini, M.T.; Cardone, R.A.; Guerra, L.; Monterisi, S.; Castellani, S.; Laselva, O.; di Sole, F.; Conese, M.; et al. Correctors of mutant CFTR enhance subcortical cAMP-PKA signaling through modulating ezrin phosphorylation and cytoskeleton organization. J. Cell Sci. 2016, 129, 1128–1140. [Google Scholar] [CrossRef]
- Tosco, A.; De Gregorio, F.; Esposito, S.; De Stefano, D.; Sana, I.; Ferrari, E.; Sepe, A.; Salvadori, L.; Buonpensiero, P.; di Pasqua, A.; et al. A novel treatment of cystic fibrosis acting on-target: Cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016, 23, 1380–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galietta, L.; Jayaraman, S.; Verkman, A.S. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am. J. Physiol. Physiol. 2001, 281, C1734–C1742. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.-J.; Lee, S.; Verkman, A.S. Hollow Micropillar Array Method for High-Capacity Drug Screening on Filter-Grown Epithelial Cells. Anal. Chem. 2018, 90, 7675–7681. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Vetrivel, L.; Yang, H.; Pedemonte, N.; Zegarra-Moran, O.; Galietta, L.J.; Verkman, A.S. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J. Biol. Chem. 2002, 277, 37235–37241. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.; Giuliano, K.A.; Shumate, J.; Baillargeon, P.; McEwan, B.; Cullen, M.D.; Miller, J.P.; Drew, L.; Scampavia, L.; Spicer, T.P. A Homogeneous Cell-Based Halide-Sensitive Yellow Fluorescence Protein Assay to Identify Modulators of the Cystic Fibrosis Transmembrane Conductance Regulator Ion Channel. ASSAY Drug Dev. Technol. 2017, 15, 395–406. [Google Scholar] [CrossRef]
- Van Der Plas, S.E.; Kelgtermans, H.; De Munck, T.; Martina, S.L.X.; Dropsit, S.; Quinton, E.; De Blieck, A.; Joannesse, C.; Tomaskovic, L.; Jans, M.; et al. Discovery of N-(3-Carbamoyl-5,5,7,7-tetramethyl-5,7-dihydro-4H-thieno[2,3-c]pyran-2-yl)-lH-pyrazole-5-carboxamide (GLPG1837), a Novel Potentiator Which Can Open Class III Mutant Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Channels to a High. J. Med. Chem. 2018, 61, 1425–1435. [Google Scholar] [CrossRef] [Green Version]
- Ensinck, M.; De Keersmaecker, L.; Heylen, L.; Ramalho, A.S.; Gijsbers, R.; Farré, R.; De Boeck, K.; Christ, F.; Debyser, Z.; Carlon, M.S. Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells 2020, 9, 754. [Google Scholar] [CrossRef] [Green Version]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Vertex Announces FDA Approvals of TRIKAFTA® (Elexacaftor/Tezacaftor/Ivacaftor and Ivacaftor), SYMDEKO® (Tezacaftor/Ivacaftor and Ivacaftor) and KALYDECO® (Ivacaftor) for Use in People with CF with Certain Rare Mutations|Vertex Pharmaceuticals n.d. Available online: https://investors.vrtx.com/news-releases/news-release-details/vertex-announces-fda-approvals-trikaftar (accessed on 16 December 2021).
