
Targeting the WNK-SPAK/OSR1 Pathway and Cation-Chloride Cotransporters for the Therapy of Stroke


Abstract
1. Introduction of Cation-Chloride Cotransporter Family

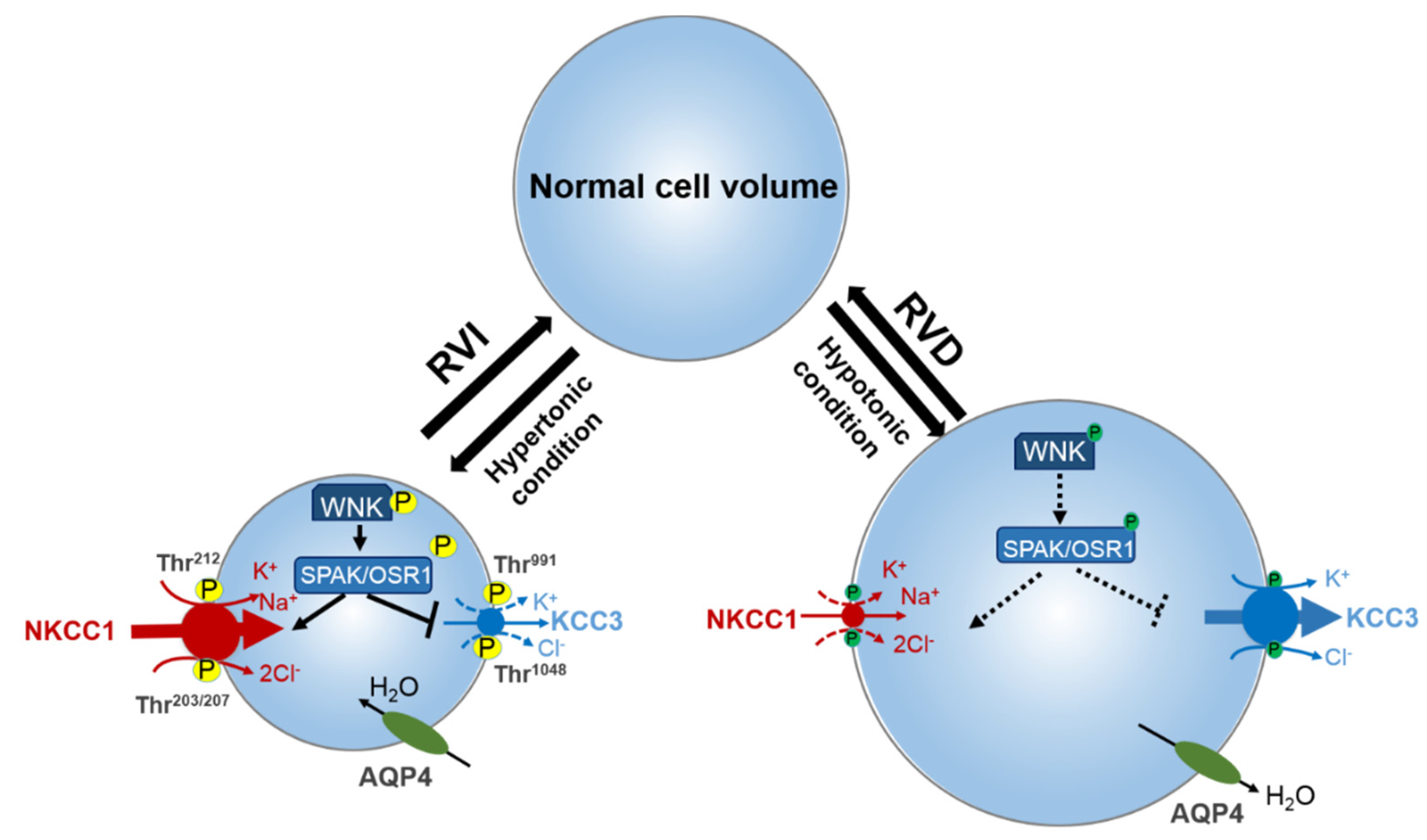{kind=link}
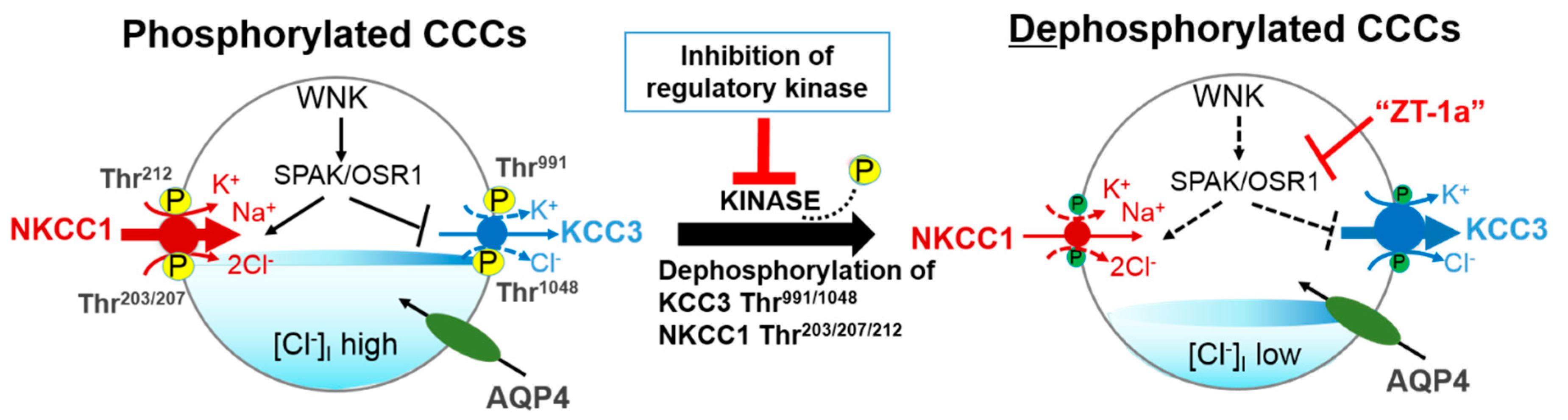{kind=link}
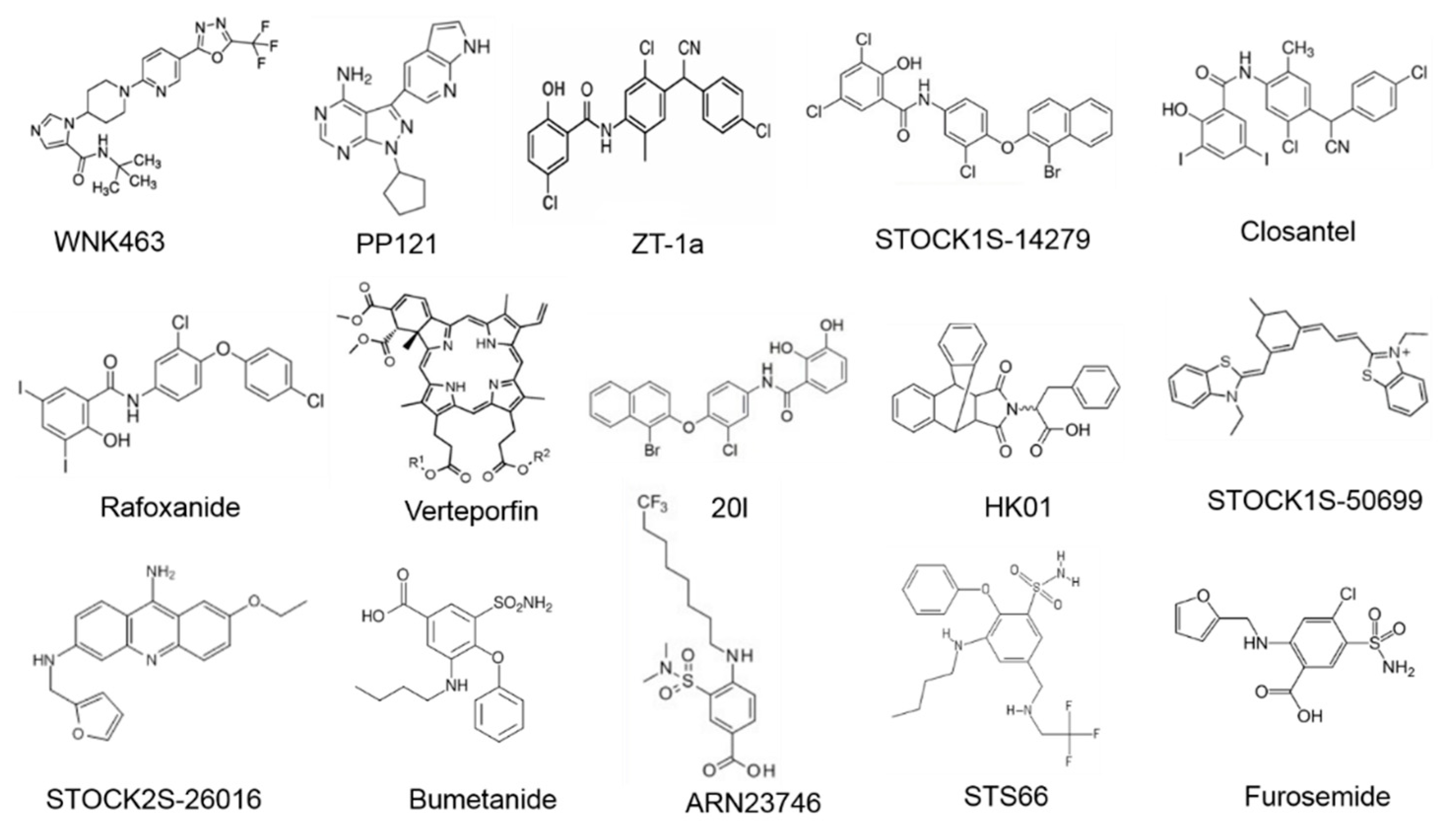{kind=link}
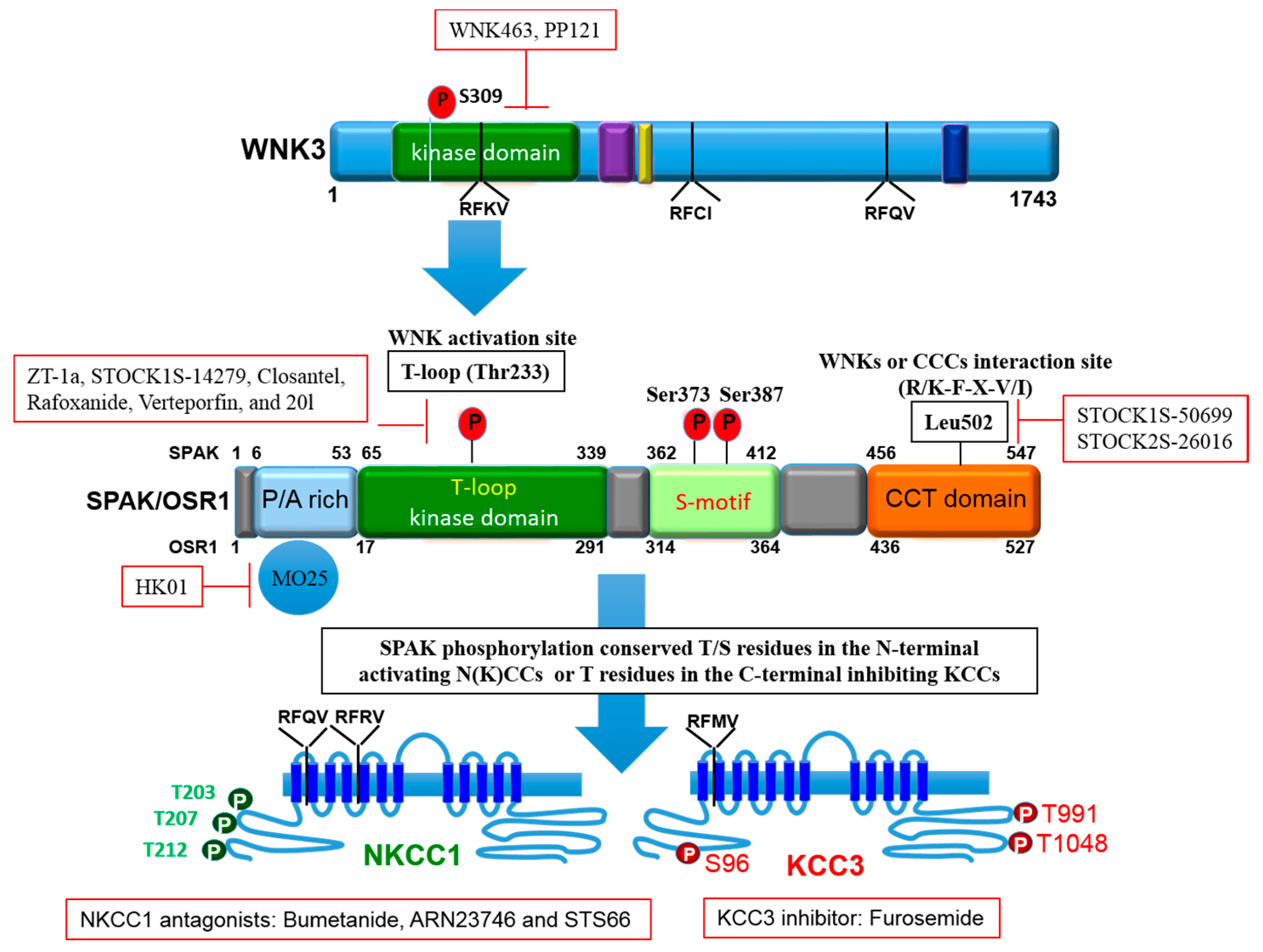{kind=link}
| Encoding Gene (Protein) | Co-Transport Ions | Tissue Distribution | Physiological Functions | Genetic Disorders | References |
|---|---|---|---|---|---|
| SLC12A1 (NKCC2) | Na+, K+, Cl− | Kidney-specific (TAL) | NaCl reabsorption in the TAL; regulation of Ca2+ excretion; urine concentration | Bartter’s syndrome | [2,35,36,37] |
| SLC12A2 (NKCC1) | Na+, K+, Cl− | Ubiquitous | Cell volume regulation (RVI); provide ions for secretion | Potential role in human schizophrenia multi-organ system failure, congenital hydrocephalus, hearing, and neurodevelopmental disorder | [2,38,39,40,41,42,43] |
| SLC12A3 (NCC) | Na+, Cl− | Kidney-specific (DCT) | NaCl reabsorption in the DCT; regulation of Ca2+ and K+ renal excretion; | Gitelman’s syndrome | [9,44,45] |
| SLC12A4 (KCC1) | K+, Cl− | Ubiquitous | cell volume regulation (RVD), KCl epithelial Transport | ND | [2,12,45,46] |
| SLC12A5 (KCC2) | K+, Cl− | Neuron-specific | Intraneuronal Cl− Concentration regulation | Idiopathic generalized epilepsy, developmental apoptosis, neurodevelopmental pathology, Rett syndrome | [2,13,47,48,49,50,51] |
| SLC12A6 (KCC3) | K+, Cl− | Widespread | Volume regulation in the brain; K+ recycling in the kidney | Anderman’s syndrome, Charcot–Marie–Tooth disease, hydrocephalus, sensorimotor neuropathy | [2,14,15,52,53,54,55,56,57] |
| SLC12A7 (KCC4) | K+, Cl− | Widespread | Participates in acid excretion in alpha intercalated cells of collecting duct | ND | [2,15] |
| SLC12A8 (CCC9) | Unknown | Widespread | No function ascribed yet | Psoriasis, dyslipidemia | [2,16,17,58,59] |
| SLC12A9 (CIP) | Unknown | Widespread | No function ascribed yet | May be involved in feather pecking and aggressive behavior | [2,16,18,58] |
2. Role of NNKCC1 in Stroke
3. Role of KCC3 in Stroke
4. Role of Regulatory WNK-SPAK/OSR1 Pathway in Stroke
5. Current Pharmacological Treatments for Stroke
5.1. Inhibitors of WNK-SPAK/OSR1 Pathway
5.2. Inhibitors of NKCC1
5.3. Activator of KCC3
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, D.C.-Y.; Hannemann, A.; Wadud, R.; Rees, D.C.; Brewin, J.N.; Low, P.S.; Gibson, J.S. The role of WNK in modulation of KCl cotransport activity in red cells from normal individuals and patients with sickle cell anaemia. Pflügers Arch. Eur. J. Physiol. 2019, 471, 1539–1549. [Google Scholar] [CrossRef]
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef]
- Meor Azlan, N.; Koeners, M.; Zhang, J. Regulatory control of the Na-Cl co-transporter NCC and its therapeutic potential for hypertension. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- Lauf, P.; Theg, B. A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem. Biophys. Res. Commun. 1980, 92, 1422–1428. [Google Scholar] [CrossRef]
- Dunham, P.B.; Stewart, G.W.; Ellory, J.C. Chloride-activated passive potassium transport in human erythrocytes. Proc. Natl. Acad. Sci. USA 1980, 77, 1711–1715. [Google Scholar] [CrossRef]
- Hoffmann, E.; Sjoholm, C.; Simonsen, L. Anion-Cation Cotransport and Volume Regulation in Ehrlich Ascites Tumor-Cells. Proc. J. Physiol. Lond. 1981, 319, P94–P95. [Google Scholar]
- Gibson, J.S.; Ellory, J.C.; Adragna, N.C.; Lauf, P.K. Pathophysiology of the K+-Cl−cotransporters: Paths to discovery and overview. In Physiology and Pathology of Chloride Transporters and Channels in the Nervous System: From Molecules to Disease; Academic Press: London, UK, 2009; pp. 27–42. [Google Scholar]
- Xu, J.-C.; Lytle, C.; Zhu, T.T.; Payne, J.A.; Benz, E.; Forbush, B. Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc. Natl. Acad. Sci. USA 1994, 91, 2201–2205. [Google Scholar] [CrossRef]
- Gamba, G.; Saltzberg, S.N.; Lombardi, M.; Miyanoshita, A.; Lytton, J.; Hediger, M.A.; Brenner, B.M.; Hebert, S.C. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc. Natl. Acad. Sci. USA 1993, 90, 2749–2753. [Google Scholar] [CrossRef]
- Pellegrino, C.M.; Rybicki, A.C.; Musto, S.; Nagel, R.L.; Schwartz, R.S. Molecular identification and expression of erythroid K: Cl cotransporter in human and mouse erythroleukemic cells. Blood Cells Mol. Dis. 1998, 24, 31–40. [Google Scholar] [CrossRef]
- Piechotta, K.; Lu, J.; Delpire, E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J. Biol. Chem. 2002, 277, 50812–50819. [Google Scholar] [CrossRef]
- Gillen, C.M.; Brill, S.; Payne, J.A.; Forbush, B. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human A new member of the cation-chloride cotransporter family. J. Biol. Chem. 1996, 271, 16237–16244. [Google Scholar] [CrossRef]
- Payne, J.A. Functional characterization of the neuronal-specific K-Cl cotransporter: Implications for [K+] oregulation. Am. J. Physiol. Cell Physiol. 1997, 273, C1516–C1525. [Google Scholar] [CrossRef]
- Hiki, K.; D’Andrea, R.J.; Furze, J.; Crawford, J.; Woollatt, E.; Sutherland, G.R.; Vadas, M.A.; Gamble, J.R. Cloning, characterization, and chromosomal location of a novel human K+-Cl− cotransporter. J. Biol. Chem. 1999, 274, 10661–10667. [Google Scholar] [CrossRef]
