
An Updated PAH Mutational Spectrum of Phenylketonuria in Mexican Patients Attending a Single Center: Biochemical, Clinical-Genotyping Correlations

 , , , , , , , , and
, , , , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Subjects
2.3. Amino Acid Quantification
2.4. Genotype Analysis
2.5. Protein Modeling and Mutagenesis in Silico
2.6. Genotype–Phenotype Correlation with GPV
2.7. Genotype–Phenotype Correlation with Identical Genotypes
2.8. Theoretical BH4 Responsiveness and Recommendation to Test
2.9. Statistical Analysis
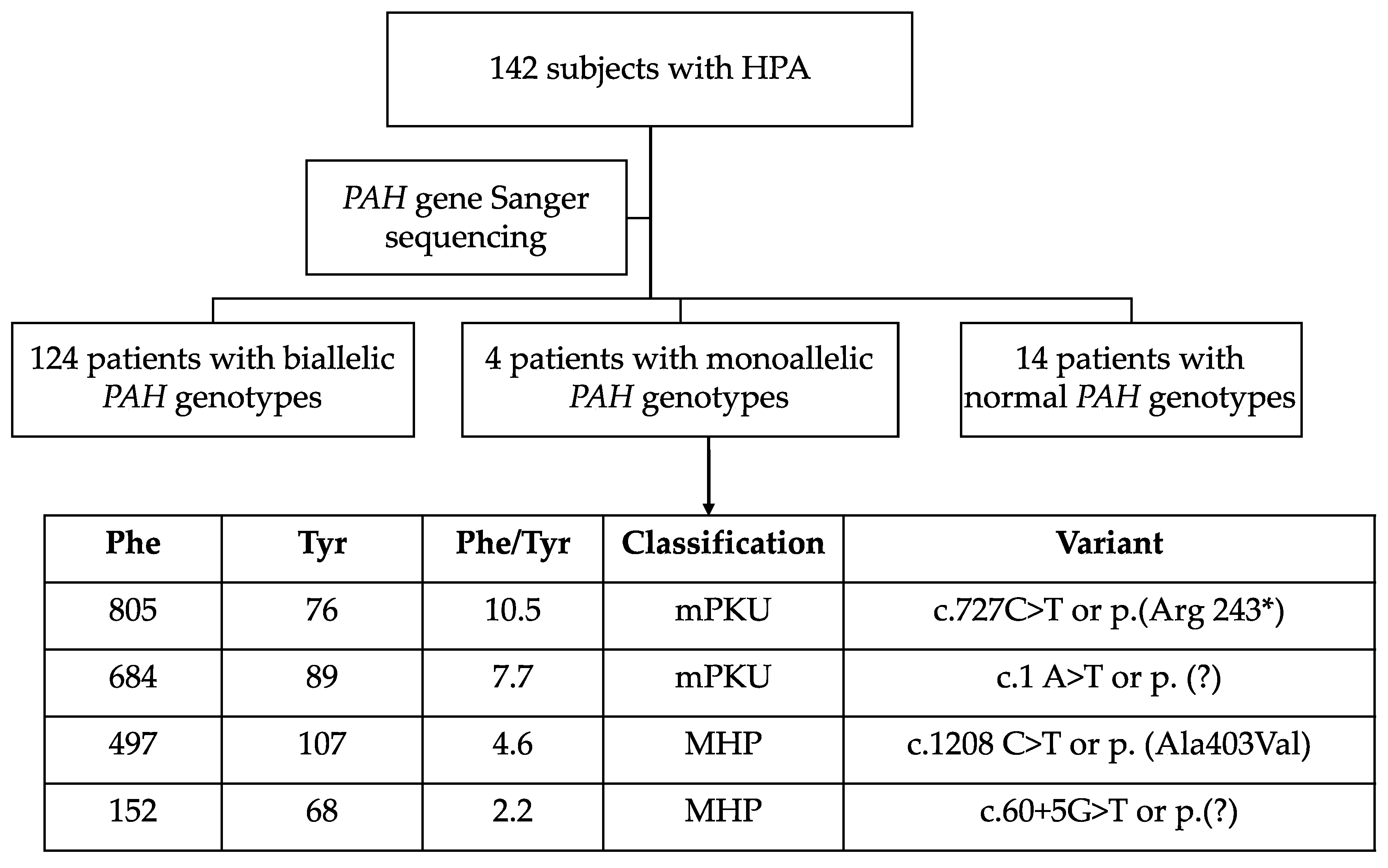
3. Results
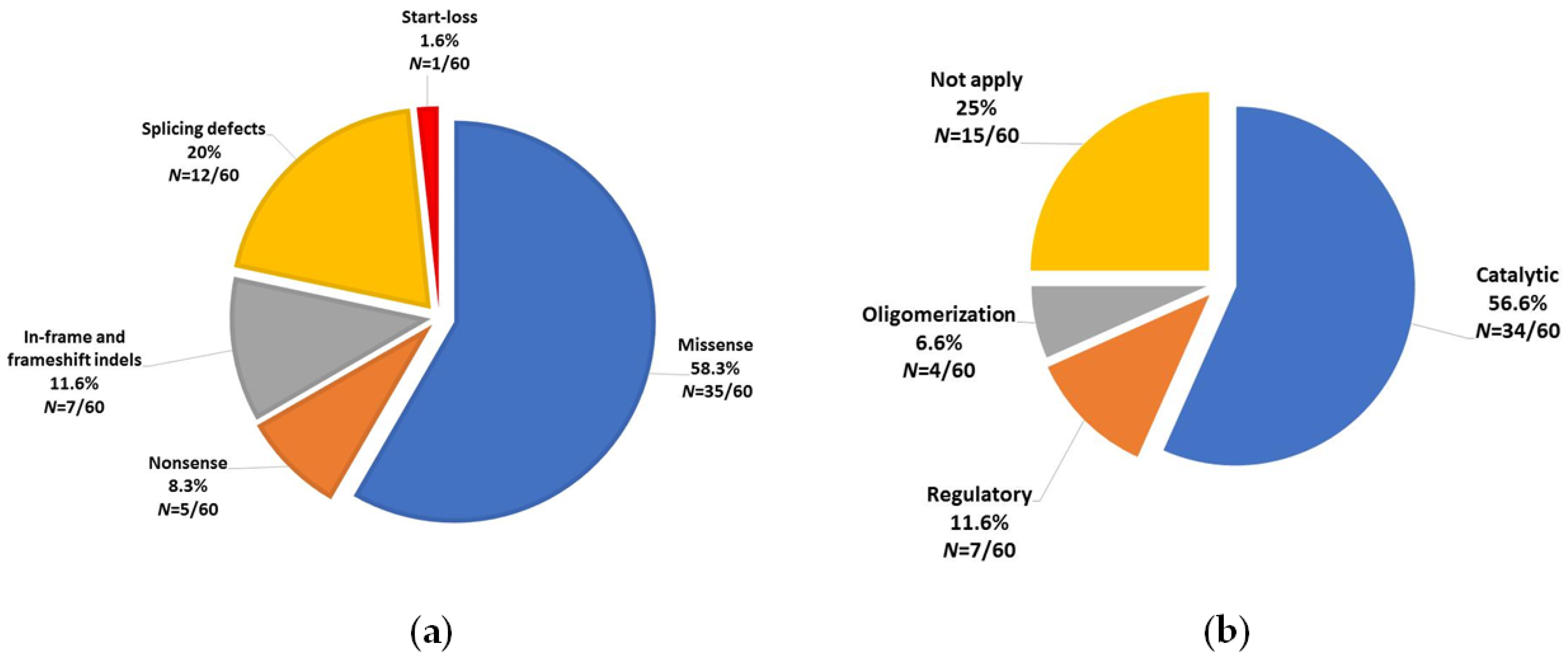
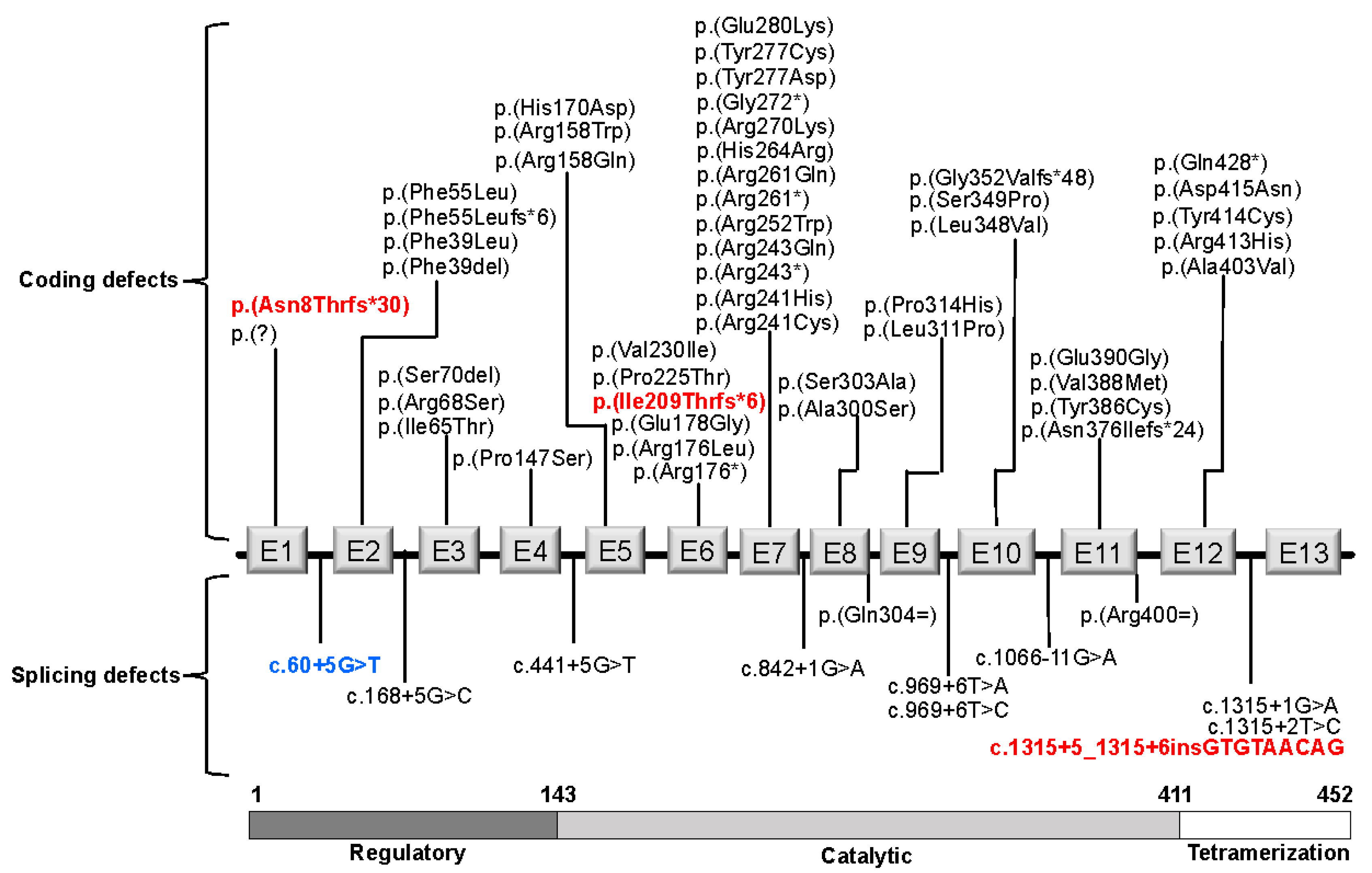
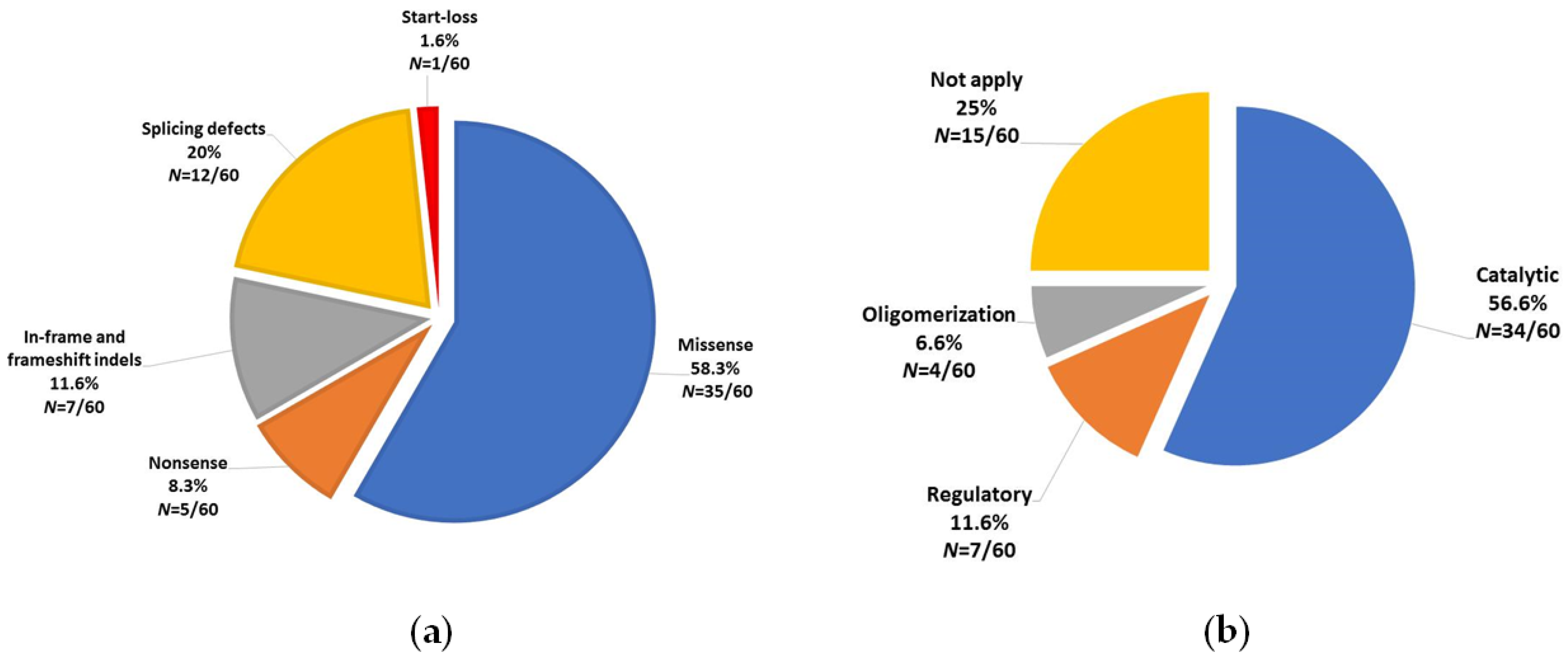
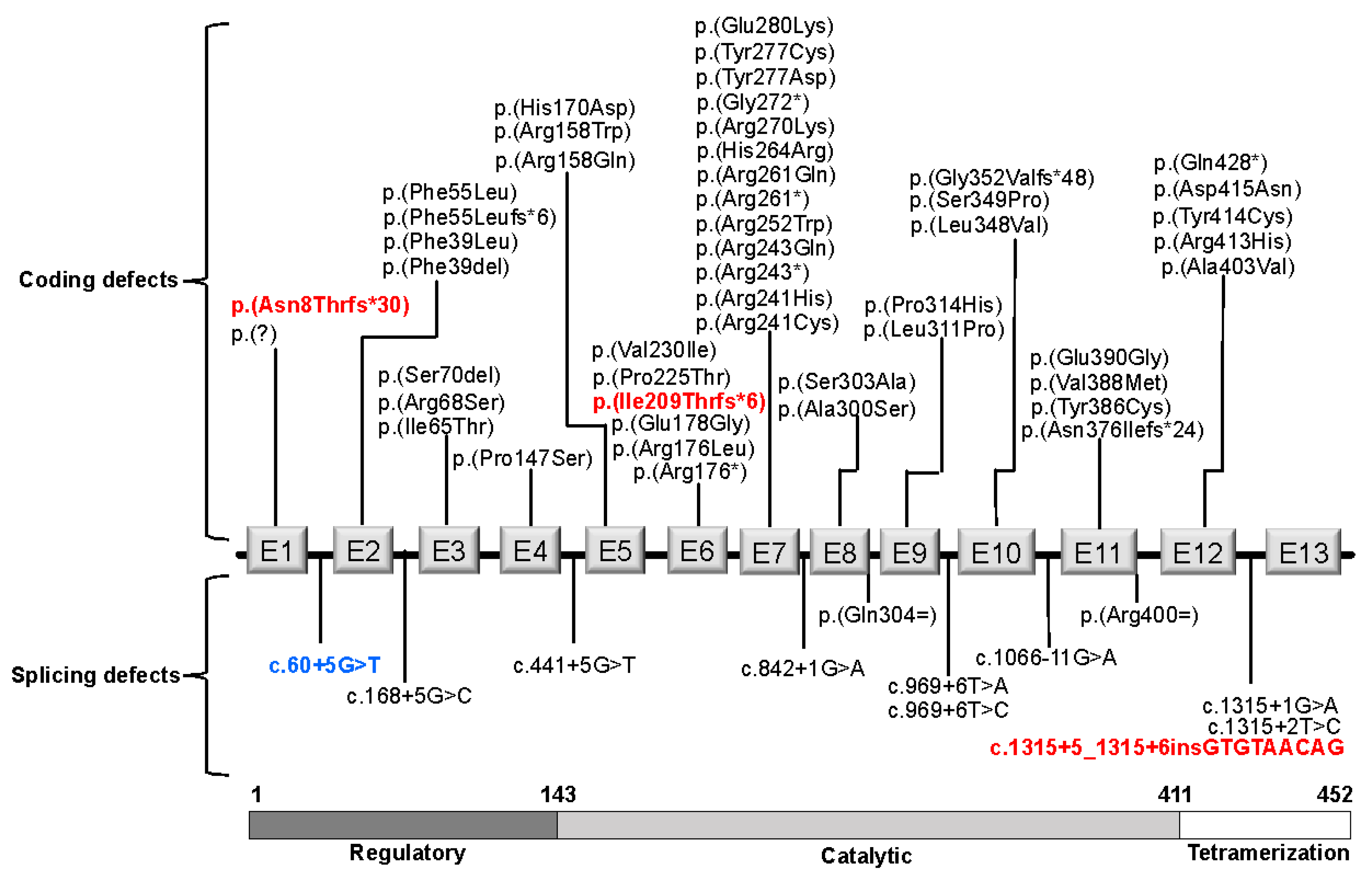
3.1. PAH Variants
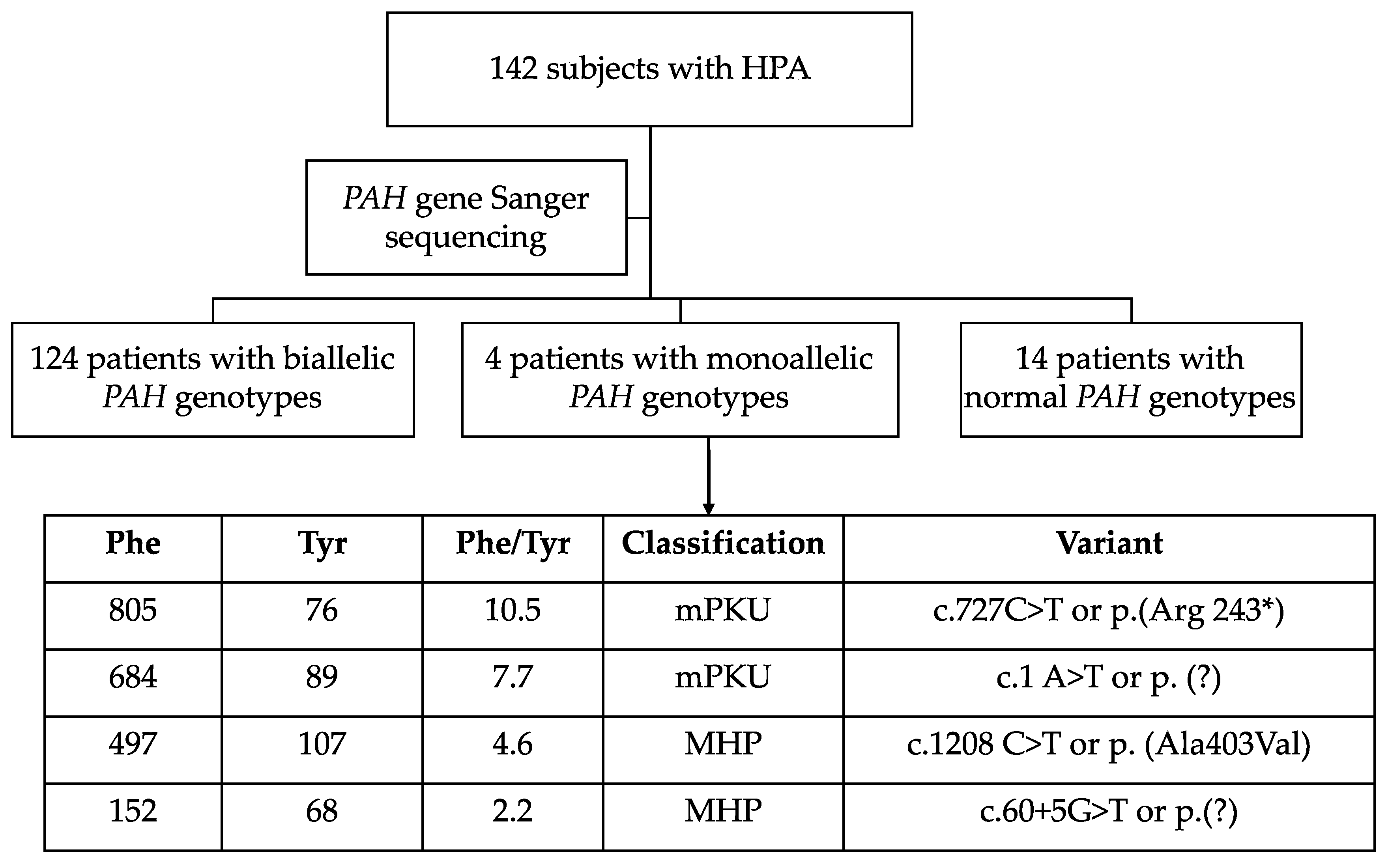
3.2. Genotypes
3.3. Genotype/Phenotype Concordance with GPV
3.4. Genotype/Phenotype Concordance with Identical Genotypes Reported in BIOPKUdb
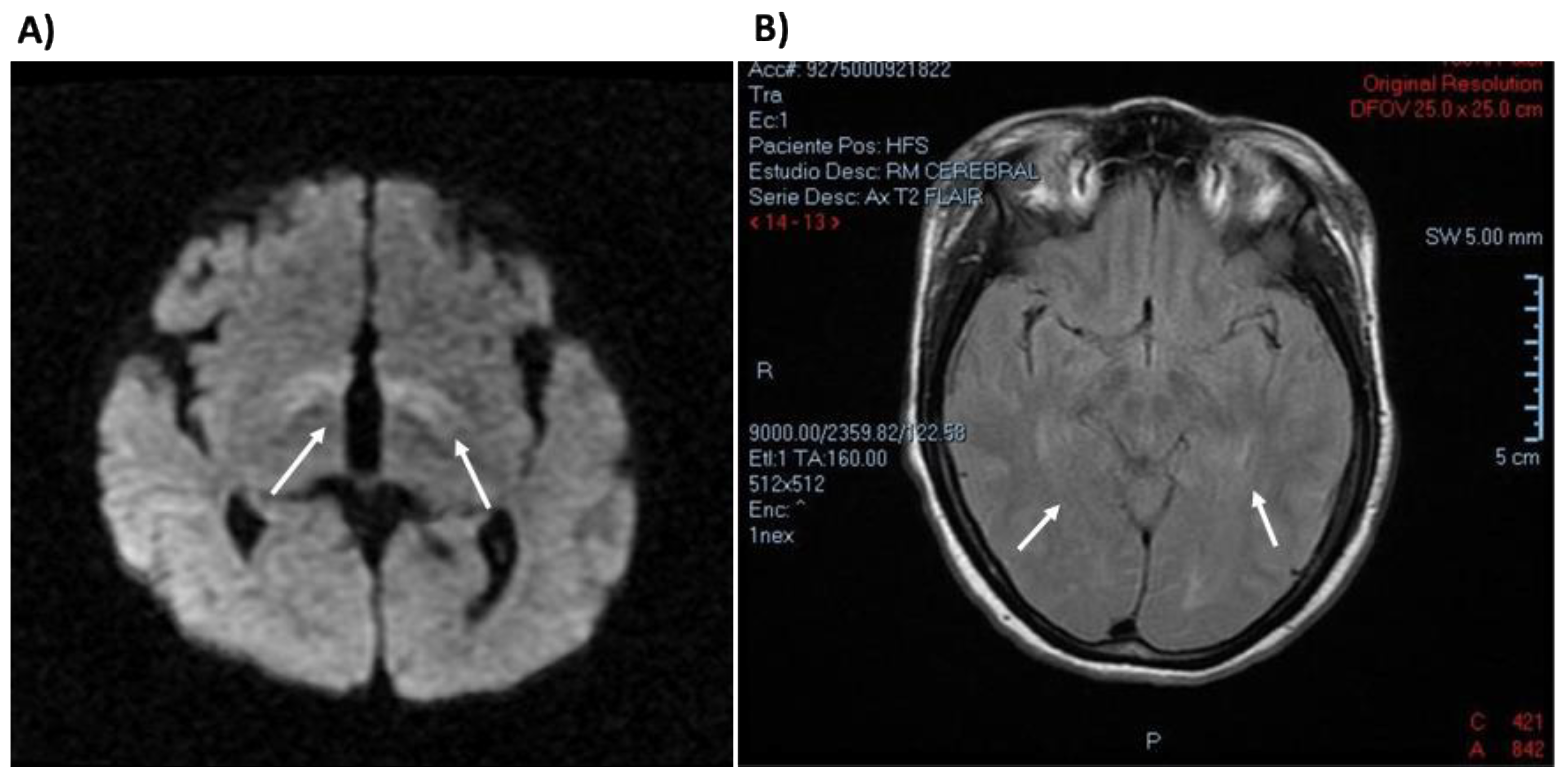
3.5. Clinical Description of Patients Bearing PAH Genotypes Containing c. 60 + 5G > T Variant
3.6. Novel PAH Variants and Description of Resulting Phenotypes
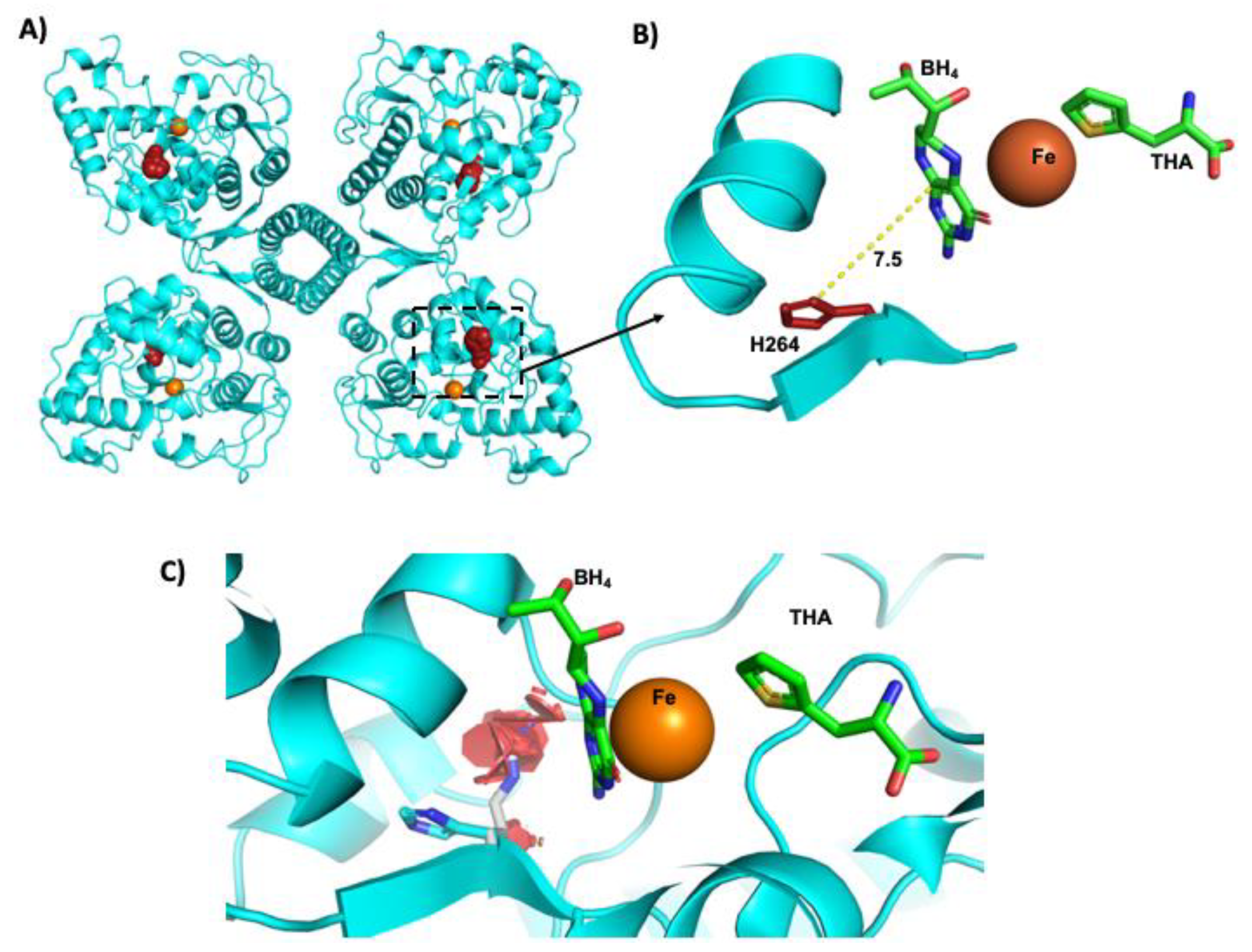
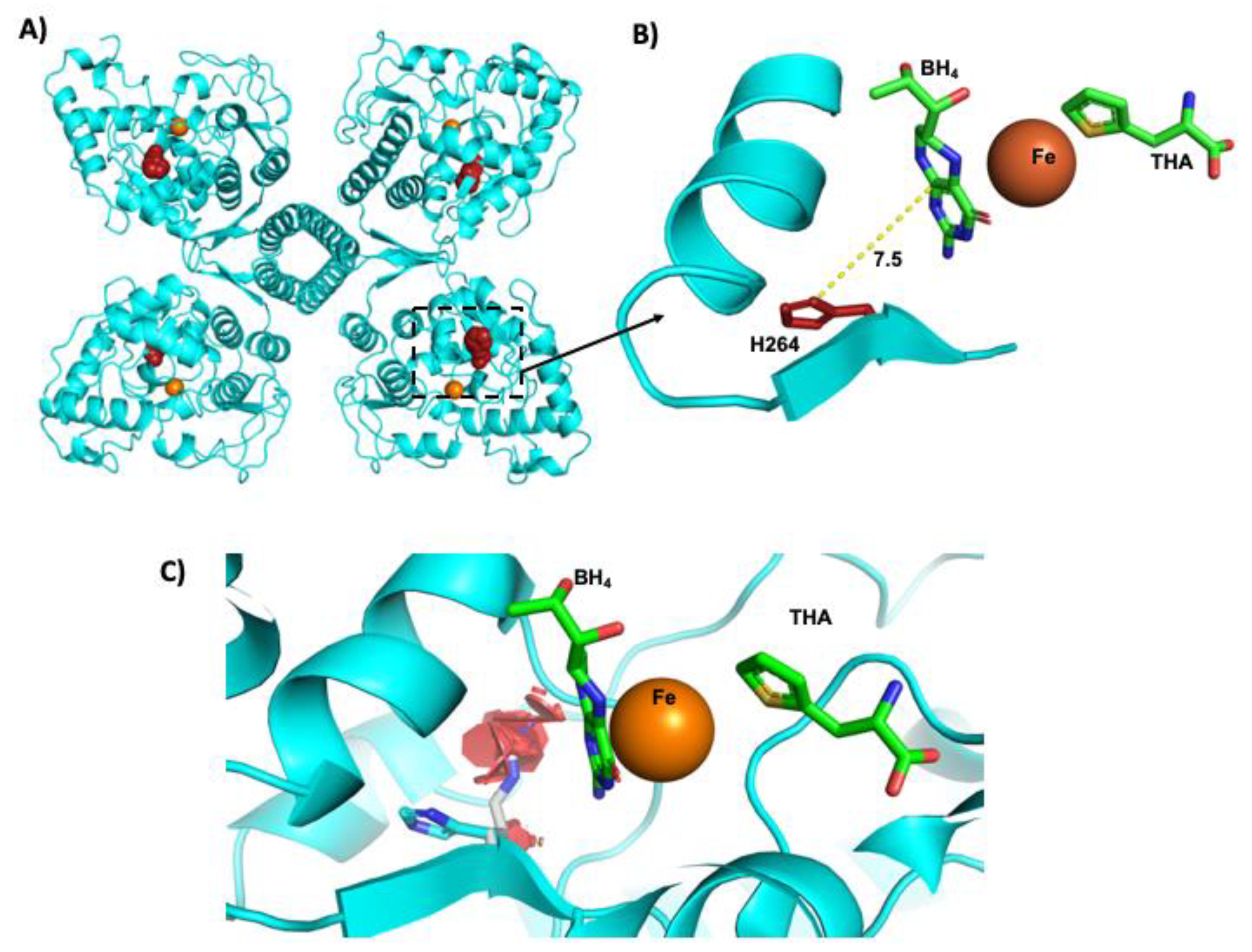
3.7. Protein in Silico Modeling of the p. (His264Arg) Variant
3.8. Theoretical BH4 Responsiveness
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 1–19. [Google Scholar] [CrossRef]
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The genetic landscape and epidemiology of phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.V.; González, I.I.; Pérez, L.D.A.H.; Caamal-Parra, G.; Martínez, L.B.; Flores, E.P.G. Epidemiología de la fenilcetonuria obtenida mediante tamiz neonatal. Acta Pediátrica Méxic 2018, 39, 25–34S. [Google Scholar] [CrossRef]
- Bortoluzzi, V.T.; Dutra Filho, C.S.; Wannmacher, C.M.D. Oxidative stress in phenylketonuria—Evidence from human studies and animal models, and possible implications for redox signaling. Metab. Brain Dis. 2021, 36, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Vela-Amieva, M.; Abreu-Gonzalez, M.; Gonzalez-del Angel, A.; Ibarra-Gonzalez, I.; Fernandez-Lainez, C.; Barrientos-Rios, R.; Monroy-Santoyo, S.; Guillén-López, S.; Alcántara-Ortigoza, M. Phenylalanine hydroxylase deficiency in Mexico: Genotype–phenotype correlations, BH4 responsiveness and evidence of a founder effect. Clin. Genet. 2015, 88, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Camp, K.M.; Parisi, M.A.; Acosta, P.B.; Berry, G.T.; Bilder, D.A.; Blau, N.; Bodamer, O.A.; Brosco, J.P.; Brown, C.S.; Burlina, A.B.; et al. Phenylketonuria scientific review conference: State of the science and future research needs. Mol. Genet. Metab. 2014, 112, 87–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wegberg, A.M.J.; Macdonald, A.; Ahring, K.; BãLanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 1–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo-Estavillo, C.; Vázquez-Avelar, S.; García-Ortíz, J.E.; Real-Guerrero, J.; Belmont-Martínez, L.; Escoto-Delgadillo, M.; Hernández-Orozco, A.A.; Torres-Mendoza, B.M. High frequency of severe phenylketonuria in Jalisco, Mexico. Int. J. Hum. Genet. 2018, 18, 1–6. [Google Scholar]
- Qureshi, G.A.; Fohlin, L.; Bergström, J. Application of high-performance liquid chromatography to the determination of free amino acids in physiological fluids. J. Chromatogr. A 1984, 297, 91–100. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Andersen, O.A.; Flatmark, T.; Hough, E. Crystal structure of the ternary complex of the catalytic domain of human phenylalanine hydroxylase with tetrahydrobiopterin and 3-(2-Thienyl)-l-alanine, and its implications for the mechanism of catalysis and substrate activation. J. Mol. Biol. 2002, 320, 1095–1108. [Google Scholar] [CrossRef]
- Garbade, S.F.; Shen, N.; Himmelreich, N.; Haas, R.; Trefz, F.K.; Hoffmann, G.F.; Burgard, P.; Blau, N. Allelic phenotype values: A model for genotype-based phenotype prediction in phenylketonuria. Genet. Med. 2018, 21, 580–590. [Google Scholar] [CrossRef]
- Van Spronsen, F.J.; van Wegberg, A.M.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017, 5, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Muntau, A.C.; Adams, D.J.; Bélanger-Quintana, A.; Bushueva, T.V.; Cerone, R.; Chien, Y.-H.; Chiesa, A.; Coşkun, T.; Heras, J.D.L.; Feillet, F.; et al. International best practice for the evaluation of responsiveness to sapropterin dihydrochloride in patients with phenylketonuria. Mol. Genet. Metab. 2019, 127, 1–11. [Google Scholar] [CrossRef]
- Buratti, E.; Chivers, M.; Královičová, J.; Romano, M.; Baralle, M.; Krainer, A.; Vořechovský, I. Aberrant 5′ splice sites in human disease genes: Mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007, 35, 4250–4263. [Google Scholar] [CrossRef]
