
The Variant p.Ala84Pro Is Causative of X-Linked Hypophosphatemic Rickets: Possible Relationship with Burosumab Swinging Response in Adults

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
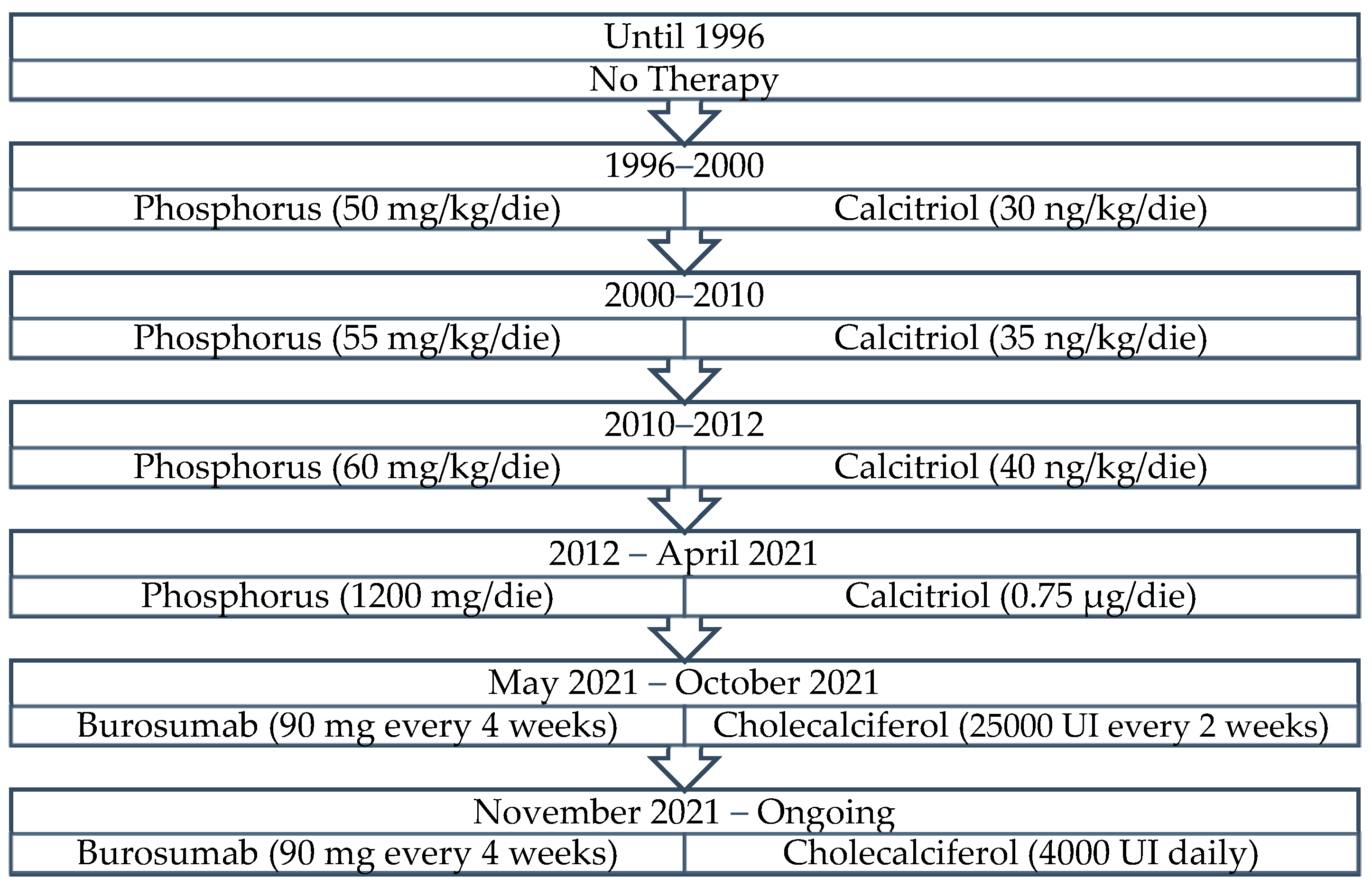
2. Patient and Methods
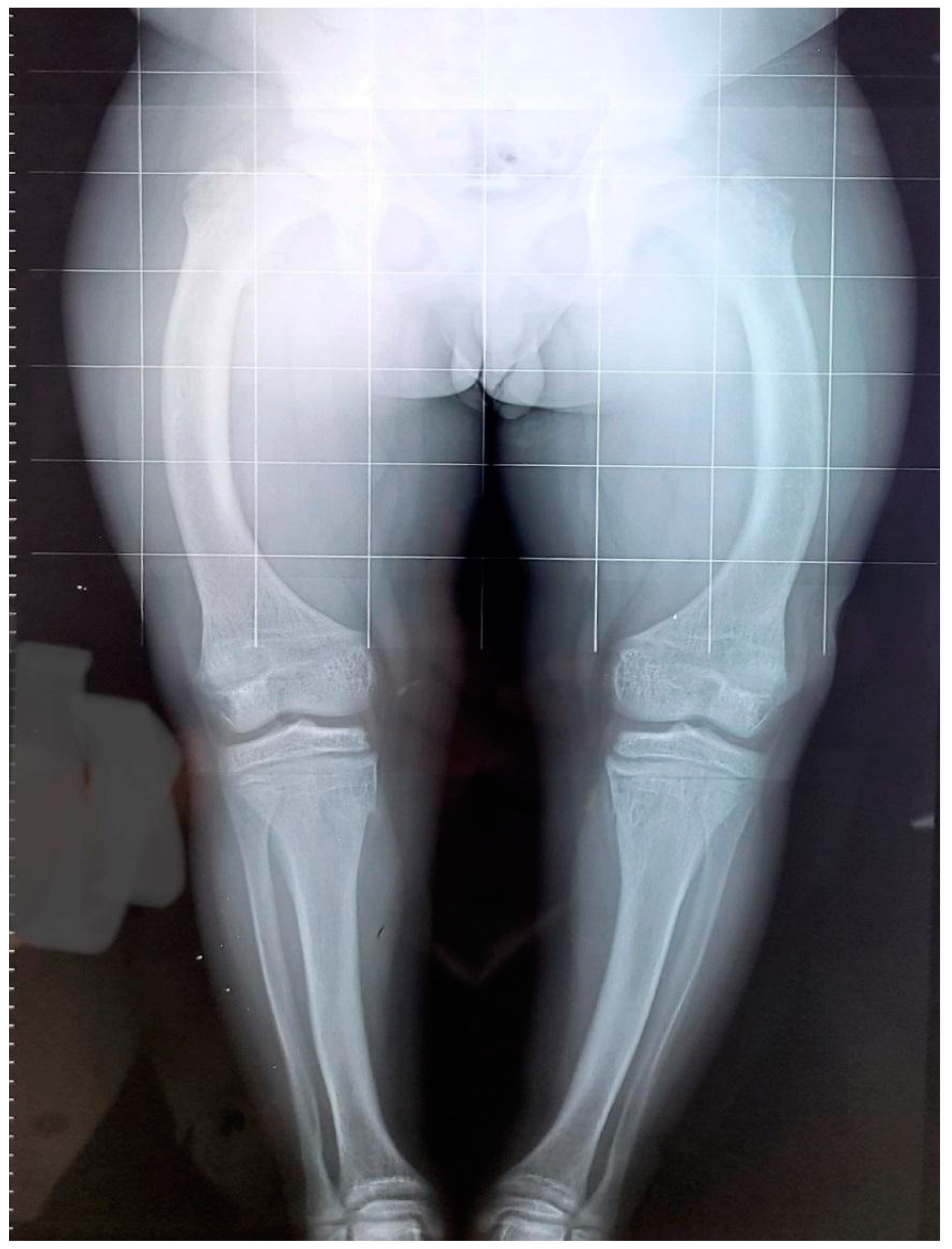
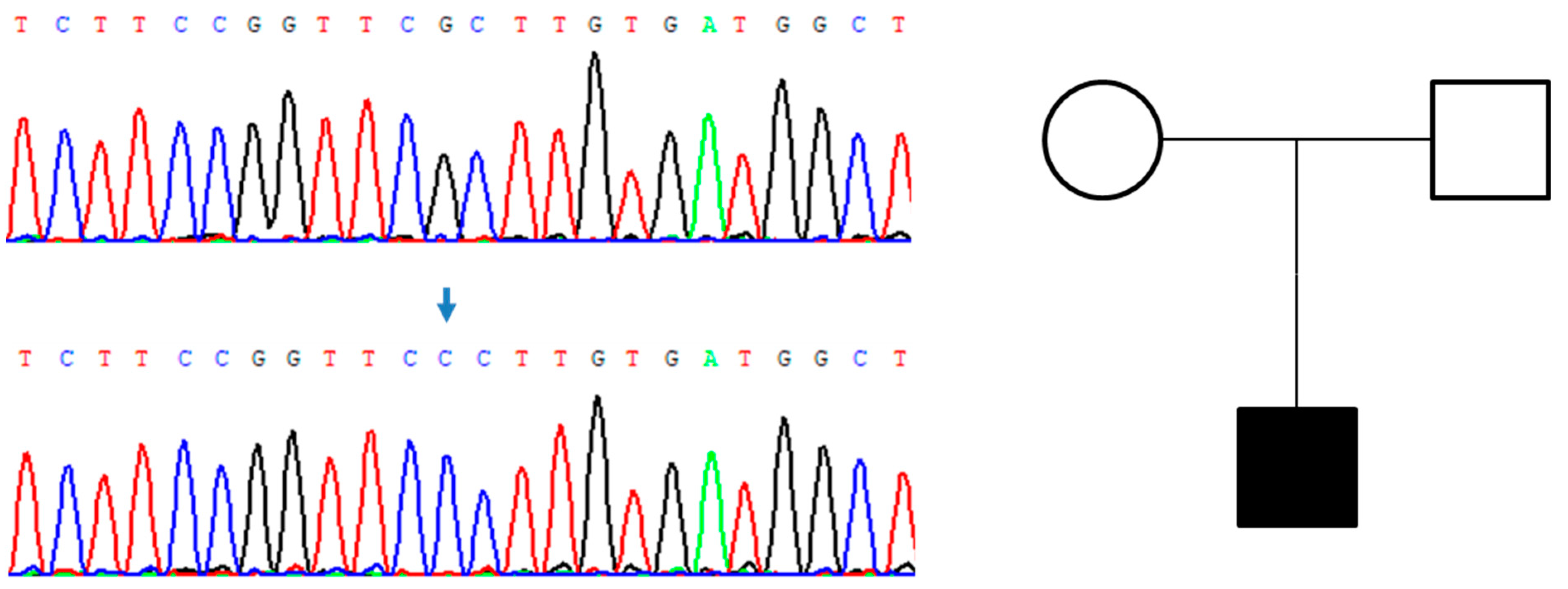
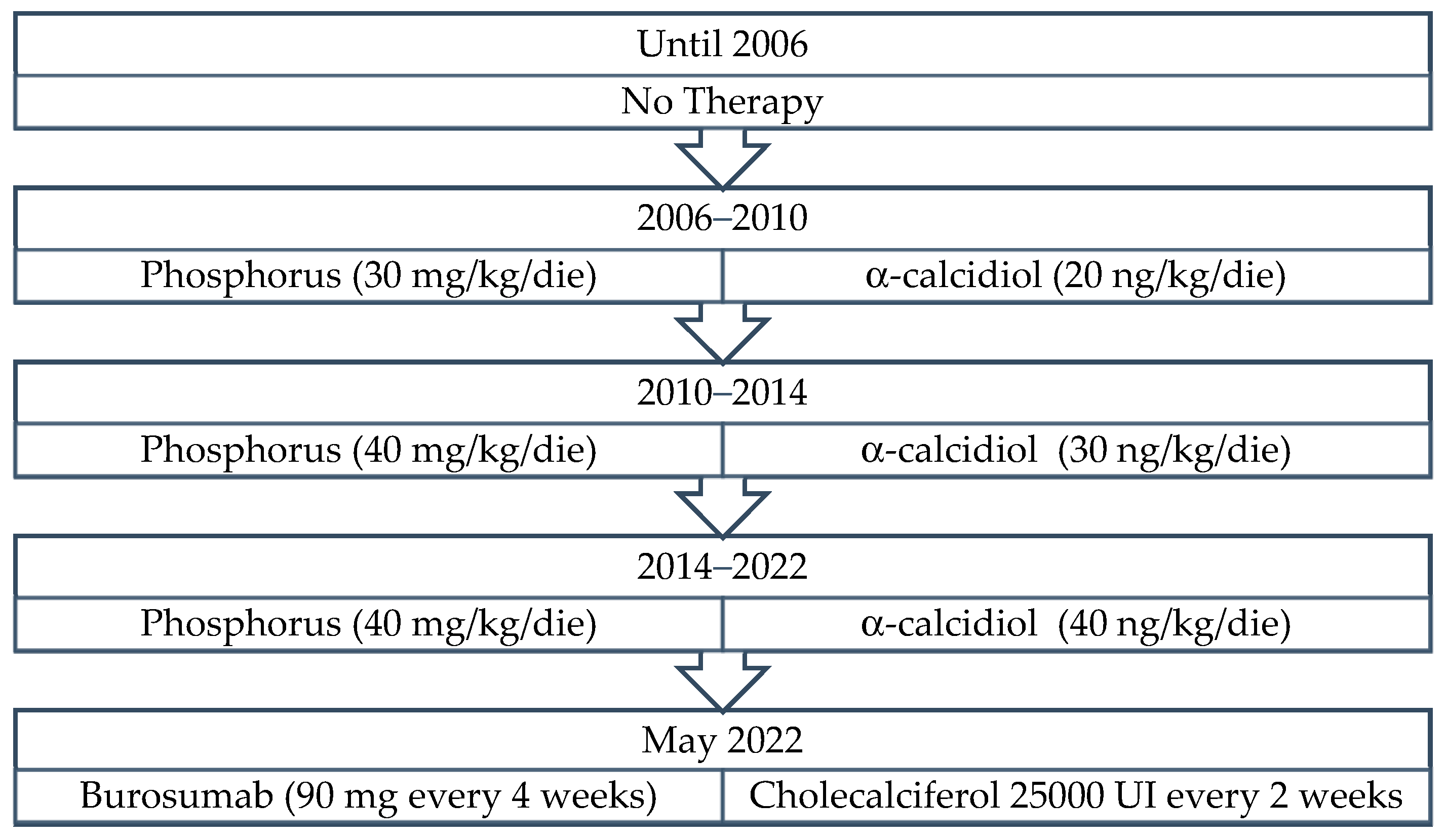
3. Results
3.1. Patient 1
3.2. Patient 2
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beck-Nielsen, S.S.; Mughal, Z.; Haffner, D.; Nilsson, O.; Levtchenko, E.; Ariceta, G.; de Lucas Collantes, C.; Schnabel, D.; Jandhyala, R.; Mäkitie, O. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J. Rare Dis. 2019, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Maio, P.; Mano, L.; Rocha, S.; Baptista, R.B.; Francisco, T.; Sousa, H.; Freixo, J.P.; Abranches, M. X-linked hypophosphatemic rickets: A new mutation. J. Bras. Nefrol. 2021, 43, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Haffner, D.; Emma, F.; Eastwood, D.M.; Duplan, M.B.; Bacchetta, J.; Schnabel, D.; Wicart, P.; Bockenhauer, D.; Santos, F.; Levtchenko, E.; et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat. Rev. Nephrol. 2019, 15, 435–455. [Google Scholar] [CrossRef]
- Beck-Nielsen, S.; Brusgaard, K.; Rasmussen, L.; Brixen, K.; Brock-Jacobsen, B.; Poulsen, M.; Vestergaard, P.; Ralston, S.; Albagha, O.; Poulsen, S.; et al. Phenotype Presentation of Hypophosphatemic Rickets in Adults. Calcif. Tissue Int. 2010, 87, 108–119. [Google Scholar] [CrossRef]
- Carpenter, T.O.; Imel, E.A.; Holm, I.A.; Jan de Beur, S.M.; Insogna, K.L. A clinician’s guide to X-linked hypophosphatemia. J. Bone Miner. Res. 2011, 26, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Linglart, A.; Biosse-Duplan, M.; Briot, K.; Chaussain, C.; Esterle, L.; Guillaume-Czitrom, S.; Kamenicky, P.; Nevoux, J.; Prié, D.; Rothenbuhler, A.; et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr. Connect. 2014, 3, R13–R30. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Whyte, M.P.; Imel, E.A.; Boot, A.M.; Högler, W.; Linglart, A.; Padidela, R.; van’t Hoff, W.; Mao, M.; Chen, C.-Y.; et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N. Engl. J. Med. 2018, 378, 1987–1998. [Google Scholar] [CrossRef]
- CHMP. Assessment Report CRYSVITA; European Medicines Agency: Amsterdam, The Netherlands, 2020; Available online: https://www.ema.europa.eu/en/documents/variation-report/crysvita-h-c-4275-ii-010-g-epar-assessment-report-variation_en.pdf (accessed on 25 October 2022).
- FDA. FDA Approves First Therapy for Rare Disease that Causes Low Phosphate Blood Levels, Bone Softening; FDA: Silver Spring, MD, USA, 2020.