- Molinski, S.V.; Gonska, T.; Huan, L.J.; Baskin, B.; Janahi, I.A.; Ray, P.N.; Bear, C.E. Genetic, cell biological, and clinical interrogation of the CFTR mutation c.3700 A>G (p.Ile1234Val) informs strategies for future medical intervention. Genet. Med. 2014, 16, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Maitra, R.; Sivashanmugam, P.; Warner, K. A Rapid Membrane Potential Assay to Monitor CFTR Function and Inhibition. J. Biomol. Screen. 2013, 18, 1132–1137. [Google Scholar] [CrossRef] [Green Version]
- Molinski, S.; Ahmadi, S.; Hung, M.; Bear, C.E. Facilitating Structure-Function Studies of CFTR Modulator Sites with Efficiencies in Mutagenesis and Functional Screening. J. Biomol. Screen. 2015, 20, 1204–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goor, F.; Straley, K.S.; Cao, D.; González, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.J.; et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Bozoky, Z.; Di Paola, M.; Xia, S.; Li, C.; Wong, A.; Wellhauser, L.; Molinski, S.; Ip, W.; Ouyang, H.; et al. Phenotypic profiling of CFTR modulators in patient-derived respiratory epithelia. NPJ Genom. Med. 2017, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.X.; Wellhauser, L.; Laselva, O.; Utkina, I.; Bozoky, Z.; Gunawardena, T.; Ngan, Z.; Xia, S.; Di Paola, M.; Eckford, P.D.; et al. A new platform for high-throughput therapy testing on iPSC-derived lung progenitor cells from cystic fibrosis patients. Stem. Cell Rep. 2021, 16, 2825–2837. [Google Scholar] [CrossRef]
- Ogawa, M.; Jiang, J.-X.; Xia, S.; Yang, D.; Ding, A.; Laselva, O.; Hernandez, M.; Cui, C.; Higuchi, Y.; Suemizu, H.; et al. Generation of functional ciliated cholangiocytes from human pluripotent stem cells. Nat. Commun. 2021, 12, 1–19. [Google Scholar] [CrossRef]
- Sondo, E.; Tomati, V.; Caci, E.; Esposito, A.I.; Pfeffer, U.; Pedemonte, N.; Galietta, L.J.V. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol. Physiol. 2011, 301, C872–C885. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, D.N.; Gray, M.; Gong, X.; Sohma, Y.; Kogan, I.; Benos, D.J.; Scott-Ward, T.S.; Chen, J.-H.; Li, H.; Cai, Z.; et al. The patch-clamp and planar lipid bilayer techniques: Powerful and versatile tools to investigate the CFTR Cl− channel. J. Cyst. Fibros. 2004, 3, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Cottrill, K.A.; McCarty, N.A. Electrophysiological Approaches for the Study of Ion Channel Function. In Structure and Function of Membrane Proteins; Humana Press Inc.: New York, NY, USA, 2021; Volume 2302, pp. 49–67. [Google Scholar] [CrossRef]
- Noel, S.; Servel, N.; Hatton, A.; Golec, A.; Rodrat, M.; Ng, D.R.S.; Li, H.; Pranke, I.; Hinzpeter, A.; Edelman, A.; et al. Correlating genotype with phenotype using CFTR-mediated whole-cell Cl—Currents in human nasal epithelial cells. J. Physiol. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Hamilton, K.L. Ussing’s “Little Chamber”: 60 Years+ Old and Counting. Front. Physiol. 2011, 2, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sheppard, D.N.; Hug, M.J. Transepithelial electrical measurements with the Ussing chamber. J. Cyst. Fibros. 2004, 3, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Silva, I.; Railean, V.; Duarte, A.; Amaral, M. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732. [Google Scholar] [CrossRef]
- Nick, H.J.; Zeitlin, P.L.; Yadav, S.; Bratcher, P.E. Measurements of spontaneous CFTR-mediated ion transport without acute channel activation in airway epithelial cultures after modulator exposure. Sci. Rep. 2021, 11, 22616. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Bel, N.S.-L.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef]