- Foreman, P.K.; Margulis, A.V.; Alexander, K.; Shediac, R.; Calingaert, B.; Harding, A.; Pladevall-Vila, M.; Landis, S. Birth prevalence of phenylalanine hydroxylase deficiency: A systematic literature review and meta-analysis. Orphanet J. Rare Dis. 2021, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Enacán, R.E.; Miñana, M.N.; Fernandez, L.; Valle, M.G.; Salerno, M.; Fraga, C.I.; Santos-Simarro, F.; Prieto, L.; Lapunzina, P.; Specola, N.; et al. Phenylalanine hydroxylase (PAH) genotyping in PKU argentine patients. J. Inborn Errors Metab. Screen. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Acosta, A.; Silva Jr, W.; Carvalho, T.; Gomes, M.; Zago, M. Mutations of the phenylalanine hydroxylase (PAH) gene in Brazilian patients with phenylketonuria. Hum. Mutat. 2001, 17, 122–130. [Google Scholar] [CrossRef]
- García-Flores, E.P.; Herrera-Maldonado, N.; Hinojosa-Trejo, M.A.; Vergara-Vázquez, M.; Halley-Castillo, M.E. Avances y logros del programa de tamiz metabólico neonatal (2012–2018). Acta Pediátrica México 2018, 39, 57–65S. [Google Scholar] [CrossRef]
- Ohlsson, A.; Bruhn, H.; Nordenström, A.; Zetterström, R.H.; Wedell, A.; von Döbeln, U. The spectrum of PAH mutations and increase of milder forms of phenylketonuria in sweden during 1965–2014. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2016; Volume 34, pp. 19–26. [Google Scholar] [CrossRef] [Green Version]
- Odagiri, S.; Kabata, D.; Tomita, S.; Kudo, S.; Sakaguchi, T.; Nakano, N.; Yamamoto, K.; Shintaku, H.; Hamazaki, T. Clinical and genetic characteristics of patients with mild hyperphenylalaninemia identified by newborn screening program in Japan. Int. J. Neonatal Screen. 2021, 7, 17. [Google Scholar] [CrossRef]
- Viall, S.; Ayyub, O.; Rasberry, M.; Lyons, K.; Mew, N.A. “Mild” hyperphenylalaninemia? A case series of seven treated patients following newborn screening. Mol. Genet. Metab. 2017, 122, 153–155. [Google Scholar] [CrossRef]
- Neto, E.V.; Laranjeira, F.; Quelhas, D.; Ribeiro, I.; Seabra, A.; Mineiro, N.; Carvalho, L.D.M.; Lacerda, L.; Ribeiro, M.G. Mutation analysis of the PAH gene in phenylketonuria patients from Rio de Janeiro, Southeast Brazil. Mol. Genet. Genom. Med. 2018, 6, 575–591. [Google Scholar] [CrossRef]
- Aldámiz-Echevarría, L.; Llarena, M.; Bueno, M.A.; Dalmau, J.; Vitoria, I.; Fernández-Marmiesse, A.; Andrade, F.; Blasco, J.; Alcalde, C.; Gil, D. Molecular epidemiology, genotype–phenotype correlation and BH 4 responsiveness in Spanish patients with phenylketonuria. J. Hum. Genet. 2016, 61, 731–744. [Google Scholar] [CrossRef]
- Hamilton, V.; María, L.S.; Fuenzalida, K.; Morales, P.; Desviat, L.R.; Ugarte, M.; Pérez, B.; Cabello, J.F.; Cornejo, V. Characterization of phenyalanine hydroxylase gene mutations in chilean PKU patients. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2017; Volume 42, pp. 71–77. [Google Scholar] [CrossRef]
- Santos, L.; Castro-Magalhaes, M.; Fonseca, C.; Starling, A.; Januário, J.; Aguiar, M.; Carvalho, M. PKU in minas Gerais state, Brazil: Mutation analysis. Ann. Hum. Genet. 2008, 72, 774–779. [Google Scholar] [CrossRef]
- Kuznetcova, I.; Gundorova, P.; Ryzhkova, O.; Polyakov, A. The study of the full spectrum of variants leading to hyperphenylalaninemia have revealed 10 new variants in the PAH gene. Metab. Brain Dis. 2019, 34, 1547–1555. [Google Scholar] [CrossRef]
- Guldberg, P.; Henriksen, K.F.; Güttler, F. Molecular analysis of phenylketonuria in Denmark: 99% of the mutations detected by denaturing gradient gel electrophoresis. Genomics 1993, 17, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.A.; González-Lamuño, D.; Delgado-Pecellín, C.; Aldámiz-Echevarría, L.; Pérez, B.; Desviat, L.R.; Couce, M.L. Molecular epidemiology and genotype–phenotype correlation in phenylketonuria patients from South Spain. J. Hum. Genet. 2013, 58, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-García, E.; Barrios-García, B.; Gutiérrez-Gutiérrez, R.; Damiani-Rossel, A. Caracterización molecular de fenilcetonúricos cubanos. Revista Cubana de Pediatría. Rev. Cuba. Pediatría 2002, 74, 101–105. [Google Scholar]
- Santos, M.; Kuzmin, A.I.; Eisensmith, R.C.; Goltsov, A.A.; Woo, S.L.; Barrantes, R.; De Céspedes, C. Phenylketonuria in costa rica: Preliminary spectrum of PAH mutations and their associations with highly polymorphic haplotypes. Hum. Hered. 1996, 46, 128–131. [Google Scholar] [CrossRef]
- Vieira Neto, E.; Laranjeira, F.; Quelhas, D.; Ribeiro, I.; Seabra, A.; Mineiro, N.; Carvalho, L.M.; Lacerda, L.; Ribeiro, M.G. Genotype-phenotype correlations and BH4 estimated responsiveness in patients with phenylketonuria from Rio de Janeiro, Southeast Brazil. Mol. Genet. Genom. Med. 2019, 7, e610. [Google Scholar] [CrossRef] [Green Version]
