
A SNP Mutation in Homeodomain-DDT (HD-DDT) Transcription Factor Results in Multiple Trichomes (mt) in Cucumber (Cucumis sativus L.)


 and
and 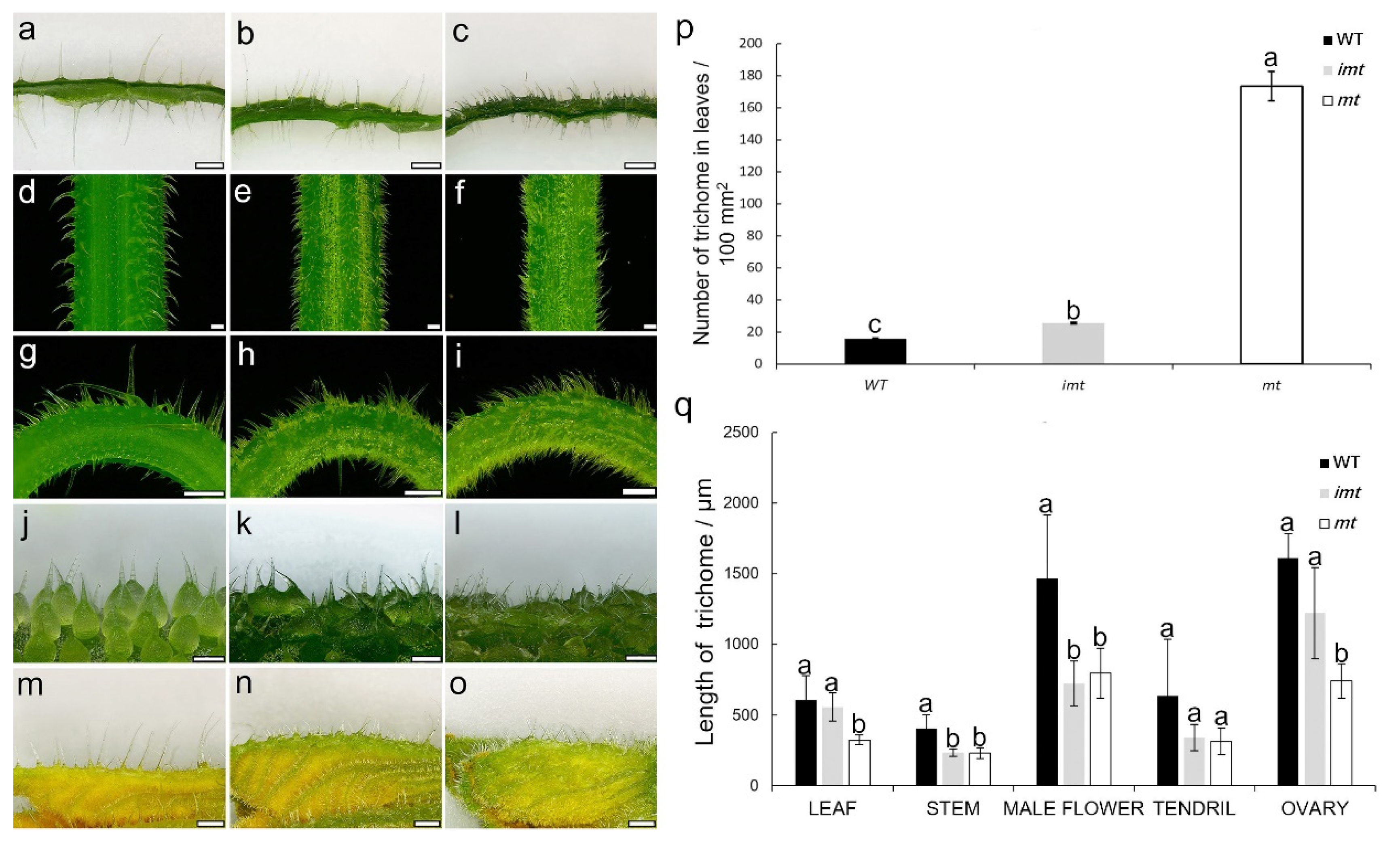{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Mapping Populations