- EMA. New medicine for rare bone disease. In Crysvita, a Medicine for the Treatment of X-Linked Hypophosphataemia, Recommended for Conditional Approval; EMA: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Li, S.S.; Gu, J.M.; Yu, W.J.; He, J.W.; Fu, W.Z.; Zhang, Z.L. Seven novel and six de novo PHEX gene mutations in patients with hypophosphatemic rickets. Int. J. Mol. Med. 2016, 38, 1703–1714. [Google Scholar] [CrossRef]
- Paduano, F.; Colao, E.; Fabiani, F.; Rocca, V.; Dinatolo, F.; Dattola, A.; D’Antona, L.; Amato, R.; Trapasso, F.; Baudi, F.; et al. Germline Testing in a Cohort of Patients at High Risk of Hereditary Cancer Predisposition Syndromes: First Two-Year Results from South Italy. Genes 2022, 13, 1286. [Google Scholar] [CrossRef]
- ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: Guidelines for the six-minute walk test. Am. J. Respir. Crit. Care Med. 2002, 166, 111–117. [Google Scholar] [CrossRef]
- Bohannon, R.W.; Peolsson, A.; Massy-Westropp, N.; Desrosiers, J.; Bear-Lehman, J. Reference values for adult grip strength measured with a Jamar dynamometer: A descriptive meta-analysis. Physiotherapy 2006, 92, 11–15. [Google Scholar] [CrossRef]
- Salaffi, F.; Leardini, G.; Canesi, B.; Mannoni, A.; Fioravanti, A.; Caporali, R.; Lapadula, G.; Punzi, L. Reliability and validity of the Western Ontario and McMaster Universities (WOMAC) Osteoarthritis Index in Italian patients with osteoarthritis of the knee. Osteoarthr. Cartil. 2003, 11, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Caraceni, A.; Mendoza, T.R.; Mencaglia, E.; Baratella, C.; Edwards, K.; Forjaz, M.J.; Martini, C.; Serlin, R.C.; de Conno, F.; Cleeland, C.S. A validation study of an Italian version of the Brief Pain Inventory (Breve Questionario per la Valutazione del Dolore). Pain 1996, 65, 87–92. [Google Scholar] [CrossRef] [PubMed]
- gnomAD, T.G.A.D. Available online: https://gnomad.broadinstitute.org/about (accessed on 28 July 2022).
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Mumm, S.; Huskey, M.; Cajic, A.; Wollberg, V.; Zhang, F.; Madson, K.L.; Wenkert, D.; McAlister, W.H.; Gottesman, G.S.; Whyte, M.P. PHEX 3′-UTR c.*231A>G near the polyadenylation signal is a relatively common, mild, American mutation that masquerades as sporadic or X-linked recessive hypophosphatemic rickets. J. Bone Miner. Res. 2015, 30, 137–143. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ohata, Y.; Ishihara, Y. Pathogenic Variants of the PHEX Gene. Endocrines 2022, 3, 498–511. [Google Scholar] [CrossRef]