- Tresbach, R.H.; Sperb-Ludwig, F.; Ligabue-Braun, R.; Tonon, T.; Cardoso, M.T.D.O.; Heredia, R.S.; Rosa, M.T.A.D.S.; Martins, B.C.; Poubel, M.O.; Da Silva, L.C.S.; et al. Phenylketonuria diagnosis by massive parallel sequencing and genotype-phenotype association in Brazilian patients. Genes 2020, 12, 20. [Google Scholar] [CrossRef]
- Gundorova, P.; Stepanova, A.A.; Kuznetsova, I.A.; Kutsev, S.I.; Polyakov, A.V. Genotypes of 2579 patients with phenylketonuria reveal a high rate of BH4 non-responders in Russia. PLoS ONE 2019, 14, e0211048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, J.; Shen, N.; Tao, Y.; Han, L.; Qiu, W.; Zhang, H.; Liang, L.; Fan, Y.; Wang, J.; et al. In vitro residual activities in 20 variants of phenylalanine hydroxylase and genotype-phenotype correlation in phenylketonuria patients. Gene 2019, 707, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Bsc, R.A.F.E.; Wegberg, A.M.J.; Anjema, K.; Lubout, C.M.A.; Dam, E.; van Vliet, D.; Blau, N.; Spronsen, F.J. The first European guidelines on phenylketonuria: Usefulness and implications for BH 4 responsiveness testing. J. Inherit. Metab. Dis. 2019, 43, 244–250. [Google Scholar] [CrossRef]
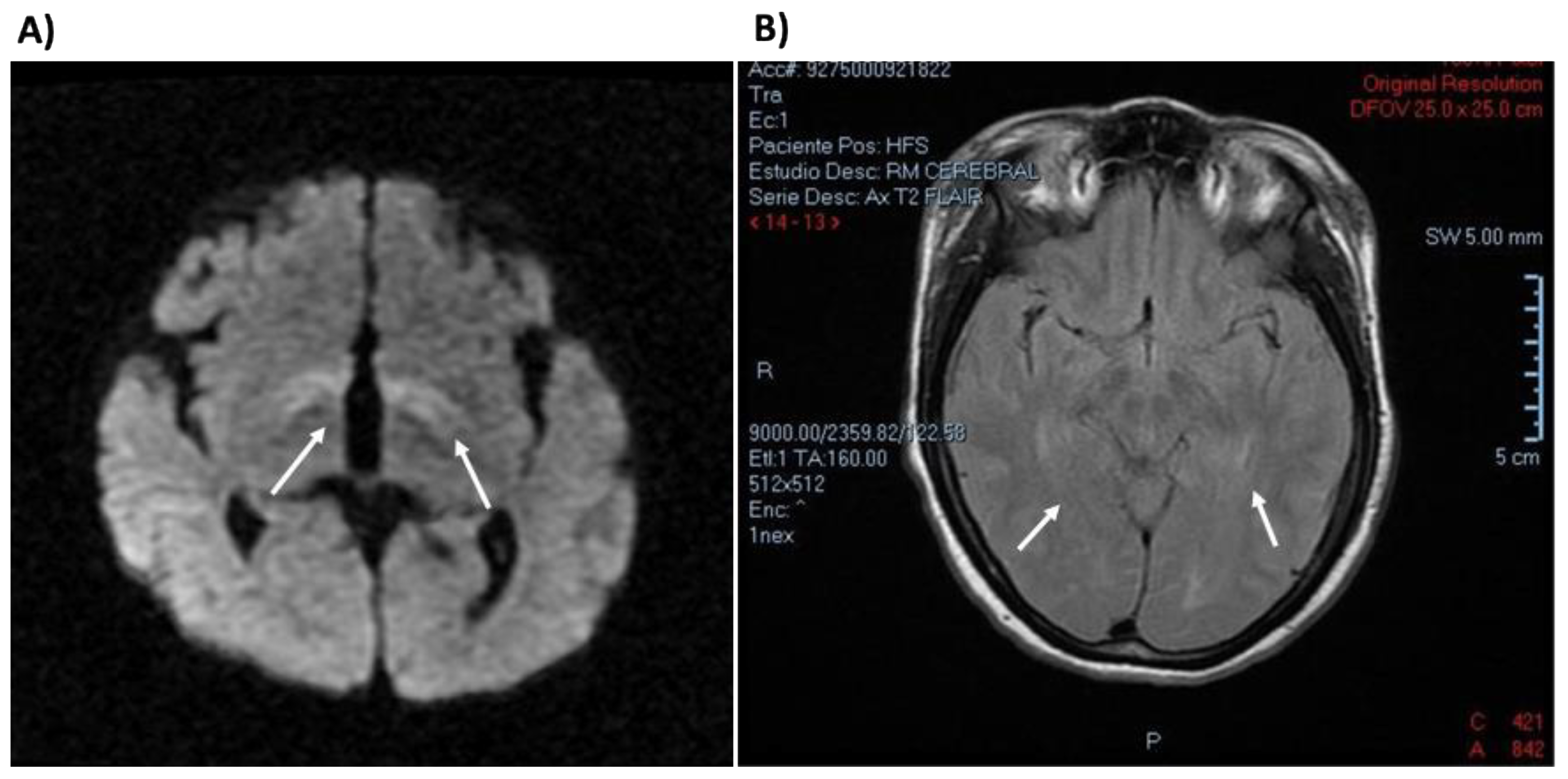
- Jiménez-Pérez, M.O.; Gómez-Garza, G.; Fernández-Lainez, C.; Ibarra-González, I.; Vela-Amieva, M.; Ruiz-García, M. Resonancia magnética nuclear de encéfalo en pacientes con fenilcetonuria diagnosticada tardíamente. Acta Pediátrica México 2015, 36, 9. [Google Scholar] [CrossRef] [Green Version]
- Velázquez, A.; Bilbao, G.; González-Trujillo, J.L.; Hernández, D.; Pérez-Andrade, M.E.; Vela, M.; Cicerón, I.; Loera-Luna, A.; Cederbaum, S.; Phoenix, B. Apparent higher frequency of phenylketonuria in the Mexican state of Jalisco. Hum. Genet. 1996, 97, 99–102. [Google Scholar] [CrossRef]
- Wang, R.; Shen, N.; Ye, J.; Han, L.; Qiu, W.; Zhang, H.; Liang, L.; Sun, Y.; Fan, Y.; Wang, L.; et al. Mutation spectrum of hyperphenylalaninemia candidate genes and the genotype-phenotype correlation in the Chinese population. Clin. Chim. Acta 2018, 481, 132–138. [Google Scholar] [CrossRef]
- Quirk, M.E.; Dobrowolski, S.F.; Nelson, B.E.; Coffee, B.; Singh, R.H. Utility of phenylalanine hydroxylase genotype for tetrahydrobiopterin responsiveness classification in patients with phenylketonuria. Mol. Genet. Metab. 2012, 107, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Muntau, A.C.; Röschinger, W.; Habich, M.; Demmelmair, H.; Hoffmann, B.; Sommerhoff, C.P.; Roscher, A.A. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N. Engl. J. Med. 2002, 347, 2122–2132. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BIOPKUdb | |||||||
|---|---|---|---|---|---|---|---|
| Classification According APV 1 | PAH Variant | Protein Change | % (Present Work) | % | APV 2 | EA (%) | Protein Domain |
| cPKU (N = 42) | c. 60 + 5G > T ▪ | p. (?) | 14.516 ♦ | 0.320 | 0.0 | NR | NA |
| c. 1162G > A | p. (Val388Met) | 11.290 ♦ | 1.800 | 1.9 | 28 | Catalytic | |
| c. 441 + 5G > T ▪ | p. (?) | 5.645 | 0.980 | 0.0 | NR | NA | |
| c. 1066-11G > A ▪ | p. (Gln355_Tyr356insGlyLeuGln) | 5.645 | 6.800 | 0.0 | 5 | Catalytic | |
| c. 1045T > C ▪ | p. (Ser349Pro) | 4.435 | 1.000 | 0.0 | 1 | Catalytic | |
| c. 782G > A | p. (Arg261Gln) | 2.823 | 5.500 | 1.6 | 44 | Catalytic | |
| c. 194T > C | p. (Ile65Thr) | 2.419 | 4.000 | 1.0 | 33 | Regulatory | |
| c. 809G > A ▪ | p. (Arg270Lys) | 2.419 | 0.220 | 0.0 | 11 | Catalytic | |
| c. 1A > T ▪ | p. (?) | 2.016 ♦ | 0.026 | 0.0 | NR | NA | |
| c. 1315 + 1G > A ▪ | p. (?) | 2.016 | 4.400 | 0.0 | NR | NA | |
| c. 728G > A ▪ | p. (Arg243Gln) | 1.613 | 2.700 | 0.0 | 14 | Catalytic | |
| c. 838G > A ▪ | p. (Glu280Lys) | 1.613 | 1.300 | 0.0 | 11 | Catalytic | |
| c. 842 + 1G > A ▪ | p. (?) | 1.613 | 0.440 | 0.0 | NR | NA | |
| c. 1055del ▪ | p. (Gly352Valfs * 48) | 1.613 | 0.590 | 0.0 | NR | Catalytic | |
| c. 208_210delTCT | p. (Ser70del) | 1.210 | 0.026 | 0.7 | NR | Regulatory | |
| c. 673C > A ▪ | p. (Pro225Thr) | 1.210 | 0.170 | 0.0 | NR | Catalytic | |
| c. 754C > T ▪ | p. (Arg252Trp) | 1.210 | 1.400 | 0.0 | 15 | Catalytic | |
| c. 781C > T ▪ | p. (Arg261 *) | 1.210 | 0.770 | 0.0 | NR | Catalytic | |
| c. 830A > G ▪ | p. (Tyr277Cys) | 1.210 | 0.016 | 0.0 | NR | Catalytic | |
| c. 969 + 6T > A | p. (?) | 1.210 ♦ | 0.005 | NR | NR | NA | |
| c. 1042C > G | p. (Leu348Val) | 1.210 | 0.940 | 1.5 | 25 | Catalytic | |
| c. 117C > G | p. (Phe39Leu) | 0.806 | 0.420 | 0.2 | 49 | Regulatory | |
| c. 165del ▪ | p. (Phe55Leufs * 6) | 0.806 | 0.910 | 0.0 | NR | Regulatory | |