- Mercado, A.; Song, L.; George, A.; Delpire, E.; Mount, D. Molecular, functional, and genomic characterization of KCC3 and KCC4. J. Am. Soc. Nephrol. 1999, 10, 38A. [Google Scholar]
- Gagnon, K.B.; Delpire, E. Physiology of SLC12 transporters: Lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am. J. Physiol. Cell Physiol. 2013, 304, C693–C714. [Google Scholar] [CrossRef]
- Wang, G.; Huang, H.; He, Y.; Ruan, L.; Huang, J. Bumetanide protects focal cerebral ischemia-reperfusion injury in rat. Int. J. Clin. Exp. Pathol. 2014, 7, 1487. [Google Scholar]
- Wilkinson, C.M.; Fedor, B.A.; Aziz, J.R.; Nadeau, C.A.; Brar, P.S.; Clark, J.J.; Colbourne, F. Failure of bumetanide to improve outcome after intracerebral hemorrhage in rat. PLoS ONE 2019, 14, e0210660. [Google Scholar] [CrossRef]
- De los Heros, P.; Pacheco-Alvarez, D.; Gamba, G. Role of WNK kinases in the modulation of cell volume. In Current Topics in Membranes; Elsevier: Amsterdam, The Netherlands, 2018; Volume 81, pp. 207–235. [Google Scholar]
- Yang, T.; Zhao, K.; Shu, H.; Chen, X.; Cheng, J.; Li, S.; Zhao, Z.; Kuang, Y.; Yu, S. The Nogo receptor inhibits proliferation, migration and axonal extension by transcriptionally regulating WNK1 in PC12 cells. Neuroreport 2017, 28, 533–539. [Google Scholar] [CrossRef]
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. Bull. World Health Organ. 2016, 94, 634. [Google Scholar] [CrossRef] [PubMed]
- Martín-Aragón Baudel, M.A.; Poole, A.V.; Darlison, M.G. Chloride co-transporters as possible therapeutic targets for stroke. J. Neurochem. 2017, 140, 195–209. [Google Scholar] [CrossRef]
- Schulte, J.T.; Wierenga, C.J.; Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018, 90, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Mayor, D.; Tymianski, M. Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology 2018, 134, 178–188. [Google Scholar] [CrossRef]
- Boscia, F.; Begum, G.; Pignataro, G.; Sirabella, R.; Cuomo, O.; Casamassa, A.; Sun, D.; Annunziato, L. Glial Na+-dependent ion transporters in pathophysiological conditions. Glia 2016, 64, 1677–1697. [Google Scholar] [CrossRef]
- Song, S.; Luo, L.; Sun, B.; Sun, D. Roles of glial ion transporters in brain diseases. Glia 2020, 68, 472–494. [Google Scholar] [CrossRef] [PubMed]
- Zagrean, A.-M.; Grigoras, I.-F.; Iesanu, M.I.; Ionescu, R.-B.; Chitimus, D.M.; Haret, R.M.; Ianosi, B.; Ceanga, M.; Zagrean, L. Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation. Brain Sci. 2019, 9, 360. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Costa, R.O.; Duarte, C.B. Alterations in GABAA-receptor trafficking and synaptic dysfunction in brain disorders. Front. Cell. Neurosci. 2019, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Muir, J.; Rao, Y.; Browarski, M.; Gruenig, M.C.; Sheehan, D.F.; Haucke, V.; Kittler, J.T. Stabilization of GABAA receptors at endocytic zones is mediated by an AP2 binding motif within the GABAA receptor β3 subunit. J. Neurosci. 2012, 32, 2485–2498. [Google Scholar] [CrossRef]
- Mielke, J.G.; Wang, Y.T. Insulin exerts neuroprotection by counteracting the decrease in cell-surface GABAA receptors following oxygen–glucose deprivation in cultured cortical neurons. J. Neurochem. 2005, 92, 103–113. [Google Scholar] [CrossRef]
- Fu, C.-Y.; He, X.-Y.; Li, X.-F.; Zhang, X.; Huang, Z.-W.; Li, J.; Chen, M.; Duan, C.-Z. Nefiracetam attenuates pro-inflammatory cytokines and GABA transporter in specific brain regions of rats with post-ischemic seizures. Cell. Physiol. Biochem. 2015, 37, 2023–2031. [Google Scholar] [CrossRef]
- Begum, G.; Yuan, H.; Kahle, K.T.; Li, L.; Wang, S.; Shi, Y.; Shmukler, B.E.; Yang, S.S.; Lin, S.H.; Alper, S.L.; et al. Inhibition of WNK3 Kinase Signaling Reduces Brain Damage and Accelerates Neurological Recovery After Stroke. Stroke 2015, 46, 1956–1965. [Google Scholar] [CrossRef]
- Russell, J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 2012, 18, 467–486. [Google Scholar] [CrossRef]
- Adachi, M.; Asakura, Y.; SATO, Y.; Tajima, T.; Nakajima, T.; Yamamoto, T.; Fujieda, K. Novel SLC12A1 (NKCC2) mutations in two families with Bartter syndrome type 1. Endocr. J. 2007, 54, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Konopacka, A.; Qiu, J.; Yao, S.T.; Greenwood, M.P.; Greenwood, M.; Lancaster, T.; Inoue, W.; de Souza Mecawi, A.; Vechiato, F.M.; de Lima, J.B. Osmoregulation requires brain expression of the renal Na-K-2Cl cotransporter NKCC2. J. Neurosci. 2015, 35, 5144–5155. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.K.; Jelks, K.A.; O’Donnell, M.E. Ischemia-induced stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport involves p38 and JNK MAP kinases. Am. J. Physiol. Cell Physiol. 2012, 302, C505–C517. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J.; Gazzard, J.; Chaudhry, S.S.; Sampson, N.; Schulte, B.A.; Steel, K.P. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum. Mol. Genet. 1999, 8, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Nezu, A.; Parvin, M.N.; Turner, R.J. A conserved hydrophobic tetrad near the C terminus of the secretory Na+-K+-2Cl− cotransporter (NKCC1) is required for its correct intracellular processing. J. Biol. Chem. 2009, 284, 6869–6876. [Google Scholar] [CrossRef]
- Orlov, S.N.; Koltsova, S.V.; Kapilevich, L.V.; Gusakova, S.V.; Dulin, N.O. NKCC1 and NKCC2: The pathogenetic role of cation-chloride cotransporters in hypertension. Genes Dis. 2015, 2, 186–196. [Google Scholar] [CrossRef]