- Lin, X.; Li, S.; Zhang, Z.; Yue, H. Clinical and Genetic Characteristics of 153 Chinese Patients With X-Linked Hypophosphatemia. Front. Cell Dev. Biol. 2021, 9, 617738. [Google Scholar] [CrossRef]
- Che, H.; Roux, C.; Etcheto, A.; Rothenbuhler, A.; Kamenicky, P.; Linglart, A.; Briot, K. Impaired quality of life in adults with X-linked hypophosphatemia and skeletal symptoms. Eur. J. Endocrinol. 2016, 174, 325–333. [Google Scholar] [CrossRef]
- Briot, K.; Portale, A.A.; Brandi, M.L.; Carpenter, T.O.; Cheong, H.I.; Cohen-Solal, M.; Crowley, R.K.; Eastell, R.; Imanishi, Y.; Ing, S.; et al. Burosumab treatment in adults with X-linked hypophosphataemia: 96-week patient-reported outcomes and ambulatory function from a randomised phase 3 trial and open-label extension. RMD Open 2021, 7, e001714. [Google Scholar] [CrossRef]
- Ito, N. Adult Presentation of X-Linked Hypophosphatemia. Endocrines 2022, 3, 375–390. [Google Scholar] [CrossRef]
- Ito, N.; Prideaux, M.; Wijenayaka, A.R.; Yang, D.; Ormsby, R.T.; Bonewald, L.F.; Atkins, G.J. Sclerostin Directly Stimulates Osteocyte Synthesis of Fibroblast Growth Factor-23. Calcif. Tissue Int. 2021, 109, 66–76. [Google Scholar] [CrossRef]
- Jurca, C.; Iuhas, O.; Kozma, K.; Petchesi, C.D.; Zaha, D.; Bembea, M.; Jurca, S.; Paul, C.; Jurca, C. Effects of Burosumab Treatment on Two Siblings with X-Linked Hypophosphatemia. Case Report and Literature Review. Genes 2022, 13, 1392. [Google Scholar] [CrossRef]
- Xiao, L.; Homer-Bouthiette, C.; Hurley, M.M. FGF23 Neutralizing Antibody Partially Improves Bone Mineralization Defect of HMWFGF2 Isoforms in Transgenic Female Mice. J. Bone Miner. Res. 2018, 33, 1347–1361. [Google Scholar] [CrossRef]
- Riminucci, M.; Colangelo, L.; Ungari, C.; Cassoni, A.; Minisola, S.; Corsi, A. Naso-Ethmoidal Phosphaturic Mesenchymal Tumor: A Rare Tumor Site for an Uncommon Paraneoplastic Syndrome. Ear Nose Throat J. 2022, 101, 289–291. [Google Scholar] [CrossRef]
- Goljanek-Whysall, K.; Tridimas, A.; McCormick, R.; Russell, N.-J.; Sloman, M.; Sorani, A.; Fraser, W.D.; Hannan, F.M. Identification of a novel loss-of-function PHEX mutation, Ala720Ser, in a sporadic case of adult-onset hypophosphatemic osteomalacia. Bone 2018, 106, 30–34. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Z.; Sun, Y.; Xu, L.; JiaJue, R.; Cui, L.; Pang, Q.; Jiang, Y.; Li, M.; Wang, O.; et al. Clinical and genetic analysis in a large Chinese cohort of patients with X-linked hypophosphatemia. Bone 2019, 121, 212–220. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
