

The Expanding Phenotype of ZTTK Syndrome Due to the Heterozygous Variant of SON Gene Focusing on Liver Involvement: Patient Report and Literature Review

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Patients and Methods
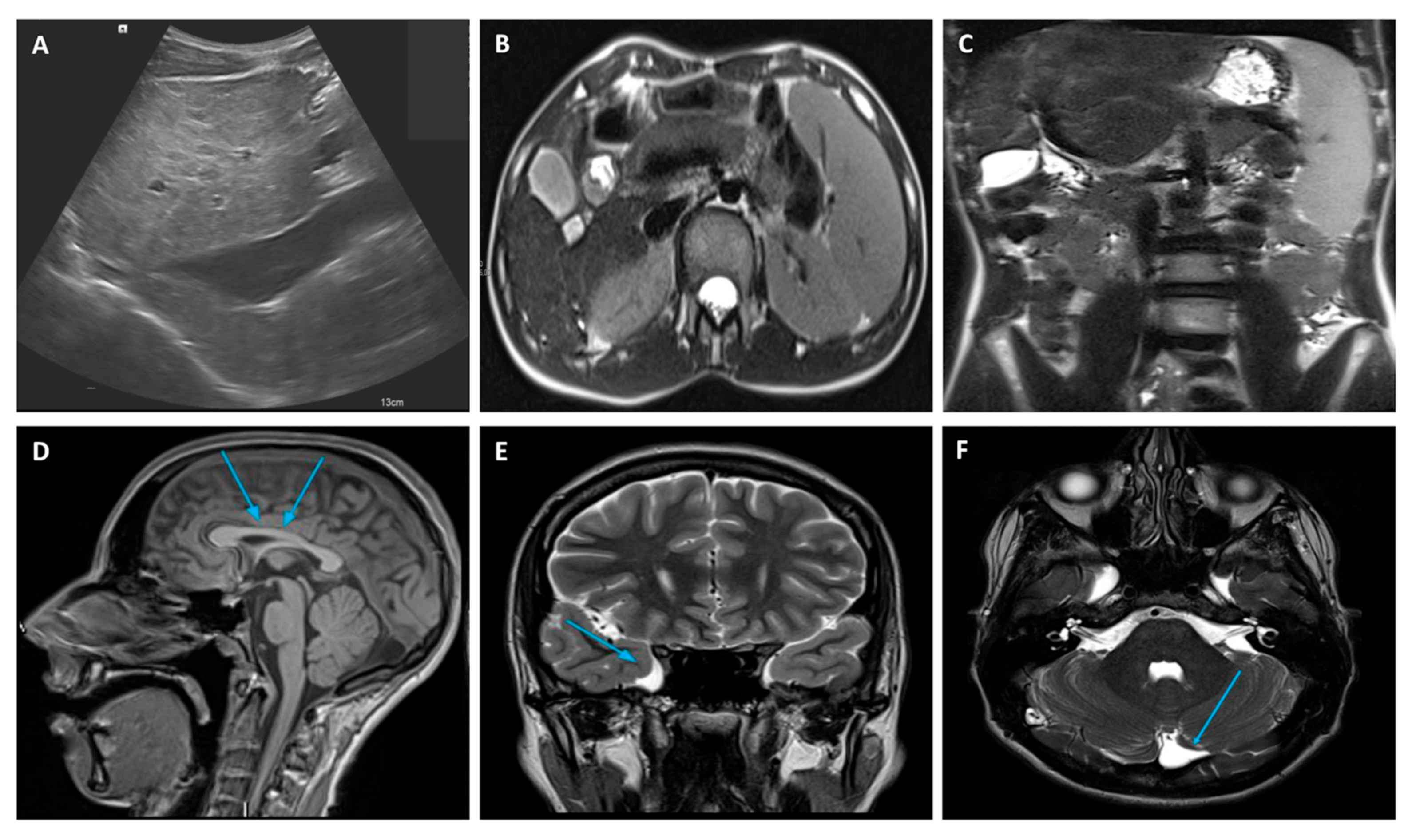
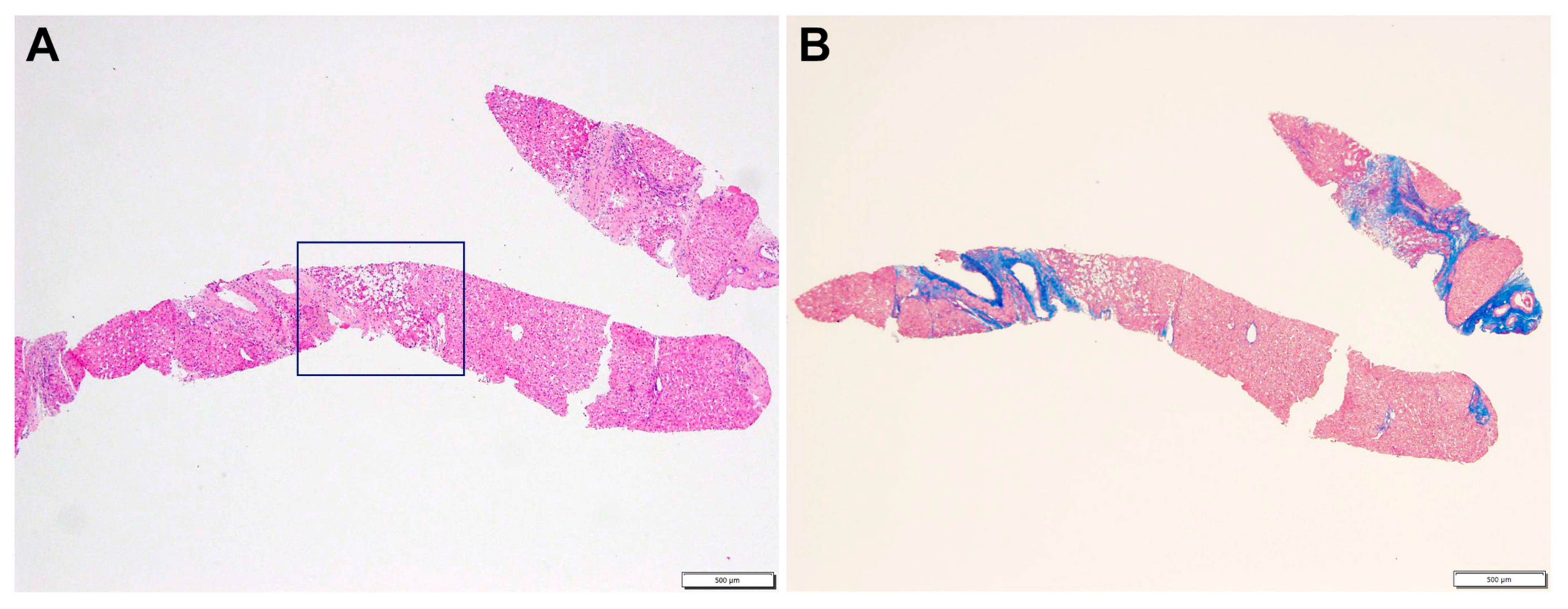
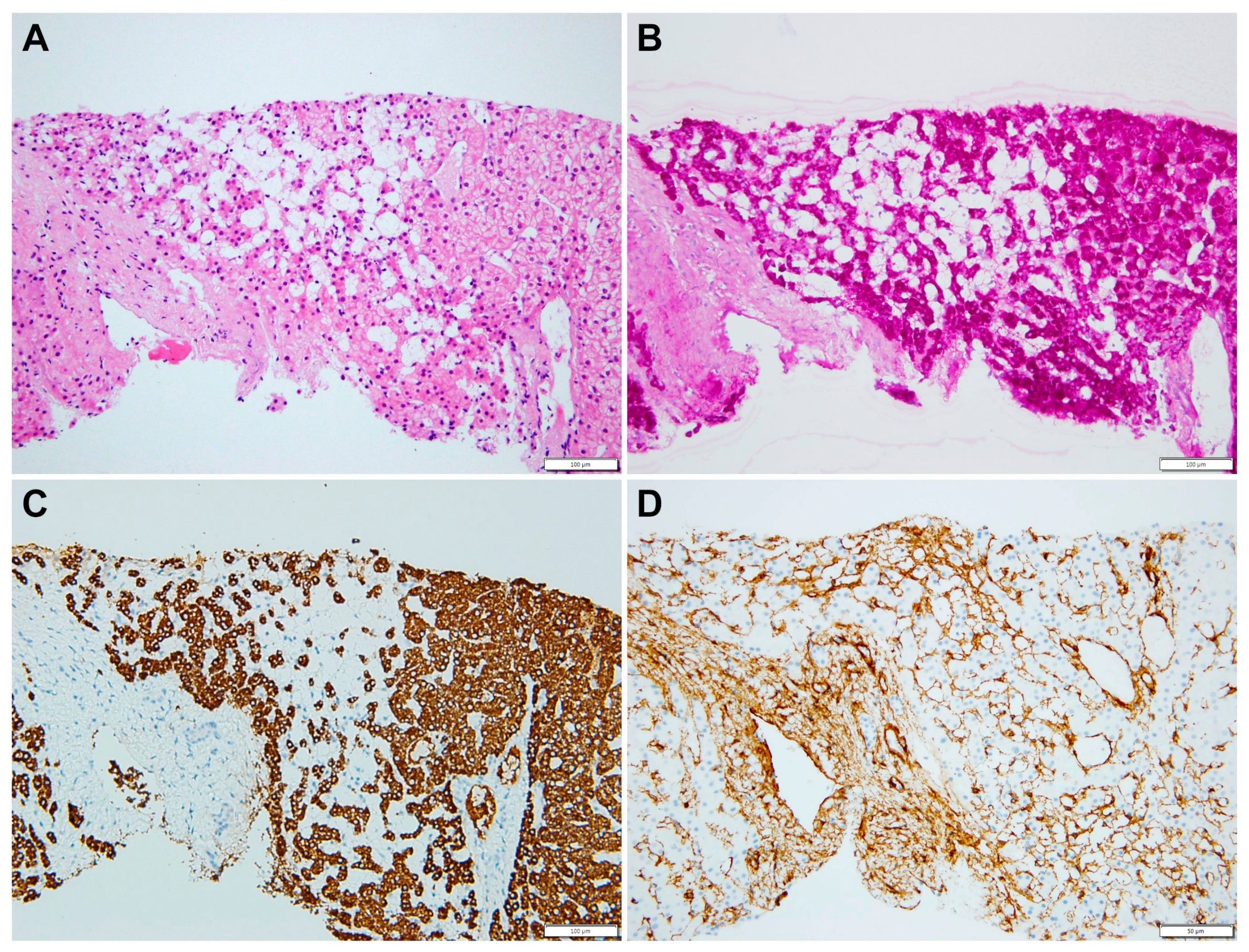
2.1. Case Description
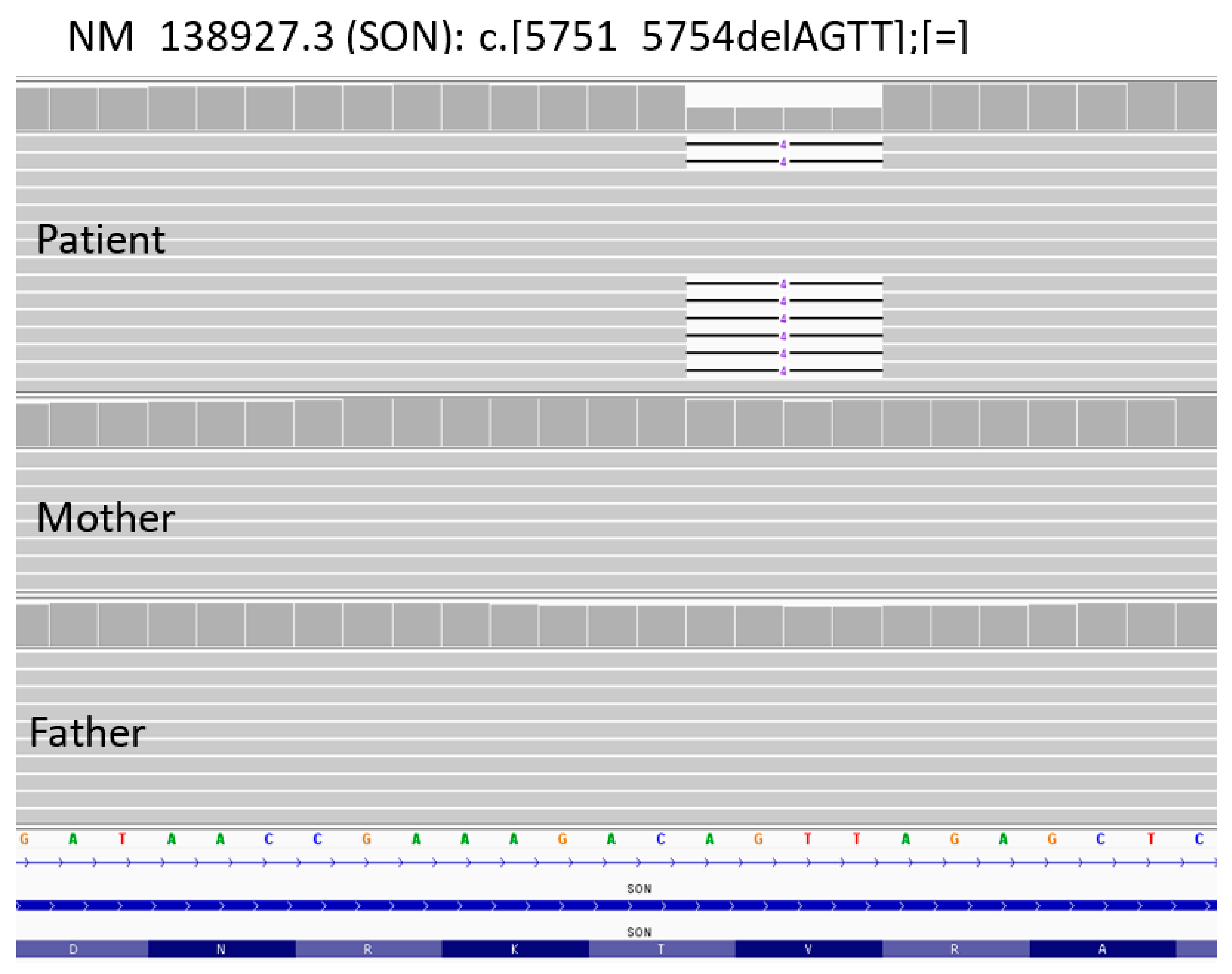
2.2. Whole-Exome Sequencing (WES)
3. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Zhu, X.; Petrovski, S.; Xie, P.; Ruzzo, E.K.; Lu, Y.-F.; McSweeney, K.M.; Ben-Zeev, B.; Nissenkorn, A.; Anikster, Y.; Oz-Levi, D.; et al. Whole-exome sequencing in undiagnosed genetic diseases: Interpreting 119 trios. Anesth. Analg. 2015, 17, 774–781. [Google Scholar] [CrossRef]
- Tokita, M.J.; Braxton, A.A.; Shao, Y.; Lewis, A.M.; Vincent, M.; Küry, S.; Besnard, T.; Isidor, B.; Latypova, X.; Bézieau, S.; et al. De Novo Truncating Variants in SON Cause Intellectual Disability, Congenital Malformations, and Failure to Thrive. Am. J. Hum. Genet. 2016, 99, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Shinde, D.N.; Reijnders, M.R.; Hauser, N.S.; Belmonte, R.L.; Wilson, G.R.; Bosch, D.G.; Bubulya, P.A.; Shashi, V.; Petrovski, S.; et al. De Novo Mutations in SON Disrupt RNA Splicing of Genes Essential for Brain Development and Metabolism, Causing an Intellectual-Disability Syndrome. Am. J. Hum. Genet. 