- Zomer-van Ommen, D.D.; de Poel, E.; Kruisselbrink, E.; Oppelaar, H.; Vonk, A.M.; Janssens, H.M.; van der Ent, C.K.; Hagemeijer, M.C.; Beekman, J.M. Comparison of ex vivo and in vitro intestinal cystic fibrosis models to measure CFTR-dependent ion channel activity. J. Cyst. Fibros. 2018, 17, 316–324. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.; Ouyang, H.; Eckford, P.D.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor + tezacaftor + ivacaftor mediated in part by the dual activities of elexacaftor as both corrector and potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef]
- Laselva, O.; McCormack, J.; Bartlett, C.; Ip, W.; Gunawardena, T.; Ouyang, H.; Eckford, P.; Gonska, T.; Moraes, T.; Bear, C. Preclinical Studies of a Rare CF-Causing Mutation in the Second Nucleotide Binding Domain (c.3700A>G) Show Robust Functional Rescue in Primary Nasal Cultures by Novel CFTR Modulators. J. Pers. Med. 2020, 10, 209. [Google Scholar] [CrossRef]
- La Selva, O.; Moraes, T.J.; He, G.; Bartlett, C.; Szàrics, I.; Ouyang, H.; Gunawardena, T.N.A.; Strug, L.; Bear, C.E.; Gonska, T. The CFTR Mutation c.3453G > C (D1152H) Confers an Anion Selectivity Defect in Primary Airway Tissue that Can be Rescued by Ivacaftor. J. Pers. Med. 2020, 10, 40. [Google Scholar] [CrossRef]
- Phuan, P.-W.; Haggie, P.M.; Tan, J.A.; Rivera, A.A.; Finkbeiner, W.E.; Nielson, D.W.; Thomas, M.M.; Janahi, I.A.; Verkman, A.S. CFTR modulator therapy for cystic fibrosis caused by the rare c.3700A>G mutation. J. Cyst. Fibros. 2021, 20, 452–459. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Amatngalim, G.; Rodenburg, L.; Aalbers, B.; Brunsveld, J.; Kruisselbrink, E.; Heida-Michel, S.; Groot, K.D.; Van Der Ent, C.; Beekman, J. Comparison of fluid secretion in nasal and bronchial spheroids from CF patients. Airw. Cell Biol. Immunopathol. 2019, 54, OA1901. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, L.; Favia, M.; Di Gioia, S.; Laselva, O.; Bisogno, A.; Casavola, V.; Colombo, C.; Conese, M. The preclinical discovery and development of the combination of ivacaftor + tezacaftor used to treat cystic fibrosis. Expert Opin. Drug Discov. 2020, 15, 873–891. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.D.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Vonk, A.M.; van Mourik, P.; Ramalho, A.S.; Silva, I.; Statia, M.; Kruisselbrink, E.; Suen, S.W.; Dekkers, J.F.; Vleggaar, F.P.; Houwen, R.H.; et al. Protocol for Application, Standardization and Validation of the Forskolin-Induced Swelling Assay in Cystic Fibrosis Human Colon Organoids. STAR Protoc. 2020, 1, 100019. [Google Scholar] [CrossRef]
- Cuyx, S.; Ramalho, A.S.; Corthout, N.; Fieuws, S.; Fürstová, E.; Arnauts, K.; Ferrante, M.; Verfaillie, C.; Munck, S.; Boon, M.; et al. Rectal organoid morphology analysis (ROMA) as a promising diagnostic tool in cystic fibrosis. Thorax 2021, 76, 1146–1149. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; de Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; van de Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- Ramalho, A.S.; Furstova, E.; Vonk, A.M.; Ferrante, M.; Verfaillie, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekman, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426. [Google Scholar] [CrossRef]
- Crawford, D.K.; Mullenders, J.; Pott, J.; Boj, S.F.; Landskroner-Eiger, S.; Goddeeris, M.M. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 2021, 20, 436–442. [Google Scholar] [CrossRef]