- Töllner, K.; Brandt, C.; Töpfer, M.; Brunhofer, G.; Erker, T.; Gabriel, M.; Feit, P.W.; Lindfors, J.; Kaila, K.; Löscher, W. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann. Neurol. 2014, 75, 550–562. [Google Scholar] [CrossRef]
- Walcott, B.P.; Kahle, K.T.; Simard, J.M. Novel treatment targets for cerebral edema. Neurotherapeutics 2012, 9, 65–72. [Google Scholar] [CrossRef]
- Koumangoye, R.; Bastarache, L.; Delpire, E. NKCC1: Newly Found as a Human Disease-Causing Ion Transporter. Function 2021, 2, zqaa028. [Google Scholar] [CrossRef] [PubMed]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-chloride cotransporters and neuronal function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Mount, D.B. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu. Rev. Physiol. 2002, 64, 803–843. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.B.; Faulhaber, J.; Budack, M.K.; Pfeffer, C.; Maritzen, T.; Didie, M.; Beck, F.X.; Boettger, T.; Schubert, R.; Ehmke, H.; et al. Neurogenic mechanisms contribute to hypertension in mice with disruption of the K-Cl cotransporter KCC3. Circ. Res. 2006, 98, 549–556. [Google Scholar] [CrossRef]
- Boettger, T.; Hubner, C.A.; Maier, H.; Rust, M.B.; Beck, F.X.; Jentsch, T.J. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002, 416, 874–878. [Google Scholar] [CrossRef]
- Kahle, K.T.; Schmouth, J.F.; Lavastre, V.; Latremoliere, A.; Zhang, J.; Andrews, N.; Omura, T.; Laganiere, J.; Rochefort, D.; Hince, P.; et al. Inhibition of the kinase WNK1/HSN2 ameliorates neuropathic pain by restoring GABA inhibition. Sci. Signal. 2016, 9, ra32. [Google Scholar] [CrossRef]
- Mavrovic, M.; Uvarov, P.; Delpire, E.; Vutskits, L.; Kaila, K.; Puskarjov, M. Loss of non-canonical KCC2 functions promotes developmental apoptosis of cortical projection neurons. EMBO Rep. 2020, 21, e48880. [Google Scholar] [CrossRef]
- Hinz, L.; Torrella Barrufet, J.; Heine, V.M. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol. Commun. 2019, 7, 196. [Google Scholar] [CrossRef]
- Pisella, L.I.; Gaiarsa, J.L.; Diabira, D.; Zhang, J.; Khalilov, I.; Duan, J.; Kahle, K.T.; Medina, I. Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci. Signal. 2019, 12, eaay0300. [Google Scholar] [CrossRef]
- Garneau, A.P.; Marcoux, A.A.; Frenette-Cotton, R.; Mac-Way, F.; Lavoie, J.L.; Isenring, P. Molecular insights into the normal operation, regulation, and multisystemic roles of K(+)-Cl(-) cotransporter 3 (KCC3). Am. J. Physiol. Cell Physiol. 2017, 313, C516–C532. [Google Scholar] [CrossRef]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef]
- Torchia, J.; Lytle, C.; Pon, D.; Forbush, B.; Sen, A. The Na-K-Cl cotransporter of avian salt gland. Phosphorylation in response to cAMP-dependent and calcium-dependent secretogogues. J. Biol. Chem. 1992, 267, 25444–25450. [Google Scholar] [CrossRef]
- Al Shibli, N.; Al-Maawali, A.; Elmanzalawy, A.; Al-Nabhani, M.; Koul, R.; Gabr, A.; Al Murshedi, F. A Novel Splice-Site Variant in SLC12A6 Causes Andermann Syndrome without Agenesis of the Corpus Callosum. J. Pediatr. Genet. 2020, 9, 293–295. [Google Scholar] [CrossRef]
- Jin, S.C.; Furey, C.G.; Zeng, X.; Allocco, A.; Nelson-Williams, C.; Dong, W.; Karimy, J.K.; Wang, K.; Ma, S.; Delpire, E.; et al. SLC12A ion transporter mutations in sporadic and familial human congenital hydrocephalus. Mol. Genet. Genom. Med. 2019, 7, e892. [Google Scholar] [CrossRef]
- Park, J.; Flores, B.R.; Scherer, K.; Kuepper, H.; Rossi, M.; Rupprich, K.; Rautenberg, M.; Deininger, N.; Weichselbaum, A.; Grimm, A.; et al. De novo variants in SLC12A6 cause sporadic early-onset progressive sensorimotor neuropathy. J. Med. Genet. 2020, 57, 283–288. [Google Scholar] [CrossRef]
- Flatman, P.W. Cotransporters, WNKs and hypertension: Important leads from the study of monogenetic disorders of blood pressure regulation. Clin. Sci. 2007, 112, 203–216. [Google Scholar] [CrossRef]
- Richardson, C.; Alessi, D.R. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J. Cell Sci. 2008, 121, 3293–3304. [Google Scholar] [CrossRef]
- Heubl, M.; Zhang, J.; Pressey, J.C.; Al Awabdh, S.; Renner, M.; Gomez-Castro, F.; Moutkine, I.; Eugene, E.; Russeau, M.; Kahle, K.T.; et al. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl(-)-sensitive WNK1 kinase. Nat. Commun. 2017, 8, 1776. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- Delpire, E. Cation-chloride cotransporters in neuronal communication. Physiology 2000, 15, 309–312. [Google Scholar] [CrossRef]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Huberfeld, G.; Wittner, L.; Clemenceau, S.; Baulac, M.; Kaila, K.; Miles, R.; Rivera, C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 2007, 27, 9866–9873. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Song, S.; Banerjee, S.; Jiang, T.; Zhang, J.; Kahle, K.T.; Sun, D.; Zhang, Z. The WNK-SPAK/OSR1 Kinases and the Cation-Chloride Cotransporters as Therapeutic Targets for Neurological Diseases. Aging Dis. 2019, 10, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Khanna, A.R.; Alper, S.L.; Adragna, N.C.; Lauf, P.K.; Sun, D.; Delpire, E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 2015, 21, 513–523. [Google Scholar] [CrossRef]
- Alessi, D.R.; Zhang, J.; Khanna, A.; Hochdorfer, T.; Shang, Y.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: Master regulator of cation-chloride cotransporters. Sci. Signal. 2014, 7, re3. [Google Scholar] [CrossRef]