| c. 439C > T ▪ | p. (Pro147Ser) | 0.806 | 0.042 | 0.0 | NR | Catalytic | |
| c. 625_626insC | p. (Ile209Thrfs * 6) | 0.806 | NR | NR | NR | NA | |
| c. 727C > T ▪ | p. (Arg243 *) | 0.806 | 1.200 | 0.0 | NR | Catalytic | |
| c. 791A > G | p. (His264Arg) | 0.806 | NR | NR | NR | Catalytic | |
| c. 1157A > G ▪ | p. (Tyr386Cys) | 0.806 | 0.074 | 0.0 | NR | Catalytic | |
| c. 23del | p. (Asn8Thrfs * 30) | 0.403 | NR | NR | NR | NA | |
| c. 116_118delTCT | p. (Phe39del) | 0.403 | 0.130 | 0.9 | NR | Regulatory | |
| c. 168 + 5G > C ▪ | p. (?) | 0.403 | 0.600 | 0.0 | NR | NA | |
| c. 472C > T ▪ | p. (Arg158Trp) | 0.403 | 0.110 | 0.0 | 2 | Catalytic | |
| c. 473G > A ▪ | p. (Arg158Gln) | 0.403 | 2.900 | 0.0 | 10 | Catalytic | |
| c. 526C > T ▪ | p. (Arg176 *) | 0.403 | 0.410 | 0.0 | NR | Catalytic | |
| c. 814G > T ▪ | p. (Gly272 *) | 0.403 | 0.790 | 0.0 | NR | Catalytic | |
| c. 829T > G | p. (Tyr277Asp) | 0.403 | 0.240 | 0.4 | NR | Catalytic | |
| c. 932T > C ▪ | p. (Leu311Pro) | 0.403 | 0.170 | 0.0 | 10 | Catalytic | |
| c. 1127del | p. (Asn376Ilefs * 24) | 0.403 | 0.005 | 1.0 | NR | Catalytic | |
| c. 1198A > C | p. (Arg400=) | 0.403 | 0.005 | NR | NR | NA | |
| c. 1282C > T ▪ | p. (Gln428 *) | 0.403 ♦ | 0.000 | 0 | NR | Oligomerization | |
| c. 1315 + 2T > C ▪ | p. (?) | 0.403 | 0.026 | 0.0 | NR | NA | |
| c. 1315 + 5_1315 + 6insGTGTAACAG | p. (?) | 0.403 | NR | NR | NR | NA | |
| mPKU (N = 6) | c. 204A > T | p. (Arg68Ser) | 1.210 ♦ | 0.600 | 5.4 | 25 | Regulatory |
| c. 721C > T | p. (Arg241Cys) | 0.403 | 1.100 | 5.5 | 57 | Catalytic | |
| c. 722G > A | p. (Arg241His) | 2.419 | 0.460 | 5.2 | 23 | Catalytic | |
| c. 912G > A | p. (Gln304=) | 0.806 | 0.095 | 3.3 | NR | NA | |
| c. 969 + 6T > C | p. (?) | 0.403 | 0.005 | NR | NR | NA | |
| c. 1241A > G | p. (Tyr414Cys) | 0.806 | 3.100 | 5.1 | 57 | Oligomerization | |
| MHP (N = 12) | c. 165T > G | p. (Phe55Leu) | 0.403 | 0.130 | 8.2 | NR | Regulatory |
| c. 508C > G | p. (His170Asp) | 1.613 | 0.053 | 10.0 | 43 | Catalytic | |
| c. 527G > T | p. (Arg176Leu) | 0.403 | 0.150 | 9.8 | 42 | Catalytic | |
| c. 533A > G | p. (Glu178Gly) | 2.419 | 0.300 | 7.6 | 39 | Catalytic | |
| c. 688G > A | p. (Val230Ile) | 0.403 | 0.270 | 10.0 | 63 | Catalytic | |
| c. 898G > T | p. (Ala300Ser) | 0.403 | 1.500 | 9.7 | 65 | Catalytic | |
| c. 907T > G | p. (Ser303Ala) | 0.403 | 0.047 | 9.3 | NR | Catalytic | |
| c. 941C > A | p. (Pro314His) | 0.403 | 0.037 | 10.0 | NR | Catalytic | |
| c. 1169A > G | p. (Glu390Gly) | 2.419 | 1.300 | 6.9 | 62 | Catalytic | |
| c. 1243G > A | p. (Asp415Asn) | 3.226 | 0.440 | 10.0 | 72 | Oligomerization | |
| c. 1208C > T | p. (Ala403Val) | 2.016 | 2.400 | 9.7 | 66 | Catalytic | |
| c. 1238G > A | p. (Arg413His) | 0.403 | NR | 9.0 | NR | Oligomerization | |
| Patient ID | PAH Genotype | Protein Change | Diagnosis | Sex | Geographical Origin | Max Historical Phe (μmol/L) | Observed Biochemical Phenotype | Genotypic Phenotype Value (GPV) |
|---|---|---|---|---|---|---|---|---|
| ♦ 1 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | NBS | M | Jalisco | 3591 | cPKU | 0 |
| ♦ 2 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | CD | F | CDMX | 2421 | cPKU | 0 |
| 3 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | CD | M | Jalisco | 1896 | cPKU | 0 |
| ♦ 4 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | CD | M | Jalisco | 1654 | cPKU | 0 |
| 5 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | CD | M | Jalisco | 1876 | cPKU | 0 |
| 6 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | NBS | F | Jalisco | 1573 | cPKU | 0 |
| 7 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | NBS | M | Jalisco | 1311 | cPKU | 0 |
| 8 | c. [60 + 5G > T; 60 + 5G > T] | p. [?]; [?] | NBS | M | Jalisco | 1712 | cPKU | 0 |
| 9 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | NBS | F | Edo Mex | 1581 | cPKU | 0 |
| 10 | c. [60 + 5G > T]; [60 + 5G > T] | p. [?]; [?] | NBS | F | Jalisco | 1453 | cPKU | 0 |
| ♦ 11 | c. [60 + 5G > T]; [1162G > A] | p. [?]; [Val388Met] | CD | F | Puebla | 1900 | cPKU | 1.8 |
| ♦ 12 | c. [60 + 5G > T]; [1162G > A] | p. [?]; [Val388Met] | NBS | M | Jalisco | 3832 | cPKU | 1.8 |
| ♦ 13 | c. [60 + 5G > T]; c. [1162G > A] | p. [?]; [Val388Met] | NBS | M | Aguascalientes | 1876 | cPKU | 1.8 |
| ♦ 14 | c. [60 + 5G > T]; [1162G > A] | p. [?]; [Val388Met] | NBS | M | Jalisco | 1715 | cPKU | 1.8 |
| 15 | c. [60 + 5G > T]; [441 + 5G > T] | p. [?]; [?] | NBS | F | Edo Mex | 1682 | cPKU | 0 |
| ♦ 16 | c. [60 + 5G > T]; [441 + 5G > T] | p. [?]; [?] | NBS | F | Baja California Sur | 1815 | cPKU | 0 |
| ♦ 17 | c. [60 + 5G > T]; [441 + 5G > T] | p. [?]; [?] | CD | M | Jalisco | 1346 | cPKU | 0 |
| ♦ 18 | c. [60 + 5G > T]; [1315 + 1G > A] | p. [?]; [?] | NBS | F | Guanajuato | 1385 | cPKU | 0 |