- De Winter Groot, K.M.; Janssens, H.M.; Van Uum, R.T.; Dekkers, J.F.; Berkers, G.; Vonk, A.; Kruisselbrink, E.; Oppelaar, H.; Vries, R.; Clevers, H.; et al. Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur. Respir. J. 2018, 52, 1702529. [Google Scholar] [CrossRef] [PubMed]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; Groot, K.M.D.W.-D.; Arets, H.G.; der Wilt, R.E.M.-V.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, M.D.; de Boeck, K.; Davies, J.C.; Drevinek, P.; Elborn, S.; Kerem, E.; Lee, T. Theranostics by testing CFTR modulators in patient-derived materials: The current status and a proposal for subjects with rare CFTR mutations. J. Cyst. Fibros. 2019, 18, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Ommen, D.D.Z.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Amatngalim, G.D.; Rodenburg, L.W.; Aalbers, B.L.; Raeven, H.H.M.; Aarts, E.M.; Silva, I.A.L.; Nijenhuis, W.; Vrendenbarg, S.; Kruisselbrink, E.; Brunsveld, J.E.; et al. Measuring cystic fibrosis drug responses in organoids derived from 2D differentiated nasal epithelia. BioRxiv 2021. [Google Scholar] [CrossRef]
- De Jonge, H.R.; Ballmann, M.; Veeze, H.; Bronsveld, I.; Stanke, F.; Tümmler, B.; Sinaasappel, M. Ex vivo CF diagnosis by intestinal current measurements (ICM) in small aperture, circulating Ussing chambers. J. Cyst. Fibros. 2004, 3, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Mall, M.; Hirtz, S.; Gonska, T.; Kunzelmann, K. Assessment of CFTR function in rectal biopsies for the diagnosis of cystic fibrosis. J. Cyst. Fibros. 2004, 3, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Graeber, S.Y.; Hug, M.J.; Sommerburg, O.; Hirtz, S.; Hentschel, J.; Heinzmann, A.; Dopfer, C.; Schulz, A.; Mainz, J.G.; Tümmler, B.; et al. Intestinal Current Measurements Detect Activation of Mutant CFTR in Patients with Cystic Fibrosis with the G551D Mutation Treated with Ivacaftor. Am. J. Respir. Crit. Care Med. 2015, 192, 1252–1255. [Google Scholar] [CrossRef]
- Derichs, N.; Sanz, J.; Von Kanel, T.; Stolpe, C.; Zapf, A.; Tümmler, B.; Gallati, S.; Ballmann, M. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: Validation and reference data. Thorax 2010, 65, 594–599. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1. [Google Scholar] [CrossRef] [Green Version]
- Roth, E.K.; Hirtz, S.; Duerr, J.; Wenning, D.; Eichler, I.; Seydewitz, H.H.; Amaral, M.D.; Mall, M.A. The K+ Channel Opener 1-EBIO Potentiates Residual Function of Mutant CFTR in Rectal Biopsies from Cystic Fibrosis Patients. PLoS ONE 2011, 6, e24445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, J.P.; Szczesniak, R.D.; Ashlock, M.A.; Ernst, S.E.; Fan, L.; Hornick, D.; Karp, P.H.; Khan, U.; Lymp, J.; Ostmann, A.J.; et al. Multicenter Intestinal Current Measurements in Rectal Biopsies from CF and Non-CF Subjects to Monitor CFTR Function. PLoS ONE 2013, 8, e73905. [Google Scholar] [CrossRef] [PubMed]
- McCague, A.F.; Raraigh, K.S.; Pellicore, M.J.; Davis-Marcisak, E.F.; Evans, T.A.; Han, S.; Lu, Z.; Joynt, A.T.; Sharma, N.; Castellani, C.; et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- LeGrys, V.A.; Yankaskas, J.R.; Quittell, L.M.; Marshall, B.C.; Mogayzel, P.J., Jr. Diagnostic Sweat Testing: The Cystic Fibrosis Foundation Guidelines. J. Pediatr. 2007, 151, 85–89. [Google Scholar] [CrossRef]