- Kahle, K.T.; Khanna, A.R.; Duan, J.; Staley, K.J.; Delpire, E.; Poduri, A. The KCC2 Cotransporter and Human Epilepsy: Getting Excited About Inhibition. Neuroscientist 2016, 22, 555–562. [Google Scholar] [CrossRef]
- Hartmann, A.-M.; Nothwang, H.G. Molecular and evolutionary insights into the structural organization of cation chloride cotransporters. Front. Cell. Neurosci. 2015, 8, 470. [Google Scholar] [CrossRef]
- Zhang, J.; Bhuiyan, M.I.H.; Zhang, T.; Karimy, J.K.; Wu, Z.; Fiesler, V.M.; Zhang, J.; Huang, H.; Hasan, M.N.; Skrzypiec, A.E.; et al. Modulation of brain cation-Cl(-) cotransport via the SPAK kinase inhibitor ZT-1a. Nat. Commun. 2020, 11, 78. [Google Scholar] [CrossRef]
- Zhang, J.; Cordshagen, A.; Medina, I.; Nothwang, H.G.; Wisniewski, J.R.; Winklhofer, M.; Hartmann, A.M. Staurosporine and NEM mainly impair WNK-SPAK/OSR1 mediated phosphorylation of KCC2 and NKCC1. PLoS ONE 2020, 15, e0232967. [Google Scholar] [CrossRef]
- De Los Heros, P.; Alessi, D.R.; Gourlay, R.; Campbell, D.G.; Deak, M.; Macartney, T.J.; Kahle, K.T.; Zhang, J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl− co-transporters. Biochem. J. 2014, 458, 559–573. [Google Scholar] [CrossRef]
- Brown, A.; Meor Azlan, N.F.; Wu, Z.; Zhang, J. WNK-SPAK/OSR1-NCC kinase signaling pathway as a novel target for the treatment of salt-sensitive hypertension. Acta Pharmacol. Sin. 2020. [Google Scholar] [CrossRef] [PubMed]
- AlAmri, M.A.; Kadri, H.; Alderwick, L.J.; Jeeves, M.; Mehellou, Y. The Photosensitising Clinical Agent Verteporfin Is an Inhibitor of SPAK and OSR1 Kinases. Chembiochem 2018, 19, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.; Vazquez, N.; Kahle, K.T.; Hodson, C.A.; Ring, A.M.; Gulcicek, E.E.; Louvi, A.; Bobadilla, N.A.; Gamba, G.; Lifton, R.P. WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. J. Biol. Chem. 2011, 286, 30171–30180. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; McBrayer, D.; Lytle, C. [Cl−] i-dependent phosphorylation of the Na-K-Cl cotransport protein of dog tracheal epithelial cells. J. Biol. Chem. 1995, 270, 28955–28961. [Google Scholar] [CrossRef] [PubMed]
- Lytle, C.; Forbush 3rd, B. Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: Modulation by cytoplasmic Cl. Am. J. Physiol. Cell Physiol. 1996, 270, C437–C448. [Google Scholar] [CrossRef]
- Cossins, A.; Weaver, Y.; Lykkeboe, G.; Nielsen, O. Role of protein phosphorylation in control of K flux pathways of trout red blood cells. Am. J. Physiol. Cell Physiol. 1994, 267, C1641–C1650. [Google Scholar] [CrossRef]
- Flatman, P.W.; Adragna, N.C.; Lauf, P.K. Role of protein kinases in regulating sheep erythrocyte K-Cl cotransport. Am. J. Physiol. Cell Physiol. 1996, 271, C255–C263. [Google Scholar] [CrossRef]
- Jennings, M.L.; Schulz, R.K. Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either cell swelling or N-ethylmaleimide. J. Gen. Physiol. 1991, 97, 799–817. [Google Scholar] [CrossRef]
- McCormick, J.A.; Ellison, D.H. The WNKs: Atypical protein kinases with pleiotropic actions. Physiol. Rev. 2011, 91, 177–219. [Google Scholar] [CrossRef]
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol. Asp. Med. 2013, 34, 288–298. [Google Scholar] [CrossRef]
- Piala, A.T.; Moon, T.M.; Akella, R.; He, H.; Cobb, M.H.; Goldsmith, E.J. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci. Signal. 2014, 7, ra41. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, J.; Kobayashi, Y.; Umeda, T.; Vandewalle, A.; Takeda, K.; Ichijo, H.; Naguro, I. Osmotic stress induces the phosphorylation of WNK4 Ser575 via the p38MAPK-MK pathway. Sci. Rep. 2016, 6, 18710. [Google Scholar] [CrossRef] [PubMed]
- Salihu, S.; Meor Azlan, N.; Josiah, S.; Wu, Z.; Wang, Y.; Zhang, J. Role of the cation-chloride-cotransporters in the circadian system. Asian J. Pharm. Sci. 2020. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, G.; Begum, G.; Wang, J.; Khanna, A.R.; Shmukler, B.E.; Daubner, G.M.; de Los Heros, P.; Davies, P.; Varghese, J.; et al. Functional kinomics establishes a critical node of volume-sensitive cation-Cl(-) cotransporter regulation in the mammalian brain. Sci. Rep. 2016, 6, 35986. [Google Scholar] [CrossRef] [PubMed]
- Adragna, N.C.; Ravilla, N.B.; Lauf, P.K.; Begum, G.; Khanna, A.R.; Sun, D.; Kahle, K.T. Regulated phosphorylation of the K-Cl cotransporter KCC3 is a molecular switch of intracellular potassium content and cell volume homeostasis. Front. Cell Neurosci. 2015, 9, 255. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.; Risinger, M.; Pan, W.; Wu, D.; Colangelo, C.M. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 2009, 138, 525–536. [Google Scholar] [CrossRef]
- Thastrup, J.O.; Rafiqi, F.H.; Vitari, A.C.; Pozo-Guisado, E.; Deak, M.; Mehellou, Y.; Alessi, D.R. SPAK/OSR1 regulate NKCC1 and WNK activity: Analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem. J. 2012, 441, 325–337. [Google Scholar] [CrossRef]
- Markkanen, M.; Ludwig, A.; Khirug, S.; Pryazhnikov, E.; Soni, S.; Khiroug, L.; Delpire, E.; Rivera, C.; Airaksinen, M.S.; Uvarov, P. Implications of the N-terminal heterogeneity for the neuronal K-Cl cotransporter KCC2 function. Brain Res. 2017, 1675, 87–101. [Google Scholar] [CrossRef]
- Vitari, A.C.; Thastrup, J.; Rafiqi, F.H.; Deak, M.; Morrice, N.A.; Karlsson, H.K.; Alessi, D.R. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem. J. 2006, 397, 223–231. [Google Scholar] [CrossRef]