| 19 | c. [60 + 5G > T]; [1315 + 1G > A] | p. [?]; [?] | NBS | F | Guanajuato | 1206 | cPKU | 0 |
| 20 | c. [60 + 5G > T]; [673C > A] | p. [?]; [Pro225Thr] | NBS | F | Yucatan | 2326 | cPKU | 0 |
| ♦ 21 | c. [60 + 5G > T]; [727C > T] | p. [?]; [Arg243 *] | CD | F | Jalisco | 832 | cPKU | 0 |
| ♦ 22 | c. [60 + 5G > T]; [1066-11G > A] | p. [?]; [Gln355_Tyr356insGlyLeuGln] | NBS | F | Jalisco | 1498 | cPKU | 0 |
| 23 | c. [60 + 5G > T]; [1169A > G] | p. [?]; [Glu390Gly] | NBS | F | Baja California | 1616 | MHP | 6.9 |
| ♦ 24 | c. [508C > G]; [60 + 5G > T] | p. [His170Asp]; [?] | NBS | M | Jalisco | 1423 | MHP | 10 |
| ♦ 25 | c. [754C > T]; [60 + 5G > T] | p. [Arg252Trp]; [?] | CD | M | CDMX | 2075 | cPKU | 0 |
| ♦ 26 | c. [838G > A]; [60 + 5G > T] | p. [Glu280Lys]; [?] | CD | F | Jalisco | 1949 | cPKU | 0 |
| 27 | c. [1162G > A]; [1162G > A] | p. [Val388Met]; [Val388Met] | NBS | M | Puebla | 1900 | mPKU | 1.8 |
| 28 | c. [1162G > A]; [1162G > A] | p. [Val388Met]; [Val388Met] | NBS | F | Puebla | 1516 | mPKU | 1.8 |
| 29 | c. [1162G > A]; [1162G > A] | p. [Val388Met]; [Val388Met] | CD | M | Hidalgo | 1529 | cPKU | 1.8 |
| 30 | c. [1162G > A]; [1162G > A] | p. [Val388Met]; [Val388Met] | NBS | M | Queretaro | 2265 | mPKU | 1.8 |
| 31 | c. [1162G > A]; [1162G > A] | p. [Val388Met]; [Val388Met] | NBS | F | Queretaro | 2258 | mPKU | 1.8 |
| 32 | c. [194T > C]; [1241A > G] | p. [Ile65Thr]; [Tyr414Cys] | NBS | M | Zacatecas | 790 | mPKU | 5.1 |
| 33 | c. [194T > C]; [1241A > G] | p. [Ile65Thr]; [Tyr414Cys] | NBS | M | Queretaro | 1070 | mPKU | 5.1 |
| ♦ 34 | c. [441 + 5G > T]; [441 + 5G > T] | p. [?]; [?] | NBS | F | Tamaulipas | 1640 | mPKU | 0 |
| 35 | c. [441 + 5G > T]; [441 + 5G > T] | p. [?]; [?] | CD | M | Coahuila | 1460 | cPKU | 0 |
| ♦ 36 Δ | c. [969 + 6T > A]; [1162G > A] | p. [?]; [Val388Met] | NBS | F | CDMX | 966 | MHP | 1.8 |
| 37 Δ | c. [969 + 6T > A]; [1162G > A] | p. [?]; [Val388Met] | NBS | F | Jalisco | 424 | MHP | 1.8 |
| 38 | c. [1066-11G > A]; [1162G > A] | p. [Gln355_Tyr356insGlyLeuGln]; [Val388Met] | CD | F | CDMX | 674 | cPKU | 1.8 |
| 39 | c. [1066-11G > A]; [1162G > A] | p. [Gln355_Tyr356insGlyLeuGln]; [Val388Met] | NBS | F | Puebla | 1304 | mPKU | 1.8 |
| 40 | c. [1208C > T]; [1208C > T] | p. [Ala403Val]; [Ala403Val] | NBS | M | Queretaro | 1748 | MHP | 9.7 |
| 41 | c. [1208C > T]; [1208C > T] | p. [Ala403Val]; [Ala403Val] | NBS | F | CDMX | 1477 | MHP | 9.7 |
| ♦ 42 | c. [1A > T]; c. [1A > T] | p. [?]; [?] | CD | M | Veracruz | 1431 | cPKU | 0 |
| ♦ 43 Δ | c. [1A > T]; [722G > A] | p. [?]; [Arg241His] | NBS | M | Veracruz | 1225 | MHP | 5.2 |
| 44 | c. [1A > T]; [1042C > G] | p. [?]; [Leu348Val] | NBS | F | CDMX | 1852 | cPKU | 1.5 |
| 45 | c. [1A > T]; [1243G > A] | p. [?]; [Asp415Asn] | NBS | M | Veracruz | 652 | MHP | 10 |
| 46 | c. [23del]; [1162G > A] | p. [Asn8Thrfs * 30]; [Val388Met] | NBS | F | Chihuahua | 1187 | cPKU | Unknown |
| 47 | c. [117C > G]; [441 + 5G > T] | p. [Phe39Leu]; [?] | NBS | M | Chihuahua | 1075 | mPKU | 0.2 |
| 48 | c. [117C > G]; [1157A > G] | p. [Phe39Leu]; [Tyr386Cys] | NBS | F | Durango | 1184 | mPKU | 0.2 |
| 49 | c. [165del]; [194T > C] | p. [Phe55Leufs * 6]; [Ile65Thr] | NBS | M | Durango | 1066 | cPKU | 1.1 |
| 50 | c. [165T > G]; [208_210del] | p. [Phe55Leu]; [Ser70del] | NBS | M | Chihuahua | 2825 | MHP | 8.2 |
| 51 | c. [168 + 5G > C]; [1066-11G > A] | p. [?]; [Gln355_Tyr356insGlyLeuGln] | NBS | M | Chihuahua | 1222 | cPKU | 0 |
| 52 | c. [194T > C]; [208_210del] | p. [Ile65Thr]; [Ser70del] | NBS | M | Zacatecas | 1791 | cPKU | 1 |
| 53 Δ | c. [194T > C]; [533A > G] | p. [Ile65Thr]; [Glu178Gly] | NBS | F | Sonora | 1527 | MHP | 7.6 |
| 54 | c. [204A > T]; [527G > T] | p. [Arg68Ser]; [Arg176Leu] | NBS | M | CDMX | 799 | MHP | 9.7 |
| ♦ 55 | c. [204A > T]; [782G > A] | p. [Arg68Ser]; [Arg261Gln] | NBS | M | Queretaro | 1187 | mPKU | 5.4 |
| 56 | c. [204A > T]; [829T > G] | p. [Arg68Ser]; [Tyr277Asp] | NBS | M | Veracruz | 1255 | MHP | 5.4 |
| 57 | c. [208_210del]; [728G > A] | p. [Ser70del]; [Arg243Gln] | NBS | F | Nuevo León | 1970 | mPKU | 0.7 |
| ♦ 58 | c. [439C > T]; [1045T > C] | p. [Pro147Ser]; [Ser349Pro] | NBS | M | Jalisco | 2098 | cPKU | 0 |
| ♦ 59 | c. [439C > T]; [782G > A] | p. [Pro147Ser]; [Arg261Gln] | NBS | F | Michoacan | 1923 | mPKU | 1.6 |
| ♦ 60 | c. [441 + 5G > T]; [1045T > C] | p. [?]; [Ser349Pro] | NBS | F | Edo Mex | 1452 | cPKU | 0 |
| 61 | c. [441 + 5G > T]; [1055del] | p. [?]; [Gly352Valfs * 48] | NBS | M | CDMX | 1756 | cPKU | 0 |
| ♦ 62 | c. [441 + 5G > T]; [165del] | p. [?]; [Phe55Leufs * 6] | CD | F | Chihuahua | 1634 | cPKU | 0 |
| ♦ 63 | c. [441 + 5G > T]; [782G > A] | p. [?]; [Arg261Gln] | CD | M | CDMX | 890 | cPKU | 1.6 |