- Quinton, P.M. Both Ways at Once: Keeping Small Airways Clean. Physiology 2017, 32, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accurso, F.J.; Van Goor, F.; Zha, J.; Stone, A.J.; Dong, Q.; Ordonez, C.L.; Rowe, S.M.; Clancy, J.P.; Konstan, M.W.; Hoch, H.E.; et al. Sweat chloride as a biomarker of CFTR activity: Proof of concept and ivacaftor clinical trial data. J. Cyst. Fibros. 2014, 13, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; Van Der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Sagel, S.D.; Khan, U.; Heltshe, S.L.; Clancy, J.P.; Borowitz, D.; Gelfond, D.; Donaldson, S.H.; Moran, A.; Ratjen, F.; VanDalfsen, J.M.; et al. Clinical Effectiveness of Lumacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for F508del-CFTR. A Clinical Trial. Ann. Am. Thorac. Soc. 2021, 18, 75–83. [Google Scholar] [CrossRef]
- Durmowicz, A.G.; Witzmann, K.A.; Rosebraugh, C.J.; Chowdhury, B.A. Change in Sweat Chloride as a Clinical End Point in Cystic Fibrosis Clinical Trials: The ivacaftor experience. Chest 2013, 143, 14–18. [Google Scholar] [CrossRef]
- Fidler, M.C.; Beusmans, J.; Panorchan, P.; Van Goor, F. Correlation of sweat chloride and percent predicted FEV1 in cystic fibrosis patients treated with ivacaftor. J. Cyst. Fibros. 2017, 16, 41–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, K. Trying to find a cure for cystic fibrosis: CFTR biomarkers as outcomes. Eur. Respir. J. 2013, 42, 1156–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarry, M.E.; Illek, B.; Ly, N.P.; Zlock, L.; Bs, S.O.; Bs, C.M.; Finkbeiner, W.E.; Nielson, D.W. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr. Pulmonol. 2017, 52, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, K.; Yang, F.; Brewington, J.J.; McPhail, G.; Cortez, A.R.; Sundaram, N.; Ramananda, Y.; Ogden, H.; Helmrath, M.A.; Clancy, J.P.; et al. Patient personalized translational tools in cystic fibrosis to transform data from bench to bed-side and back. Am. J. Physiol. Liver Physiol. 2021, 320, G1123–G1130. [Google Scholar] [CrossRef]
- Sato, K.; Sato, F. Defective beta adrenergic response of cystic fibrosis sweat glands in vivo and in vitro. J. Clin. Investig. 1984, 73, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Quinton, P.; Molyneux, L.; Ip, W.; Dupuis, A.; Avolio, J.; Tullis, E.; Conrad, D.; Shamsuddin, A.K.; Durie, P.; Gonska, T. β-Adrenergic Sweat Secretion as a Diagnostic Test for Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 732–739. [Google Scholar] [CrossRef]
- Wine, J.J.; Char, J.E.; Chen, J.; Cho, H.-J.; Dunn, C.; Frisbee, E.; Joo, N.S.; Milla, C.; Modlin, S.E.; Park, I.-H.; et al. In Vivo Readout of CFTR Function: Ratiometric Measurement of CFTR-Dependent Secretion by Individual, Identifiable Human Sweat Glands. PLoS ONE 2013, 8, e77114. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Farahmand, M.; Dunn, C.; Davies, Z.; Frisbee, E.; Milla, C.; Wine, J.J. Evaporimeter and Bubble-Imaging Measures of Sweat Gland Secretion Rates. PLoS ONE 2016, 11, e0165254. [Google Scholar] [CrossRef] [Green Version]
- Salinas, D.B.; Peng, Y.-H.; Horwich, B.; Wee, C.P.; Frisbee, E.; Maarek, J.-M. Image-based β-adrenergic sweat rate assay captures minimal cystic fibrosis transmembrane conductance regulator function. Pediatr. Res. 2019, 87, 137–145. [Google Scholar] [CrossRef]
- Char, J.E.; Wolfe, M.H.; Cho, H.-J.; Park, I.-H.; Jeong, J.H.; Frisbee, E.; Dunn, C.; Davies, Z.; Milla, C.; Moss, R.B.; et al. A Little CFTR Goes a Long Way: CFTR-Dependent Sweat Secretion from G551D and R117H-5T Cystic Fibrosis Subjects Taking Ivacaftor. PLoS ONE 2014, 9, e88564. [Google Scholar] [CrossRef] [Green Version]