- Zhao, H.; Nepomuceno, R.; Gao, X.; Foley, L.M.; Wang, S.; Begum, G.; Zhu, W.; Pigott, V.M.; Falgoust, L.M.; Kahle, K.T.; et al. Deletion of the WNK3-SPAK kinase complex in mice improves radiographic and clinical outcomes in malignant cerebral edema after ischemic stroke. J. Cereb. Blood Flow Metab. 2017, 37, 550–563. [Google Scholar] [CrossRef]
- Cuomo, O.; Vinciguerra, A.; Cerullo, P.; Anzilotti, S.; Brancaccio, P.; Bilo, L.; Scorziello, A.; Molinaro, P.; Di Renzo, G.; Pignataro, G. Ionic homeostasis in brain conditioning. Front. Neurosci. 2015, 9, 277. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.; Liu, F.; Sun, X.; Qu, H.; Zhao, S.; Zhou, Z.; Xiao, T.; Zhao, C.; Zhao, M. Bumetanide: A review of its neuroplasticity and behavioral effects after stroke. Restor. Neurol. Neurosci. 2019, 37, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, N.; Witte, O.W.; Frahm, C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke 2010, 41, e151–e159. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kumada, T.; Morishima, T.; Iwata, S.; Kaneko, T.; Yanagawa, Y.; Yoshida, S.; Fukuda, A. Accumulation of GABAergic neurons, causing a focal ambient GABA gradient, and downregulation of KCC2 are induced during microgyrus formation in a mouse model of polymicrogyria. Cereb. Cortex 2014, 24, 1088–1101. [Google Scholar] [CrossRef]
- Yan, Y.; Dempsey, R.J.; Flemmer, A.; Forbush, B.; Sun, D. Inhibition of Na+–K+–Cl− cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003, 961, 22–31. [Google Scholar] [CrossRef]
- Yan, Y.; Dempsey, R.J.; Sun, D. Na+-K+-Cl− cotransporter in rat focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 711–721. [Google Scholar] [CrossRef]
- Chen, X.; Kintner, D.B.; Luo, J.; Baba, A.; Matsuda, T.; Sun, D. Endoplasmic reticulum Ca2+ dysregulation and endoplasmic reticulum stress following in vitro neuronal ischemia: Role of Na+-K+-Cl−cotransporter. J. Neurochem. 2008, 106, 1563–1576. [Google Scholar] [CrossRef]
- Chen, H.; Sun, D. The role of Na–K–Cl co–transporter in cerebral ischemia. Neurol. Res. 2005, 27, 280–286. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, G.; Zhao, C.; Yang, Y.; Miao, Z.; Xu, X. Interleukin-18 from neurons and microglia mediates depressive behaviors in mice with post-stroke depression. Brain Behav. Immun. 2020, 88, 411–420. [Google Scholar] [CrossRef]
- Huang, L.-Q.; Zhu, G.-F.; Deng, Y.-Y.; Jiang, W.-Q.; Fang, M.; Chen, C.-B.; Cao, W.; Wen, M.-Y.; Han, Y.-L.; Zeng, H.-K. Hypertonic saline alleviates cerebral edema by inhibiting microglia-derived TNF-α and IL-1β-induced Na-K-Cl Cotransporter up-regulation. J. Neuroinflamm. 2014, 11, 1–20. [Google Scholar] [CrossRef][Green Version]
- Su, G.; Kintner, D.B.; Flagella, M.; Shull, G.E.; Sun, D. Astrocytes from Na+-K+-Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am. J. Physiol. Cell Physiol. 2002, 282, C1147–C1160. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Luo, J.; Kintner, D.B.; Shull, G.E.; Sun, D. Na+-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2005, 25, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.A.; Hong, S.H.; Kim, J.W.; Jang, I.S. Possible involvement of DNA methylation in NKCC1 gene expression during postnatal development and in response to ischemia. J. Neurochem. 2010, 114, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.I.H.; Song, S.; Yuan, H.; Begum, G.; Kofler, J.; Kahle, K.T.; Yang, S.-S.; Lin, S.-H.; Alper, S.L.; Subramanya, A.R. WNK-Cab39-NKCC1 signaling increases the susceptibility to ischemic brain damage in hypertensive rats. J. Cereb. Blood Flow Metab. 2017, 37, 2780–2794. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Xu, J.; Chen, Y.; Gibbs, M.E.; Du, T. Antagonists of the Vasopressin V1 Receptor and of the β1-Adrenoceptor Inhibit Cytotoxic Brain Edema in Stroke by Effects on Astrocytes-but the Mechanisms Differ. Curr. Neuropharmacol. 2014, 12, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.-T.; Huang, T.-C.; Wang, J.-Y.; You, Y.-S.; Chou, J.-L.; Chan, M.W.; Wo, P.Y.; Amstislavskaya, T.G.; Tikhonova, M.A.; Yang, Y.-L. NKCC1 mediates traumatic brain injury-induced hippocampal neurogenesis through CREB phosphorylation and HIF-1α expression. Pflügers Arch. Eur. J. Physiol. 2015, 467, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Tang, R.; Yu, Z.; Huang, S.; Xie, M.; Luo, X.; Wang, W. Bumetanide-induced NKCC1 inhibition attenuates oxygen–glucose deprivation-induced decrease in proliferative activity and cell cycle progression arrest in cultured OPCs via p-38 MAPKs. Brain Res. 2015, 1613, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fu, P.; Yu, Z.; Xie, M.; Wang, W.; Luo, X. NKCC1 inhibition attenuates chronic cerebral hypoperfusion-induced white matter lesions by enhancing progenitor cells of oligodendrocyte proliferation. J. Mol. Neurosci. 2018, 64, 449–458. [Google Scholar] [CrossRef]
- Mu, X.; Wang, H.; Cheng, X.; Yang, L.; Sun, X.; Qu, H.; Zhao, S.; Zhou, Z.; Liu, T.; Xiao, T. Inhibition of Nkcc1 promotes axonal growth and motor recovery in ischemic rats. Neuroscience 2017, 365, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Yuen, N.Y.; Chechneva, O.V.; Chen, Y.-J.; Tsai, Y.-C.; Little, L.K.; Dang, J.; Tancredi, D.J.; Conston, J.; Anderson, S.E.; O’Donnell, M.E. Exacerbated brain edema in a rat streptozotocin model of hyperglycemic ischemic stroke: Evidence for involvement of blood–brain barrier Na–K–Cl cotransport and Na/H exchange. J. Cereb. Blood Flow Metab. 2019, 39, 1678–1692. [Google Scholar] [CrossRef]