| 64 | c. [441 + 5G > T]; [791A > G] | p. [?]; [His264Arg] | NBS | F | Edo Mex | 921 | cPKU | 0 |
| 65 | c. [473G > A]; [1045T > C] | p. [Arg158Gln]; [Ser349Pro] | NBS | M | CDMX | 1656 | cPKU | 0 |
| 66 | c. [508C > G]; [1243G > A] | p. [His170Asp]; [Asp415Asn] | NBS | M | Jalisco | 1098 | MHP | 10 |
| ♦ 67 | c. [526C > T]; [1066-11G > A] | p. [Arg176 *]; [Gln355_Tyr356insGlyLeuGln] | CD | M | CDMX | 1113 | cPKU | 0 |
| 68 | c. [533A > G]; [1162G > A] | p. [Glu178Gly]; [Val388Met] | NBS | F | Hidalgo | 502 | MHP | 7.6 |
| 69 | c. [533A > G]; [1169A > G] | p. [Glu178Gly]; [Glu390Gly] | NBS | M | CDMX | 179 | MHP | 7.6 |
| 70 | c. [533A > G]; [533A > G] | p. [Glu178Gly]; [Glu178Gly] | CD | M | Queretaro | 1277 | MHP | 7.6 |
| 71 | c. [533A > G]; [809G > A] | p. [Glu178Gly]; [Arg270Lys] | NBS | F | Edo Mex | 1154 | MHP | 7.6 |
| 72 | c. [625_626insC]; [625_626insC] | p. [?]; [?] | NBS | M | Veracruz | 1008 | cPKU | Unknown |
| 73 | c. [673C > A]; [830A > G] | p. [Pro225Thr]; [Tyr277Cys] | NBS | M | Quintana Roo | 1022 | mPKU | 0 |
| 74 | c. [688G > A]; [754C > T] | p. [Val230Ile]; [Arg252Trp] | NBS | F | Veracruz | 1608 | MHP | 10 |
| 75 | c. [721C > T]; [1066-11G > A] | p. [Arg241Cys]; [Gln355_Tyr356insGlyLeuGln] | NBS | M | CDMX | 1747 | MHP | 5.5 |
| 76 | c. [722G > A]; [782G > A] | p. [Arg241His]; [Arg261Gln] | NBS | F | CDMX | 2396 | MHP | 5.2 |
| 77 ● | c. [722G > A]; [842 + 1G > A] | p. [Arg241Cys]; [?] | NBS | M | Tabasco | 2134 | cPKU | 5.2 |
| 78 | c. [722G > A]; [722G > A] | p. [Arg241Cys]; [Arg241His] | NBS | M | Puebla | 4627 | MHP | 5.2 |
| 79 | c. [727C > T]; [1243G > A] | p. [Arg241Cys]; [Asp415Asn] | NBS | F | CDMX | 948 | MHP | 10 |
| 80 | c. [728G > A]; [1243G > A] | p. [Arg243Gln]; [Asp415Asn] | NBS | M | CDMX | 712 | MHP | 10 |
| ♦ 81 Δ | c. [728G > A]; [722G > A] | p. [Arg243Gln]; [Arg241His] | NBS | F | CDMX | 506 | MHP | 5.2 |
| 82 | c. [728G > A]; [809G > A] | p. [Arg243Gln]; [Arg270Lys] | NBS | M | Chihuahua | 587 | mPKU | 0 |
| ♦ 83 | c. [781C > T]; [1066-11G > A] | p. [Arg261 *]; [Gln355_Tyr356insGlyLeuGln] | CD | M | Jalisco | 1623 | cPKU | 0 |
| ♦ 84 | c. [781C > T]; [782G > A] | p. [Arg261 *]; [Arg261Gln] | NBS | F | CDMX | 177 | cPKU | 1.6 |
| 85 | c. [781C > T]; [1243G > A] | p. [Arg261 *]; [Asp415Asn] | NBS | M | CDMX | 470 | MHP | 10 |
| 86 | c. [782G > A]; [1045T > C] | p. [Arg261Gln]; [Ser349Pro] | NBS | M | Nuevo León | 794 | cPKU | 1.6 |
| 87 | c. [782G > A]; [1169A > G] | p. [Arg261Gln]; [Glu390Gly] | NBS | F | Guanajuato | 215 | MHP | 7 |
| 88 | c. [791A > G]; [1315 + 5_1315 + 6insGTGTAACAG] | p. [His264Arg]; [?] | NBS | M | Veracruz | 401 | cPKU | Unknown |
| 89 Δ | c. [809G > A]; [1162G > A] | p. [Arg270Lys]; [Val388Met] | NBS | M | San Luis Potosi | 467 | mPKU | 1.8 |
| 90 | c. [809G > A]; [1066-11G > A] | p. [Arg270Lys]; [Gln355_Tyr356insGlyLeuGln] | NBS | M | Puebla | 282 | cPKU | 0 |
| 91 | c. [814G > T]; [1282C > T] | p. [Gly272 *]; [Gln428 *] | NBS | F | Edo Mex | 566 | mPKU | 0 |
| 92 | c. [830A > G]; [830A > G] | p. [Tyr277Cys]; [Tyr277Cys] | NBS | M | Yucatan | 454 | MHP | 0 |
| ♦ 93 | c. [838G > A]; [838G > A] | p. [Glu280Lys]; [Glu280Lys] | NBS | F | CDMX | 283 | mPKU | 0 |
| 94 | c. [838G > A]; [1066-11G > A] | p. [Glu280Lys]; [Gln355_Tyr356insGlyLeuGln] | NBS | F | Colima | 512 | cPKU | 0 |
| ♦ 95 | c. [842 + 1G > A]; [441 + 5G > T] | p. [?]; [?] | NBS | M | Edo Mex | 276 | cPKU | 0 |
| ♦ 96 ● | c. [842 + 1G > A]; [508C > G] | p. [?]; [His170Asp] | CD | M | Guanajuato | 239 | mPKU | 10 |
| 97 | c. [842 + 1G > A]; [932T > C] | p. [?]; [Leu311Pro] | CD | F | Sonora | 239 | cPKU | 0 |
| 98 | c. [898G > T]; [1162G > A] | p. [Ala300Ser]; [Val388Met] | NBS | F | Queretaro | 289 | MHP | 9.7 |
| ♦ 99 | c. [907T > G]; [673C > A] | p. [Ser303Ala]; [Pro225Thr] | NBS | F | CDMX | 1351 | MHP | 9.3 |
| ♦ 100 | c. [912G > A]; [912G > A] | p. [Gln304=]; [Gln304=] | CD | F | Sinaloa | 218 | cPKU | 3.3 |
| ♦ 101 | c. [941C > A]; [809G > A] | p. [Pro314His]; [Arg270Lys] | NBS | M | Aguascalientes | 245 | MHP | 10 |
| 102 | c. [969 + 6T > A]; [1198A > C] | p. [?]; [Arg400=] | NBS | M | Michoacan | 278 | MHP | Undetermined |
| ♦ 103 | c. [1042C > G]; [194T > C] | p. [Leu348Val]; [Ile65Thr] | CD | M | Morelos | 180 | mPKU | 1.5 |
| 104 | c. [1042C > G]; [1238G > A] | p. [Leu348Val]; [Arg413His] | NBS | F | Tabasco | 248 | cPKU | 9 |
| ♦ 105 | c. [1045T > C]; [1066-11G > A] | p. [Ser349Pro]; [Gln355_Tyr356insGlyLeuGln] | NBS | M | Chihuahua | 242 | cPKU | 0 |
| ♦ 106 | c. [1045T > C]; [1169A > G] | p. [Ser349Pro]; [Glu390Gly] | NBS | F | CDMX | 600 | MHP | 7 |
| 107 | c. [1045T > C]; [1208C > T] | p. [Ser349Pro]; [Ala403Val] | NBS | F | Guerrero | 455 | MHP | 9.7 |