- Reynaerts, A.; Vermeulen, F.; Mottais, A.; Gohy, S.; Lebecque, P.; Frédérick, R.; Vanbever, R.; Leal, T. Needle-free iontophoresis-driven β-adrenergic sweat rate test. J. Cyst. Fibros. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Salinas, D.B.; Kang, L.; Azen, C.; Quinton, P. Low Beta-Adrenergic Sweat Responses in Cystic Fibrosis and Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome Children. Pediatr. Allergy Immunol. Pulmonol. 2017, 30, 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergamini, G.; Tridello, G.; Calcaterra, E.; Ceri, S.; Tagliasacchi, M.; Bianchi, F.; Monti, F.; Masciadri, A.; Laudanna, E.; Peserico, D.; et al. Ratiometric sweat secretion optical test in cystic fibrosis, carriers and healthy subjects. J. Cyst. Fibros. 2018, 17, 186–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, M.R.; Paradiso, A.M.; Boucher, R.C. In Vivo Nasal Potential Difference: Techniques and Protocols for Assessing Efficacy of Gene Transfer in Cystic Fibrosis. Hum. Gene Ther. 1995, 6, 445–455. [Google Scholar] [CrossRef]
- Schüler, D.; Sermet-Gaudelus, I.; Wilschanski, M.; Ballmann, M.; Dechaux, M.; Edelman, A.; Hug, M.; Leal, T.; Lebacq, J.; Lebecque, P.; et al. Basic protocol for transepithelial nasal potential difference measurements. J. Cyst. Fibros. 2004, 3, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Standaert, T.A.; Boitano, L.; Emerson, J.; Milgram, L.J.; Konstan, M.W.; Hunter, J.; Berclaz, P.-Y.; Brass, L.; Zeitlin, P.; Hammond, K.; et al. Standardized procedure for measurement of nasal potential difference: An outcome measure in multicenter cystic fibrosis clinical trials. Pediatr. Pulmonol. 2004, 37, 385–392. [Google Scholar] [CrossRef]
- Vermeulen, F.; Proesmans, M.; Boon, M.; De Boeck, K. Improved repeatability of nasal potential difference with a larger surface catheter. J. Cyst. Fibros. 2015, 14, 317–323. [Google Scholar] [CrossRef] [Green Version]
- De Wachter, E.; De Schutter, I.; Meulemans, A.; Buyl, R.; Malfroot, A.; Information, P.E.K.F.C. A semi-blinded study comparing 2 methods of measuring nasal potential difference: Subcutaneous needle versus dermal abrasion. J. Cyst. Fibros. 2015, 15, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Rowe, S.M.; Clancy, J.P.; Wilschanski, M. Nasal Potential Difference Measurements to Assess CFTR Ion Channel Activity. Methods Mol. Biol. 2011, 741, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Solomon, G.M.; Bronsveld, I.; Hayes, K.; Wilschanski, M.; Melotti, P.; Rowe, S.M.; Sermet-Gaudelus, I. Standardized Measurement of Nasal Membrane Transepithelial Potential Difference (NPD). J. Vis. Exp. 2018, 2018, e57006. [Google Scholar] [CrossRef] [Green Version]
- Wilschanski, M.; Famini, H.; Strauss-Liviatan, N.; Rivlin, J.; Blau, H.; Bibi, H.; Bentur, L.; Yahav, Y.; Springer, H.; Kramer, M.; et al. Nasal potential difference measurements in patients with atypical cystic fibrosis. Eur. Respir. J. 2001, 17, 1208–1215. [Google Scholar] [CrossRef] [Green Version]