- Kahle, K.T.; Rinehart, J.; Lifton, R.P. Phosphoregulation of the Na–K–2Cl and K–Cl cotransporters by the WNK kinases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Lucas, O.; Hilaire, C.; Delpire, E.; Scamps, F. KCC3-dependent chloride extrusion in adult sensory neurons. Mol. Cell Neurosci. 2012, 50, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Byun, N.; Delpire, E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol. Dis. 2007, 28, 39–51. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Auer, R.N.; Laganiere, J.L.; Robitaille, Y.O.; Richardson, J.; Dion, P.A.; Rouleau, G.A.; Shekarabi, M. KCC3 axonopathy: Neuropathological features in the central and peripheral nervous system. Mod. Pathol. 2016, 29, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Dupré, N.; Howard, H.C.; Rouleau, G.A. Hereditary motor and sensory neuropathy with agenesis of the corpus callosum. In GeneReviews [Internet]; University of Washington: Seattle, WA, USA, 2014. [Google Scholar]
- Uyanik, G.; Elcioglu, N.; Penzien, J.; Gross, C.; Yilmaz, Y.; Olmez, A.; Demir, E.; Wahl, D.; Scheglmann, K.; Winner, B.; et al. Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology 2006, 66, 1044–1048. [Google Scholar] [CrossRef]
- Delpire, E.; Kahle, K.T. The KCC3 cotransporter as a therapeutic target for peripheral neuropathy. Expert. Opin. Ther. Targets 2017, 21, 113–116. [Google Scholar] [CrossRef]
- Boettger, T.; Rust, M.B.; Maier, H.; Seidenbecher, T.; Schweizer, M.; Keating, D.J.; Faulhaber, J.; Ehmke, H.; Pfeffer, C.; Scheel, O.; et al. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 2003, 22, 5422–5434. [Google Scholar] [CrossRef]
- Howard, H.C.; Mount, D.B.; Rochefort, D.; Byun, N.; Dupre, N.; Lu, J.; Fan, X.; Song, L.; Riviere, J.B.; Prevost, C.; et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat. Genet. 2002, 32, 384–392. [Google Scholar] [CrossRef]
- Kahle, K.T.; Flores, B.; Bharucha-Goebel, D.; Zhang, J.; Donkervoort, S.; Hegde, M.; Hussain, G.; Duran, D.; Liang, B.; Sun, D.; et al. Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci. Signal. 2016, 9, ra77. [Google Scholar] [CrossRef]
- Inoue, K.; Furukawa, T.; Kumada, T.; Yamada, J.; Wang, T.; Inoue, R.; Fukuda, A. Taurine inhibits K+-Cl− cotransporter KCC2 to regulate embryonic Cl− homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J. Biol. Chem. 2012, 287, 20839–20850. [Google Scholar] [CrossRef]
- Huang, H.; Bhuiyan, M.I.H.; Jiang, T.; Song, S.; Shankar, S.; Taheri, T.; Li, E.; Schreppel, P.; Hintersteininger, M.; Yang, S.-S. A Novel Na+-K+-Cl− Cotransporter 1 Inhibitor STS66* Reduces Brain Damage in Mice After Ischemic Stroke. Stroke 2019, 50, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Zhang, J.; Mansuri, M.S.; Duan, J.; Karimy, J.K.; Delpire, E.; Alper, S.L.; Lifton, R.P.; Fukuda, A.; Kahle, K.T. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 2019, 12, eaaw9315. [Google Scholar] [CrossRef] [PubMed]
- Mccarthy, M.M. Estradiol and the developing brain. Physiol. Rev. 2008, 88, 91–134. [Google Scholar] [CrossRef] [PubMed]
- Nugent, B.M.; Valenzuela, C.V.; Simons, T.J.; McCarthy, M.M. Kinases SPAK and OSR1 are upregulated by estradiol and activate NKCC1 in the developing hypothalamus. J. Neurosci. 2012, 32, 593–598. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheng, J.; Zhang, P.; Li, X.; Lei, S.; Li, W.; He, X.; Zhang, J.; Wang, N.; Qi, C.; Chen, X. Post-stroke estradiol treatment enhances neurogenesis in the subventricular zone of rats after permanent focal cerebral ischemia. Neuroscience 2013, 231, 82–90. [Google Scholar] [CrossRef]
- Hannemann, A.; Flatman, P.W. Phosphorylation and transport in the Na-K-2Cl cotransporters, NKCC1 and NKCC2A, compared in HEK-293 cells. PLoS ONE 2011, 6, e17992. [Google Scholar] [CrossRef]
- Zhang, J.; Siew, K.; Macartney, T.; O’Shaughnessy, K.M.; Alessi, D.R. Critical role of the SPAK protein kinase CCT domain in controlling blood pressure. Hum. Mol. Genet. 2015, 24, 4545–4558. [Google Scholar] [CrossRef]
- Shekarabi, M.; Lafreniere, R.G.; Gaudet, R.; Laganiere, J.; Marcinkiewicz, M.M.; Dion, P.A.; Rouleau, G.A. Comparative analysis of the expression profile of Wnk1 and Wnk1/Hsn2 splice variants in developing and adult mouse tissues. PLoS ONE 2013, 8, e57807. [Google Scholar] [CrossRef]
- Shekarabi, M.; Salin-Cantegrel, A.; Laganiere, J.; Gaudet, R.; Dion, P.; Rouleau, G.A. Cellular expression of the K+-Cl− cotransporter KCC3 in the central nervous system of mouse. Brain Res. 2011, 1374, 15–26. [Google Scholar] [CrossRef]
- De Los Heros, P.; Kahle, K.T.; Rinehart, J.; Bobadilla, N.A.; Vazquez, N.; San Cristobal, P.; Mount, D.B.; Lifton, R.P.; Hebert, S.C.; Gamba, G. WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 1976–1981. [Google Scholar] [CrossRef]
- Kahle, K.T.; Rinehart, J.; Ring, A.; Gimenez, I.; Gamba, G.; Hebert, S.C.; Lifton, R.P. WNK protein kinases modulate cellular Cl− flux by altering the phosphorylation state of the Na-K-Cl and K-Cl cotransporters. Physiology 2006, 21, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Rinehart, J.; de Los Heros, P.; Louvi, A.; Meade, P.; Vazquez, N.; Hebert, S.C.; Gamba, G.; Gimenez, I.; Lifton, R.P. WNK3 modulates transport of Cl− in and out of cells: Implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. USA 2005, 102, 16783–16788. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.B.; Delpire, E. Multiple pathways for protein phosphatase 1 (PP1) regulation of Na-K-2Cl cotransporter (NKCC1) function: The N-terminal tail of the Na-K-2Cl cotransporter serves as a regulatory scaffold for Ste20-related proline/alanine-rich kinase (SPAK) AND PP1. J. Biol. Chem. 2010, 285, 14115–14121. [Google Scholar] [CrossRef] [PubMed]