| ♦ 108 | c. [1045T > C]; [472C > T] | p. [Ser349Pro]; [Arg158Trp] | CD | M | CDMX | 515 | cPKU | 0 |
| 109 | c. [1045T > C]; [1162G > A] | p. [Ser349Pro]; [Val388Met] | NBS | F | Chihuahua | 366 | mPKU | 1.8 |
| 110 | c. [1045T > C]; [1243G > A] | p. [Ser349Pro]; [Asp415Asn] | NBS | M | Veracruz | 425 | MHP | 10 |
| 111 | c. [1055del]; [1055del] | p. [Gly352Valfs * 48]; [Gly352ValfsTer48] | NBS | F | Hidalgo | 537 | cPKU | 0 |
| 112 | c. [1055del]; [1162G > A] | p. [Gly352Valfs * 48]; [Val388Met] | NBS | M | Hidalgo | 498 | cPKU | 1.8 |
| ♦ 113 | c. [1066-11G > A]; [.809G > A] | p. [Gln355_Tyr356insGlyLeuGln]; [Arg270Lys] | NBS | M | Chihuahua | 654 | mPKU | 0 |
| ♦ 114 | c. [1066-11G > A]; [969 + 6T > C] | p. [Gln355_Tyr356insGlyLeuGln]; [?] | NBS | M | Chiapas | 453 | cPKU | Undetermined |
| ♦ 115 Δ | c. [1127delA]; [1066-11G > A] | p. [Asn376Ilefs * 24]; [Gln355_Tyr356insGlyLeuGln] | CD | M | Veracruz | 244 | mPKU | Undetermined |
| ♦ 116 Δ | c. [1157A > G]; [754C > T] | p. [Tyr386Cys]; [Arg252Trp] | CD | F | Oaxaca | 365 | mPKU | 0 |
| 117 | c. [116_118del]; [1045T > C] | p. [Phe39del]; [Ser349Pro] | NBS | F | CDMX | 377 | cPKU | 0.9 |
| ♦ 118 | c. [1162G > A]; [1066-11G > A] | p. [Val388Met]; [Gln355_Tyr356insGlyLeuGln] | NBS | M | Guerrero | 628 | cPKU | 1.9 |
| 119 | c. [1162G > A]; c. [1243G > A] | p. [Val388Met]; [Asp415Asn] | NBS | F | Michoacan | 2798 | MHP | 10 |
| ♦ 120 | c. [1162G > A]; [1315 + 1G > A] | p. [Val388Met]; [?] | CD | F | Edo Mex | 1217 | cPKU | 1.9 |
| ♦ 121 | c. [1169A > G]; [1169A > G] | p. [Glu390Gly]; [Glu390Gly] | NBS | F | Michoacan | 1753 | MHP | 7 |
| 122 | c. [1243G > A]; [1315 + 1G > A] | p. [Asp415Asn]; [?] | NBS | M | Tabasco | 192 | MHP | 10 |
| ♦ 123 ● | c. [1315 + 1G > A]; [508C > G] | p. [?]; [His170Asp] | NBS | M | Guanajuato | 1779 | mPKU | 10 |
| ♦ 124 ● | c. [1315 + 2T > C]; [1162G > A] | p. [?]; [Val388Met] | CD | F | Queretaro | 1060 | cPKU | 1.8 |
| Patient ID | PAH Genotype | Observed Biochemical Phenotype | GPV | Diagnosis | Max Historical Phe (µmol/L) | Tyr (µmol/L) | Phe/Tyr Ratio |
|---|---|---|---|---|---|---|---|
| 1 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | NBS | 3591 | 70 | 41.26 |
| 2 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | CD | 2421 | 64 | 37.83 |
| 3 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | CD | 1896 | 38 | 49.89 |
| 4 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | CD | 1654 | 38 | 43.53 |
| 5 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | CD | 1876 | 42 | 44.67 |
| 6 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | NBS | 1573 | 61 | 25.79 |
| 7 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | NBS | 1311 | 33 | 39.73 |
| 8 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | NBS | 1712 | 39 | 43.90 |
| 9 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | NBS | 1581 | 35 | 45.17 |
| 10 | c. [60 + 5G > T]; [60 + 5G > T] | cPKU | 0.0 | NBS | 1453 | 109 | 13.33 |
| 11 | c. [60 + 5G > T]; [1162G > A] | cPKU | 1.8 | CD | 1923 | 124 | 15.51 |
| 12 | c. [60 + 5G > T]; [1162G > A] | cPKU | 1.8 | NBS | 1452 | 26 | 55.85 |
| 13 | c. [60 + 5G > T]; c. [1162G > A] | cPKU | 1.8 | NBS | 1756 | 38 | 46.21 |
| 14 | c. [60 + 5G > T]; [1162G > A] | cPKU | 1.8 | NBS | 1634 | 106 | 15.42 |
| 15 | c. [60 + 5G > T]; [441 + 5G > T] | cPKU | 0.0 | NBS | 1900 | 36 | 52.78 |
| 16 | c. [60 + 5G > T]; [441 + 5G > T] | cPKU | 0.0 | NBS | 3832 | 78 | 49.13 |
| 17 | c. [60 + 5G > T]; [441 + 5G > T] | cPKU | 0.0 | CD | 1876 | 58 | 32.34 |
| 18 | c. [60 + 5G > T]; [1315 + 1G > A] | cPKU | 0.0 | NBS | 1715 | 46 | 37.28 |
| 19 | c. [60 + 5G > T]; [1315 + 1G > A] | cPKU | 0.0 | NBS | 1682 | 70 | 24.03 |
| 20 | c. [60 + 5G > T]; [673C > A] | cPKU | 0.0 | NBS | 1815 | 69 | 26.30 |
| 21 | c. [60 + 5G > T]; [727C > T] | cPKU | 0.0 | CD | 1346 | 108 | 12.46 |
| 22 | c. [60 + 5G > T]; [1066-11G > A] | cPKU | 0.0 | NBS | 1385 | 113 | 12.26 |
| 23 | c. [60 + 5G > T]; [1169A > G] | MHP | 6.9 | NBS | 467 | 103 | 4.53 |
| 24 | c. [ 60 + 5G > T]; [ 508C > G] | MHP | 10.0 | NBS | 600 | 77 | 7.79 |
| 25 | c. [ 60 + 5G > T]; [ 754C > T] | cPKU | 0.0 | CD | 1206 | 33 | 36.55 |
| 26 | c. [ 60 + 5G > T]; [ 838G > A] | cPKU | 0.0 | CD | 2326 | 41 | 56.73 |
| Genotype | % BH4 Response Reported in BIOPKU | |||
|---|---|---|---|---|
| Observed Biochemical Phenotype | Allele 1 in Homozygous State Yes/Slow | Allele 2 in Homozygous State Yes/Slow | BH4 Responsiveness | |
| c. [722G > A]; [722G > A] | MHP | 100/0 | 100/0 | Identical genotype reported in BIOPKUdb as responder |
| c. [1169A > G]; [1169A > G] | MHP | 100/0 | 100/0 | |
| c. [1208C > T]; [1208C > T] | MHP | 100/0 | 100/0 | |
| c. [722G > A]; [782G > A] | MHP | 100/0 | 74/4 | |