- Sermet-Gaudelus, I.; Girodon, E.; Roussel, D.; Deneuville, E.; Bui, S.; Huet, F.; Guillot, M.; Aboutaam, R.; Renouil, M.; Munck, A.; et al. Measurement of nasal potential difference in young children with an equivocal sweat test following newborn screening for cystic fibrosis. Thorax 2010, 65, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, F. Analysis of CFTR function by NPD. 2014; unpublished data. [Google Scholar]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in Persons with Cystic Fibrosis and the G551D- CFTR Mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graeber, S.; Dopfer, C.; Naehrlich, L.; Gyulumyan, L.; Scheuermann, H.; Hirtz, S.; Wege, S.; Mairbäurl, H.; Dorda, M.; Hyde, R.; et al. Effects of Lumacaftor–Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I.; Clancy, J.P.; Nichols, D.P.; Nick, J.A.; De Boeck, K.; Solomon, G.M.; Mall, M.A.; Bolognese, J.; Bouisset, F.; Hollander, W.D.; et al. Antisense oligonucleotide eluforsen improves CFTR function in F508del cystic fibrosis. J. Cyst. Fibros. 2019, 18, 536–542. [Google Scholar] [CrossRef] [Green Version]
- Tiddens, H.A.; Puderbach, M.; Venegas, J.G.; Ratjen, F.; Donaldson, S.H.; Davis, S.D.; Rowe, S.M.; Sagel, S.D.; Higgins, M.; Waltz, D.A. Novel outcome measures for clinical trials in cystic fibrosis. Pediatr. Pulmonol. 2014, 50, 302–315. [Google Scholar] [CrossRef]
- Kyrilli, S.; Henry, T.; Wilschanski, M.; Fajac, I.; Davies, J.C.; Jais, J.-P.; Sermet-Gaudelus, I. Insights into the variability of nasal potential difference, a biomarker of CFTR activity. J. Cyst. Fibros. 2020, 19, 620–626. [Google Scholar] [CrossRef]
- Vermeulen, F.; Proesmans, M.; Feyaerts, N.; De Boeck, K. Nasal potential measurements on the nasal floor and under the inferior turbinate: Does it matter? Pediatr. Pulmonol. 2010, 46, 145–152. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Key References | |
|---|---|---|
| In vitro bio-assays | Iodide efflux | Norez et al., 2004 [22], Long, K.J.; Walsh, K.B. 1997 [24] |
| Halide-sensitive Fluorescent probes | Munkonge, F. et al., 2004 [30] | |
| Halide-sensitive yellow fluorescent protein (YFP) | Galietta, L.V.J. et al., 2001 [34] | |
| Fluorescent-base membrane potential probe | Maitra, R. et al., 2013 [43] | |
| Patch clamp CFTR functional analysis | Cai, Z. et al., 2011 [9] Sheppard, D.N. et al., 2004 [50] | |
| Transepithelial short circuit current (Isc) measurements | Li, H. et al., 2004 [54] | |
| FIS assay on rectal organoids | Dekkers, J.F. et al., 2013 [69] Vonk, A.M. et al., 2020 [70] | |
| Ex vivo bio-assays | Intestinal current measurements (ICM) | Mall, M. et al., 2004 [81] De Jonge, H.R. et al., 2004 [80] Derichs, N. et al., 2010 [83] |
| In vivo bio-assays | Sweat chloride concentration | LeGrys, V.A. et al., 2007 [88] |
| β-adrenergic sweat assays | Quinton, P. et al., 2012 [100] Wine, J.J. et al., 2013 [101] | |
| Nasal potential difference (NPD) | Knowles, M.R. et al., 1995 [108] Solomon, G.M. et al., 2018 [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramalho, A.S.; Boon, M.; Proesmans, M.; Vermeulen, F.; Carlon, M.S.; Boeck, K.D. Assays of CFTR Function In Vitro, Ex Vivo and In Vivo. Int. J. Mol. Sci. 2022, 23, 1437. https://doi.org/10.3390/ijms23031437
Ramalho AS, Boon M, Proesmans M, Vermeulen F, Carlon MS, Boeck KD. Assays of CFTR Function In Vitro, Ex Vivo and In Vivo. International Journal of Molecular Sciences. 2022; 23(3):1437. https://doi.org/10.3390/ijms23031437
Chicago/Turabian StyleRamalho, Anabela Santo, Mieke Boon, Marijke Proesmans, François Vermeulen, Marianne S. Carlon, and Kris De Boeck. 2022. "Assays of CFTR Function In Vitro, Ex Vivo and In Vivo" International Journal of Molecular Sciences 23, no. 3: 1437. https://doi.org/10.3390/ijms23031437
APA StyleRamalho, A. S., Boon, M., Proesmans, M., Vermeulen, F., Carlon, M. S., & Boeck, K. D. (2022). Assays of CFTR Function In Vitro, Ex Vivo and In Vivo. International Journal of Molecular Sciences, 23(3), 1437. https://doi.org/10.3390/ijms23031437