- Dowd, B.F.; Forbush, B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1). J. Biol. Chem. 2003, 278, 27347–27353. [Google Scholar] [CrossRef] [PubMed]
- Melo, Z.; de los Heros, P.; Cruz-Rangel, S.; Vazquez, N.; Bobadilla, N.A.; Pasantes-Morales, H.; Alessi, D.R.; Mercado, A.; Gamba, G. N-terminal serine dephosphorylation is required for KCC3 cotransporter full activation by cell swelling. J. Biol. Chem. 2013, 288, 31468–31476. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Verkman, A.S.; Curtis, K.M.; Norenberg, M.D. Aquaporin-4 deletion in mice reduces encephalopathy and brain edema in experimental acute liver failure. Neurobiol. Dis. 2014, 63, 222–228. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Zheng, H.Q.; Chen, B.Y.; Sun, L.; Ma, M.M.; Wang, G.L.; Guan, Y.Y. WNK1 is required for proliferation induced by hypotonic challenge in rat vascular smooth muscle cells. Acta Pharmacol. Sin. 2018, 39, 35–47. [Google Scholar] [CrossRef]
- Xu, W.; Mu, X.; Wang, H.; Song, C.; Ma, W.; Jolkkonen, J.; Zhao, C. Chloride co-transporter NKCC1 inhibitor bumetanide enhances neurogenesis and behavioral recovery in rats after experimental stroke. Mol. Neurobiol. 2017, 54, 2406–2414. [Google Scholar] [CrossRef]
- Shulga, A.; Magalhães, A.C.; Autio, H.; Plantman, S.; di Lieto, A.; Nykjær, A.; Carlstedt, T.; Risling, M.; Arumäe, U.; Castrén, E. The loop diuretic bumetanide blocks posttraumatic p75NTR upregulation and rescues injured neurons. J. Neurosci. 2012, 32, 1757–1770. [Google Scholar] [CrossRef]
- Yamada, K.; Park, H.M.; Rigel, D.F.; DiPetrillo, K.; Whalen, E.J.; Anisowicz, A.; Beil, M.; Berstler, J.; Brocklehurst, C.E.; Burdick, D.A.; et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat. Chem. Biol. 2016, 12, 896–898. [Google Scholar] [CrossRef]
- Apsel, B.; Blair, J.A.; Gonzalez, B.; Nazif, T.M.; Feldman, M.E.; Aizenstein, B.; Hoffman, R.; Williams, R.L.; Shokat, K.M.; Knight, Z.A. Targeted polypharmacology: Discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 4, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, E.; Mori, T.; Zeniya, M.; Isobe, K.; Ishigami-Yuasa, M.; Fujii, S.; Kagechika, H.; Ishihara, T.; Mizushima, T.; Sasaki, S.; et al. Discovery of Novel SPAK Inhibitors That Block WNK Kinase Signaling to Cation Chloride Transporters. J. Am. Soc. Nephrol. 2015, 26, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- AlAmri, M.A.; Kadri, H.; Alderwick, L.J.; Simpkins, N.S.; Mehellou, Y. Rafoxanide and Closantel Inhibit SPAK and OSR1 Kinases by Binding to a Highly Conserved Allosteric Site on Their C-terminal Domains. ChemMedChem 2017, 12, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kikuchi, E.; Watanabe, Y.; Fujii, S.; Ishigami-Yuasa, M.; Kagechika, H.; Sohara, E.; Rai, T.; Sasaki, S.; Uchida, S. Chemical library screening for WNK signalling inhibitors using fluorescence correlation spectroscopy. Biochem. J. 2013, 455, 339–345. [Google Scholar] [CrossRef]
- Kadri, H.; Alamri, M.A.; Navratilova, I.H.; Alderwick, L.J.; Simpkins, N.S.; Mehellou, Y. Towards the Development of Small-Molecule MO25 Binders as Potential Indirect SPAK/OSR1 Kinase Inhibitors. Chembiochem 2017, 18, 460–465. [Google Scholar] [CrossRef]
- Fujii, S.; Kikuchi, E.; Watanabe, Y.; Suzuyama, H.; Ishigami-Yuasa, M.; Mori, T.; Isobe, K.; Uchida, S.; Kagechika, H. Structural development of N-(4-phenoxyphenyl) benzamide derivatives as novel SPAK inhibitors blocking WNK kinase signaling. Bioorganic Med. Chem. Lett. 2020, 30, 127408. [Google Scholar] [CrossRef]
- Zhang, J.; Pu, H.; Zhang, H.; Wei, Z.; Jiang, X.; Xu, M.; Zhang, L.; Zhang, W.; Liu, J.; Meng, H. Inhibition of Na+-K+-2Cl− cotransporter attenuates blood-brain-barrier disruption in a mouse model of traumatic brain injury. Neurochem. Int. 2017, 111, 23–31. [Google Scholar] [CrossRef]
- Kahle, K.T.; Simard, J.M.; Staley, K.J.; Nahed, B.V.; Jones, P.S.; Sun, D. Molecular mechanisms of ischemic cerebral edema: Role of electroneutral ion transport. Physiology 2009, 24, 257–265. [Google Scholar] [CrossRef]
- Payne, J.A.; Rivera, C.; Voipio, J.; Kaila, K. Cation–chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003, 26, 199–206. [Google Scholar] [CrossRef]
- Hamidi, S.; Avoli, M. KCC2 function modulates in vitro ictogenesis. Neurobiol. Dis. 2015, 79, 51–58. [Google Scholar] [CrossRef]
- O’donnell, M.E.; Tran, L.; Lam, T.I.; Liu, X.B.; Anderson, S.E. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J. Cereb. Blood Flow Metab. 2004, 24, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Singh Jaggi, A.; Kaur, A.; Bali, A.; Singh, N. Expanding spectrum of sodium potassium chloride co-transporters in the pathophysiology of diseases. Curr. Neuropharmacol. 2015, 13, 369–388. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Kahle, K.T.; Gerzanich, V. Molecular mechanisms of microvascular failure in central nervous system injury—synergistic roles of NKCC1 and SUR1/TRPM4: A review. J. Neurosurg. 2010, 113, 622–629. [Google Scholar] [CrossRef]