2016, 99, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.-Y.; DeKelver, R.C.; Lo, M.-C.; Nguyen, T.A.; Matsuura, S.; Boyapati, A.; Pandit, S.; Fu, X.-D.; Zhang, D.-E. SON Controls Cell-Cycle Progression by Coordinated Regulation of RNA Splicing. Mol. Cell 2011, 42, 185–198. [Google Scholar] [CrossRef]
- Hickey, C.J.; Kim, J.-H.; Ahn, E.-Y.E. New Discoveries of Old SON: A Link Between RNA Splicing and Cancer. J. Cell. Biochem. 2014, 115, 224–231. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, E.Y.; Chitayat, D.; Stachura, D.L.; Schaper, J.; Lindstrom, K.; Jewett, T.; Wieczorek, D.; Draaisma, J.M.; Sinnema, M.; et al. SON haploinsufficiency causes impaired pre-mRNA splicing of CAKUT genes and heterogeneous renal phenotypes. Kidney Int. 2019, 95, 1494–1504. [Google Scholar] [CrossRef]
- Lu, X.; Göke, J.; Sachs, F.; Jacques, P.; Liang, H.; Feng, B.; Bourque, G.; Bubulya, P.A.; Ng, H.H. SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nature 2013, 15, 1141–1152. [Google Scholar] [CrossRef]
- Ueda, M.; Matsuki, T.; Fukada, M.; Eda, S.; Toya, A.; Iio, A.; Tabata, H.; Nakayama, A. Knockdown of Son, a mouse homologue of the ZTTK syndrome gene, causes neuronal migration defects and dendritic spine abnormalities. Mol. Brain 2020, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Dingemans, A.J.M.; Truijen, K.M.G.; Kim, J.-H.; Alaçam, Z.; Faivre, L.; Collins, K.M.; Gerkes, E.H.; van Haelst, M.; van de Laar, I.M.B.H.; Lindstrom, K.; et al. Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON. Eur. J. Hum. Genet. 2022, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Miura, K.; Uehara, T.; Mizuno, S.; Kosaki, K. Establishing SON in 21q22.11 as a cause a new syndromic form of intellectual disability: Possible contribution to Braddock-Carey syndrome phenotype. Am. J. Med. Genet. Part A 2016, 170, 2587–2590. [Google Scholar] [CrossRef]
- Halliday, B.; Baynam, G.; Ewans, L.; Greenhalgh, L.; Leventer, R.; Pilz, D.; Sachdev, R.; Scheffer, I.; Markie, D.; McGillivray, G.; et al. Distinctive Brain Malformations in Zhu-Tokita-Takenouchi-Kim Syndrome. Am. J. Neuroradiol. 2022, 43, 1660–1666. [Google Scholar] [CrossRef]
- Kushary, S.T.; Revah-Politi, A.; Barua, S.; Ganapathi, M.; Accogli, A.; Aggarwal, V.; Brunetti-Pierri, N.; Cappuccio, G.; Capra, V.; Fagerberg, C.R.; et al. ZTTK syndrome: Clinical and molecular findings of 15 cases and a review of the literature. Am. J. Med. Genet. Part A 2021, 185, 3740–3753. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, L.; Yu, Z.; Huang, H.; Yang, L. Clinical and genetic analysis of ZTTK syndrome caused by SON heterozygous mutation c.394C>T. Mol. Genet. Genom. Med. 2019, 7, e953. [Google Scholar] [CrossRef] [PubMed]