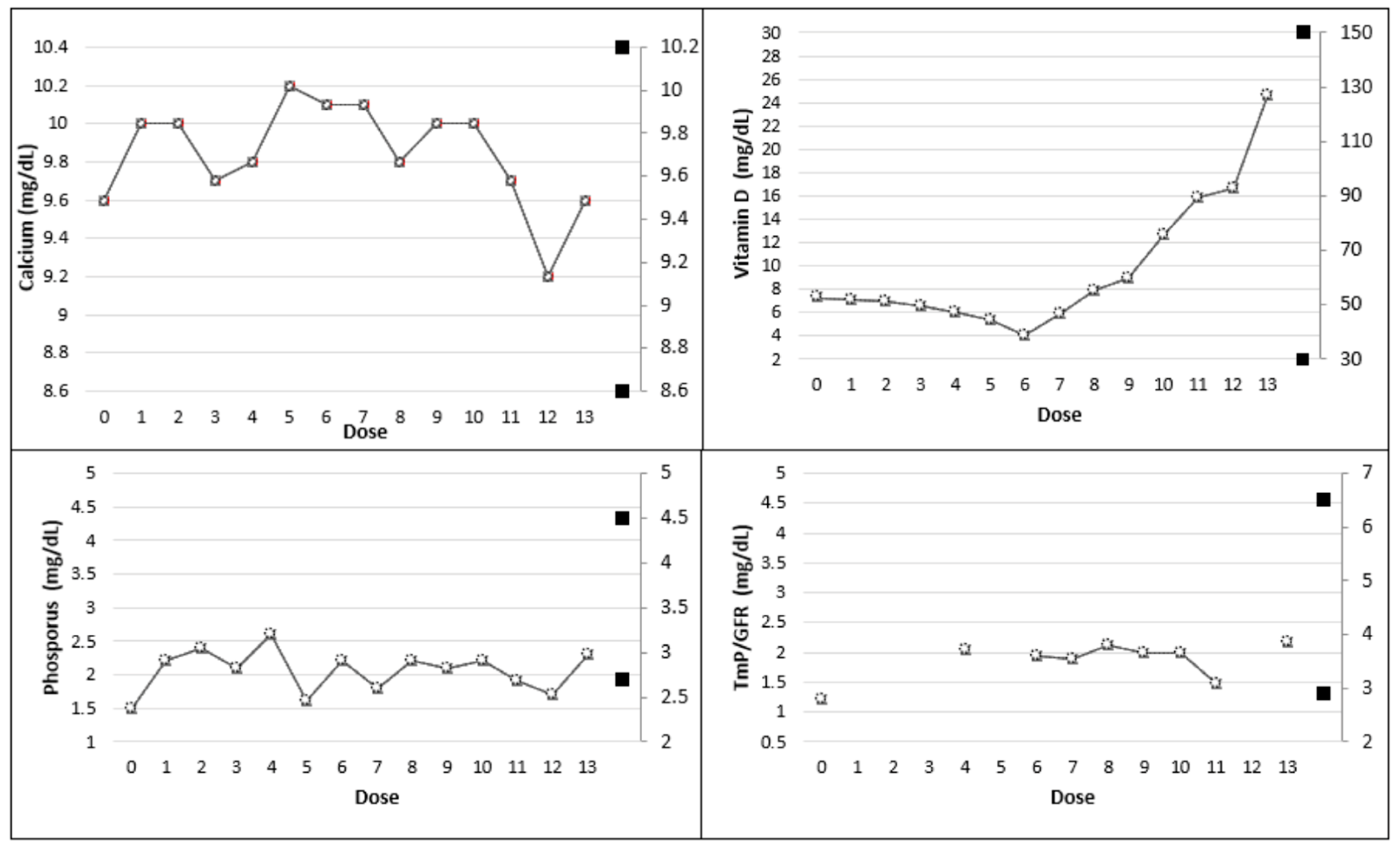
| Dose | |||||||
|---|---|---|---|---|---|---|---|
| Baseline—T0 (No therapy) | 1 | 2 | 3 | 4 | 5 | 6 | |
| Calcium (8.6–10.2 mg/dL) | 9.6 | 10.0 | 10.0 | 9.7 | 9.8 | 10.2 | 10.1 |
| Ionized Calcium (4.7–5.2 mg/dL) | 4.8 | 4.9 | 5.0 | 4.6 | 4.7 | 5.1 | 5.0 |
| Phosphorus (2.7–4.5 mg/dL) | 1.5 | 2.2 | 2.4 | 2.1 | 2.6 | 1.6 | 2.2 |
| Vitamin D (30–150 ng/mL) | 7.3 | 7.1 | 7.0 | 6.6 | 6.1 | 5.4 | <4.0 |
| 1,25-dihydroxyvitamin D (19.9–79.3 pg/dL) | 32.1 | 31.4 | 28.6 | 25.3 | 24.2 | 23.1 | 22.8 |
| PTH (14–72 pg/mL) | 274.5 | 245 | 239.7 | 189 | 148.4 | 274 | 276 |
| BAP (5.50–24.6 ug/L) | 27.7 | - | - | - | 47.5 | - | - |
| CTX (<0.58 ng/mL) | 2.91 | 2.42 | 1.92 | 1.82 | 1.89 | 1.57 | 1.44 |
| TmP/GFR (2.9–6.5 mg/dL) | 1.20 | - | - | - | 2.04 | - | 1.94 |
| Urinary Calcium (24 h) (100–321 mg/24 h) | 37 | 98 | 110 | 127 | 115 | 100 | 78 |
| Dose | |||||||
|---|---|---|---|---|---|---|---|
| 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
| Calcium (8.6–10.2 mg/dL) | 10.1 | 9.8 | 10 | 10 | 9.7 | 9.2 | 9.6 |
| Ionized Calcium (4.7–5.2 mg/dL) | 5.0 | 4.9 | 5.0 | 5.0 | 4.9 | 4.6 | 4.8 |
| Phosphorus (2.7–4.5 mg/dL) | 1.8 | 2.2 | 2.1 | 2.2 | 1.9 | 1.7 | 2.3 |
| Vitamin D (30–150 mg/dL) | 5.9 | 7.9 | 9 | 12.7 | 15.9 | 16.6 | 24.6 |
| 1,25-dihydroxyvitamin D (19.9–79.3 pg/dL) | 54.1 | 48.8 | 52.9 | 54.3 | 48.2 | 44.1 | 38.6 |
| PTH (14–72 pg/mL) | 152.2 | 142.4 | 191.2 | 174.5 | 142.8 | 181.6 | 188 |
| BAP (5.50–24.6 ug/L) | 52 | 53 | 52.4 | 57.4 | - | 69.4 | - |
| CTX (<0.58 ng/mL) | 1.42 | 1.69 | 1.89 | 1.88 | 1.82 | 2.11 | 2.13 |
| TmP/GFR (2.9–6.5 mg/dL) | 1.88 | 2.11 | 1.98 | 1.99 | 1.48 | - | 2.18 |
| Urinary Calcium (24 h) (100–321 mg/24 h) | 130 | 139 | 142 | 127 | 125 | 128 | 123 |
| Variable | T0 | T1 | T2 |
|---|---|---|---|
| 6MWT (m) | 293 | 314(+7) * | 345(+18) * |
| HGS-R (kgf) | 38 | 40 (+5) * | 42 (+11) * |
| Stiffness (score) | 3/8 | 3/8 (0) * | 2/8 (−33) * |
| Physical function (score) | 27/68 | 27/68 (0) * | 19/68 (−30) * |
| Worst pain | Severe | Moderate | Mild |
| Dose | |||||||
|---|---|---|---|---|---|---|---|
| Baseline—T0 (No therapy) | 1 | 2 | 3 | 4 | 5 | 6 | |
| Calcium (8.6–10.2 mg/dL) | 9.3 | 9.6 | 9.5 | 9.5 | 9.2 | 9.1 | 9.3 |