- Hu, J.-J.; Yang, X.-L.; Luo, W.-D.; Han, S.; Yin, J.; Liu, W.-H.; He, X.-H.; Peng, B.-W. Bumetanide reduce the seizure susceptibility induced by pentylenetetrazol via inhibition of aberrant hippocampal neurogenesis in neonatal rats after hypoxia-ischemia. Brain Res. Bull. 2017, 130, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Glykys, J.; Dzhala, V.; Egawa, K.; Kahle, K.T.; Delpire, E.; Staley, K. Chloride dysregulation, seizures, and cerebral edema: A relationship with therapeutic potential. Trends Neurosci. 2017, 40, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Grandgeorge, M.; Lemonnier, E.; Degrez, C.; Jallot, N. The effect of bumetanide treatment on the sensory behaviours of a young girl with Asperger syndrome. Case Rep. 2014, 2014, bcr2013202092. [Google Scholar] [CrossRef]
- Lemonnier, E.; Lazartigues, A.; Ben-Ari, Y. Treating schizophrenia with the diuretic bumetanide: A case report. Clin. Neuropharmacol. 2016, 39, 115–117. [Google Scholar] [CrossRef]
- Lemonnier, E.; Robin, G.; Degrez, C.; Tyzio, R.; Grandgeorge, M.; Ben-Ari, Y. Treating F ragile X syndrome with the diuretic bumetanide: A case report. Acta Paediatr. 2013, 102, e288–e290. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hammond, C.; Ben-Ari, Y. Bumetanide to treat Parkinson disease: A report of 4 cases. Clin. Neuropharmacol. 2016, 39, 57–59. [Google Scholar] [CrossRef]
- Eftekhari, S.; Mehvari Habibabadi, J.; Najafi Ziarani, M.; Hashemi Fesharaki, S.S.; Gharakhani, M.; Mostafavi, H.; Joghataei, M.T.; Beladimoghadam, N.; Rahimian, E.; Hadjighassem, M.R. Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia 2013, 54, e9–e12. [Google Scholar] [CrossRef]
- Rahmanzadeh, R.; Eftekhari, S.; Shahbazi, A.; Khodaei Ardakani, M.-r.; Rahmanzade, R.; Mehrabi, S.; Barati, M.; Joghataei, M.T. Effect of bumetanide, a selective NKCC1 inhibitor, on hallucinations of schizophrenic patients; a double-blind randomized clinical trial. Schizophr Res. 2017, 184, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Pressler, R.M.; Boylan, G.B.; Marlow, N.; Blennow, M.; Chiron, C.; Cross, J.H.; de Vries, L.S.; Hallberg, B.; Hellström-Westas, L.; Jullien, V. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): An open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015, 14, 469–477. [Google Scholar] [CrossRef]
- Kahle, K.T.; Barnett, S.M.; Sassower, K.C.; Staley, K.J. Decreased seizure activity in a human neonate treated with bumetanide, an inhibitor of the Na+-K+-2Cl− cotransporter NKCC1. J. Child Neurol. 2009, 24, 572–576. [Google Scholar] [CrossRef]
- Lemonnier, E.; Ben-Ari, Y. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. 2010, 99, 1885–1888. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, É.; Degrez, C.; Phelep, M.; Tyzio, R.; Josse, F.; Grandgeorge, M.; Hadjikhani, N.; Ben-Ari, Y. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry 2012, 2, e202. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, E.; Villeneuve, N.; Sonie, S.; Serret, S.; Rosier, A.; Roue, M.; Brosset, P.; Viellard, M.; Bernoux, D.; Rondeau, S. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 2017, 7, e1056. [Google Scholar] [CrossRef]
- Savardi, A.; Borgogno, M.; Narducci, R.; La Sala, G.; Ortega, J.A.; Summa, M.; Armirotti, A.; Bertorelli, R.; Contestabile, A.; De Vivo, M.; et al. Discovery of a Small Molecule Drug Candidate for Selective NKCC1 Inhibition in Brain Disorders. Chem 2020, 6, 2073–2096. [Google Scholar] [CrossRef]
- Delpire, E.; Weaver, C.D. Challenges of Finding Novel Drugs Targeting the K-Cl Cotransporter. ACS Chem. Neurosci. 2016, 7, 1624–1627. [Google Scholar] [CrossRef]
- Flores, B.; Schornak, C.C.; Delpire, E. A role for KCC3 in maintaining cell volume of peripheral nerve fibers. Neurochem. Int. 2019, 123, 114–124. [Google Scholar] [CrossRef]
- Delpire, E.; Days, E.; Lewis, L.M.; Mi, D.; Kim, K.; Lindsley, C.W.; Weaver, C.D. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. USA 2009, 106, 5383–5388. [Google Scholar] [CrossRef]
- Delpire, E.; Baranczak, A.; Waterson, A.G.; Kim, K.; Kett, N.; Morrison, R.D.; Daniels, J.S.; Weaver, C.D.; Lindsley, C.W. Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: Development of a highly selective and more potent in vitro probe. Bioorg. Med. Chem. Lett. 2012, 22, 4532–4535. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Josiah, S.S.; Meor Azlan, N.F.; Zhang, J. Targeting the WNK-SPAK/OSR1 Pathway and Cation-Chloride Cotransporters for the Therapy of Stroke. Int. J. Mol. Sci. 2021, 22, 1232. https://doi.org/10.3390/ijms22031232
Josiah SS, Meor Azlan NF, Zhang J. Targeting the WNK-SPAK/OSR1 Pathway and Cation-Chloride Cotransporters for the Therapy of Stroke. International Journal of Molecular Sciences. 2021; 22(3):1232. https://doi.org/10.3390/ijms22031232
Chicago/Turabian StyleJosiah, Sunday Solomon, Nur Farah Meor Azlan, and Jinwei Zhang. 2021. "Targeting the WNK-SPAK/OSR1 Pathway and Cation-Chloride Cotransporters for the Therapy of Stroke" International Journal of Molecular Sciences 22, no. 3: 1232. https://doi.org/10.3390/ijms22031232
APA StyleJosiah, S. S., Meor Azlan, N. F., & Zhang, J. (2021). Targeting the WNK-SPAK/OSR1 Pathway and Cation-Chloride Cotransporters for the Therapy of Stroke. International Journal of Molecular Sciences, 22(3), 1232. https://doi.org/10.3390/ijms22031232