- Slezak, R.; Smigiel, R.; Rydzanicz, M.; Pollak, A.; Kosinska, J.; Stawinski, P.; Sasiadek, M.M.; Ploski, R. Phenotypic expansion in Zhu-Tokita-Takenouchi-Kim syndrome caused by de novo variants in the SON gene. Mol. Genet. Genom. Med. 2020, 8, e1432. [Google Scholar] [CrossRef] [PubMed]
- Quintana Castanedo, L.; Sanchez Orta, A.; Maseda Pedrero, R.; Santos Simarro, F.; Palomares Bralo, M.; Feito Rodriguez, M.; de Lucas Laguna, R. Skin and nails abnormalities in a patient with ZTTK syndrome and a de novo mutation in SON. Pediatr. Dermatol. 2020, 37, 517–519. [Google Scholar] [CrossRef]
- Tan, Y.; Duan, L.; Yang, K.; Liu, Q.; Wang, J.; Dong, Z.; Li, Z.; He, Y.; Yan, Y.; Lin, L. A novel frameshift variant in SON causes Zhu-Tokita-Takenouchi-Kim Syndrome. J. Clin. Lab. Anal. 2020, 34, e23326. [Google Scholar] [CrossRef]
- Yang, L.; Yang, F. A de novo heterozygous variant in the SON gene is associated with Zhu-Tokita-Takenouchi-Kim syndrome. Mol. Genet. Genomic Med. 2020, 8, e1496. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kaiserling, E.; Müller, H. Neoplasm of hepatic stellate cells (spongiotic pericytoma): A new tumor entity in human liver. Pathol.-Res. Pract. 2005, 201, 733–743. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Yu, J.; Lou, J.; Peng, K.; Zhao, H.; Chen, J. Clinical and Genetic Spectra of Inherited Liver Disease in Children in China. Front. Pediatr. 2021, 9, 631620. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-T.; Lo, W.-Y.; Wang, I.-H.; Lo, Y.-H.; Shiou, S.-R.; Lai, C.-K.; Ting, L.