| c. [722G > A]; [842 + 1G > A] | cPKU | 100/0 | 0 | |
| c. [782G > A]; [1169A > G] | MHP | 74/4 | 100/0 | |
| c. [728G > A]; [1243G > A] | MHP | 0/6.67 | NR | |
| c. [60 + 5G > T]; [1169A > G] | MHP | NR | 100/0 | |
| c. [1045T > C]; [1169A > G] | MHP | 0 | 100/0 | |
| c. [1045T > C]; [1208C > T] | MHP | 0 | 100/0 | |
| c. [1045T > C]; [1243G > A] | MHP | 0 | NR | |
| c. [60 + 5G > T]; [508C > G] | MHP | NR | NR | |
| c. [204A > T]; [527G > T] | MHP | 100/0 | NR | Potential responder |
| c. [204A > T]; [782G > A] | mPKU | 100/0 | 74/4 | |
| c. [204A > T]; [829T > G] | MHP | 100/0 | 0 | |
| c. [721C > T]; [1066-11G > A] | MHP | 100/0 | 0/6.09 | |
| c. [722G > A]; [728G > A] | MHP | 100/0 | 0/6.67 | |
| c. [1042C > G]; [194T > C] | mPKU | 100/0 | 84/0 | |
| c. [1042C > G]; [1238G > A] | cPKU | 100/0 | NR | |
| c. [194T > C]; [208_210del] | cPKU | 84/0 | NR | |
| c. [194T > C]; [533A > G] | MHP | 84/0 | NR | |
| c. [194T > C]; [1241A > G] | mPKU | 84/0 | 100/0 | |
| c. [782G > A]; [1045T > C] | cPKU | 74/4 | 0 | |
| c. [1162G > A]; [1162G > A] | mPKU | 62.5/12.5 | 62.5/12.5 | |
| c. [1162G > A]; c. [1243G > A] | MHP | 62.5/12.5 | NR | |
| c. [1162G > A]; [1315 + 1G > A] | cPKU | 62.5/12.5 | 0/13 | |
| c. [1162G > A]; [1315 + 2T > C] | cPKU | 62.5/12.5 | NR | |
| c. [898G > T]; [1162G > A] | MHP | 60/40 | 62.5/12.5 | |
| c. [1055del]; [1162G > A] | cPKU | 5/5 | 62.5/12.5 | |
| c. [116_118del]; [1045T > C] | cPKU | 0/80 | 0 | |
| c. [117C > G]; [1157A > G] | mPKU | 0/50 | NR | |
| c. [117C > G]; [441 + 5G > T] | mPKU | 0/50 | NR | |
| c. [781C > T]; [782G > A] | cPKU | 0/20 | 74/4 | |
| c. [781C > T]; [1243G > A] | MHP | 0/20 | NR | |
| c. [1066-11G > A]; [1127delA] | mPKU | 0/6.09 | NR | |
| c. [1066-11G > A]; [1162G > A] | cPKU | 0/6.09 | 62.5/12.5 | |
| c. [673C > A]; [907T > G] | MHP | 0 | NR | |
| c. [1045T > C]; [1162G > A] | mPKU | 0 | 62.5/12.5 | |
| c. [1A > T]; [722G > A] | MHP | NR | 100/0 | |
| c. [1A > T]; [1042C > G] | cPKU | NR | 100/0 | |
| c. [23del]; [1162G > A] | cPKU | NR | 62.5/12.5 | |
| c. [60 + 5G > T]; [1162G > A] | cPKU | NR | 62.5/12.5 | |
| c. [165del]; [194T > C] | cPKU | NR | 84/0 | |
| c. [165T > G]; [208_210del] | MHP | NR | NR | |
| c. [208_210del]; [728G > A] | mPKU | NR | 0/6.67 | |
| c. [439C > T]; [782G > A] | mPKU | NR | 74/4 | |
| c. [441 + 5G > T]; [782G > A] | cPKU | NR | 74/4 | |
| c. [508C > G]; [842 + 1G > A] | mPKU | NR | 0 | |
| c. [508C > G]; [1243G > A] | MHP | NR | NR | |
| c. [508C > G]; [1315 + 1G > A] | mPKU | NR | 0/13 | |
| c. [533A > G]; [1162G > A] | MHP | NR | 62.5/12.5 | |
| c. [533A > G]; [1169A > G] | MHP | NR | 100/0 | |
| c. [791A > G]; [1315 + 5_1315 + 6insGTGTAACAG] | cPKU | NR | NR | |
| c. [809G > A]; [941C > A] | MHP | NR | NR | |
| c. [809G > A]; [1162G > A] | mPKU | NR | 62.5/12.5 | |
| c. [969 + 6T > C]; [1066-11G > A] | cPKU | NR | 0/6.09 | |
| c. [969 + 6T > A]; [1162G > A] | MHP | NR | 62.5/12.5 | |
| c. [1243G > A]; [1315 + 1G > A] | MHP | NR | 0/13 | |
| c. [1A > T]; [1243G > A] | MHP | NR | NR | Undetermined response |
| c. [533A > G]; [533A > G] | MHP | NR | NR | |
| c. [533A > G]; [809G > A] | MHP | NR | NR | |
| c. [625_626insC]; [625_626insC] | cPKU | NR | NR | |
| c. [688G > A]; [754C > T] | MHP | NR | 0 | |
| c. [727C > T]; [1243G > A] | MHP | No | NR | |
| c. [969 + 6T > A]; [1198A > C] | MHP | NR | NR | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vela-Amieva, M.; Alcántara-Ortigoza, M.A.; Ibarra-González, I.; González-del Angel, A.; Fernández-Hernández, L.; Guillén-López, S.; López-Mejía, L.; Carrillo-Nieto, R.I.; Belmont-Martínez, L.; Fernández-Lainez, C. An Updated PAH Mutational Spectrum of Phenylketonuria in Mexican Patients Attending a Single Center: Biochemical, Clinical-Genotyping Correlations. Genes 2021, 12, 1676. https://doi.org/10.3390/genes12111676
Vela-Amieva M, Alcántara-Ortigoza MA, Ibarra-González I, González-del Angel A, Fernández-Hernández L, Guillén-López S, López-Mejía L, Carrillo-Nieto RI, Belmont-Martínez L, Fernández-Lainez C. An Updated PAH Mutational Spectrum of Phenylketonuria in Mexican Patients Attending a Single Center: Biochemical, Clinical-Genotyping Correlations. Genes. 2021; 12(11):1676. https://doi.org/10.3390/genes12111676
Chicago/Turabian StyleVela-Amieva, Marcela, Miguel Angel Alcántara-Ortigoza, Isabel Ibarra-González, Ariadna González-del Angel, Liliana Fernández-Hernández, Sara Guillén-López, Lizbeth López-Mejía, Rosa Itzel Carrillo-Nieto, Leticia Belmont-Martínez, and Cynthia Fernández-Lainez. 2021. "An Updated PAH Mutational Spectrum of Phenylketonuria in Mexican Patients Attending a Single Center: Biochemical, Clinical-Genotyping Correlations" Genes 12, no. 11: 1676. https://doi.org/10.3390/genes12111676