| Ionized calcium (4.7–5.2 mg/dL) | 4.6 | 4.7 | 4.8 | 4.8 | - | 4.9 | 4.6 |
| Phosphorus (2.7–4.5 mg/dL) | 1.6 | 2.4 | 2.8 | 2.82 | 2.8 | 2.6 | 2.6 |
| Vitamin D (30–150 ng/mL) | 19.9 | 17.1 | 19.3 | 21.3 | 25.6 | 33.5 | 19.9 |
| 1,25-dihydroxyvitamin D (19.9–79.3 pg/dL) | 27.1 | 66.8 | - | 60.2 | 54.3 | - | 18.1 |
| PTH (14–72 pg/mL) | 109.7 | 128.9 | 121.8 | 105 | 103.2 | 100.5 | 109.7 |
| BAP (5.50–24.6 ug/L) | 67.7 | 52.2 | 54.8 | 49.6 | 48.8 | - | 67.7 |
| CTX (<0.58 ng/mL) | 1.95 | 4.36 | 4.31 | 3.43 | 2.94 | 1.84 | - |
| TmP/GFR (2.9–6.5 mg/dL) | 1.89 | 2.4 | - | 2.8 | 2.7 | 2.6 | 2.8 |
| Urinary calcium (24 h) (100–321 mg/24 h) | 115 | 195 | 102 | 127 | - | 137 | 131 |
| Variable | T0 | T1 | T2 |
|---|---|---|---|
| 6 MWT (m) | 404 | 439 (+9) * | 465 (+15) * |
| HGS-R (kgf) | 40 | 43 (+8) * | 46 (+15) * |
| Stiffness (score) | 2/8 | 2/8 (0%) * | 1/8 (+50) * |
| Physical function (score) | 24/68 | 20/68 (−17) * | 15/68 (−38) * |
| Worst pain | Severe | Moderate | Mild |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagari, M.C.; Chiarello, P.; Iuliano, S.; D’Antona, L.; Rocca, V.; Colao, E.; Perrotti, N.; Greco, F.; Iuliano, R.; Aversa, A. The Variant p.Ala84Pro Is Causative of X-Linked Hypophosphatemic Rickets: Possible Relationship with Burosumab Swinging Response in Adults. Genes 2023, 14, 80. https://doi.org/10.3390/genes14010080
Zagari MC, Chiarello P, Iuliano S, D’Antona L, Rocca V, Colao E, Perrotti N, Greco F, Iuliano R, Aversa A. The Variant p.Ala84Pro Is Causative of X-Linked Hypophosphatemic Rickets: Possible Relationship with Burosumab Swinging Response in Adults. Genes. 2023; 14(1):80. https://doi.org/10.3390/genes14010080
Chicago/Turabian StyleZagari, Maria Carmela, Paola Chiarello, Stefano Iuliano, Lucia D’Antona, Valentina Rocca, Emma Colao, Nicola Perrotti, Francesca Greco, Rodolfo Iuliano, and Antonio Aversa. 2023. "The Variant p.Ala84Pro Is Causative of X-Linked Hypophosphatemic Rickets: Possible Relationship with Burosumab Swinging Response in Adults" Genes 14, no. 1: 80. https://doi.org/10.3390/genes14010080
APA StyleZagari, M. C., Chiarello, P., Iuliano, S., D’Antona, L., Rocca, V., Colao, E., Perrotti, N., Greco, F., Iuliano, R., & Aversa, A. (2023). The Variant p.Ala84Pro Is Causative of X-Linked Hypophosphatemic Rickets: Possible Relationship with Burosumab Swinging Response in Adults. Genes, 14(1), 80. https://doi.org/10.3390/genes14010080