-P. Transcription Repression of Human Hepatitis B Virus Genes by Negative Regulatory Element-binding Protein/SON. J. Biol. Chem. 2001, 276, 24059–24067. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Findings | Current Case | Takenouchi et al. [10] | Kim et al. [3] | Tokita et al. [2] | Yang et al. [13] | Slezak et al. [14] | Quintana Castanedo et al. [15] | Tan et al. [16] | Yang et al. [17] | Dingemans et al. [9] |
|---|---|---|---|---|---|---|---|---|---|---|
| Chronic liver disease | + | |||||||||
| Developmental and/or intellectual disability | + | + | + | + | + | + | + | + | + | + |
| Seizures | + | + | + | + | + | + | + | |||
| Hypotonia | + | + | + | + | + | + | ||||
| Abnormal brain imaging | + | + | + | + | + | + | + | + | ||
| Facial dysmorphisms | + | + | + | + | + | + | + | + | + | |
| Growth delay | + | + | + | + | + | + | + | + | ||
| Eye/vision abnormality | + | + | + | + | + | |||||
| Congenital heart defects | + | + | + | + | + | + | ||||
| Feeding difficulties | + | + | + | |||||||
| Gastrointestinal malformation | + | + | + | |||||||
| Genitourinary anomalies | + | + | + | + | + | |||||
| Musculoskeletal abnormalities | + | + | + | + | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrobattista, A.; Della Volpe, L.; Francalanci, P.; Figà Talamanca, L.; Monti, L.; Lepri, F.R.; Basso, M.S.; Liccardo, D.; Della Corte, C.; Mosca, A.; et al. The Expanding Phenotype of ZTTK Syndrome Due to the Heterozygous Variant of SON Gene Focusing on Liver Involvement: Patient Report and Literature Review. Genes 2023, 14, 739. https://doi.org/10.3390/genes14030739
Pietrobattista A, Della Volpe L, Francalanci P, Figà Talamanca L, Monti L, Lepri FR, Basso MS, Liccardo D, Della Corte C, Mosca A, et al. The Expanding Phenotype of ZTTK Syndrome Due to the Heterozygous Variant of SON Gene Focusing on Liver Involvement: Patient Report and Literature Review. Genes. 2023; 14(3):739. https://doi.org/10.3390/genes14030739
Chicago/Turabian StylePietrobattista, Andrea, Luca Della Volpe, Paola Francalanci, Lorenzo Figà Talamanca, Lidia Monti, Francesca Romana Lepri, Maria Sole Basso, Daniela Liccardo, Claudia Della Corte, Antonella Mosca, and et al. 2023. "The Expanding Phenotype of ZTTK Syndrome Due to the Heterozygous Variant of SON Gene Focusing on Liver Involvement: Patient Report and Literature Review" Genes 14, no. 3: 739. https://doi.org/10.3390/genes14030739
APA StylePietrobattista, A., Della Volpe, L., Francalanci, P., Figà Talamanca, L., Monti, L., Lepri, F. R., Basso, M. S., Liccardo, D., Della Corte, C., Mosca, A., Alterio, T., Veraldi, S., Callea, F., Novelli, A., & Maggiore, G. (2023). The Expanding Phenotype of ZTTK Syndrome Due to the Heterozygous Variant of SON Gene Focusing on Liver Involvement: Patient Report and Literature Review. Genes, 14(3), 739. https://doi.org/10.3390/genes14030739