2.2. Cryo-Scanning Electron Microscopy
2.3. BSA-Seq Analysis of mt Mutant
2.4. Genetic Mapping of mt Candidate Gene
2.5. Sequencing and Annotation of the mt Candidate Gene
2.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
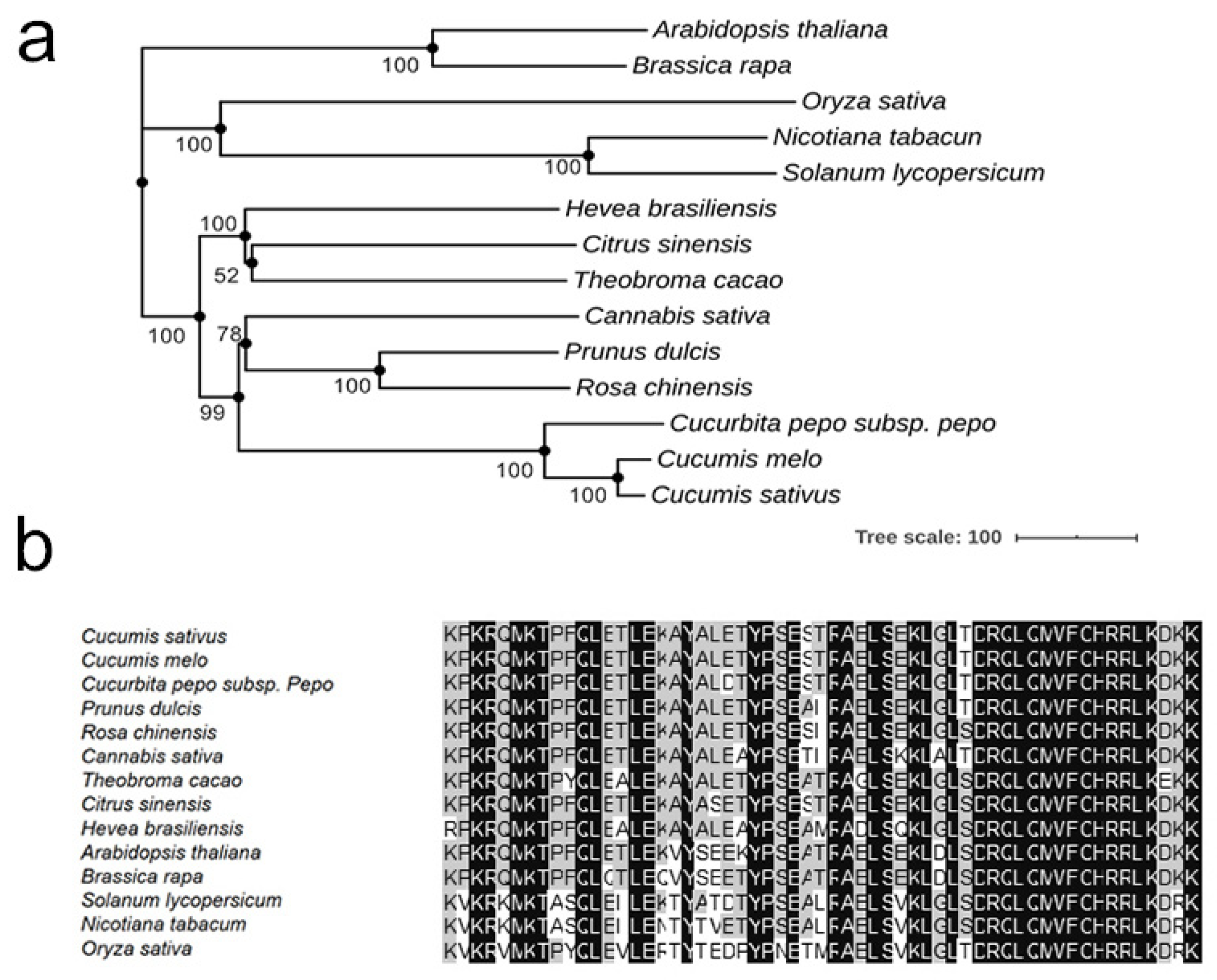
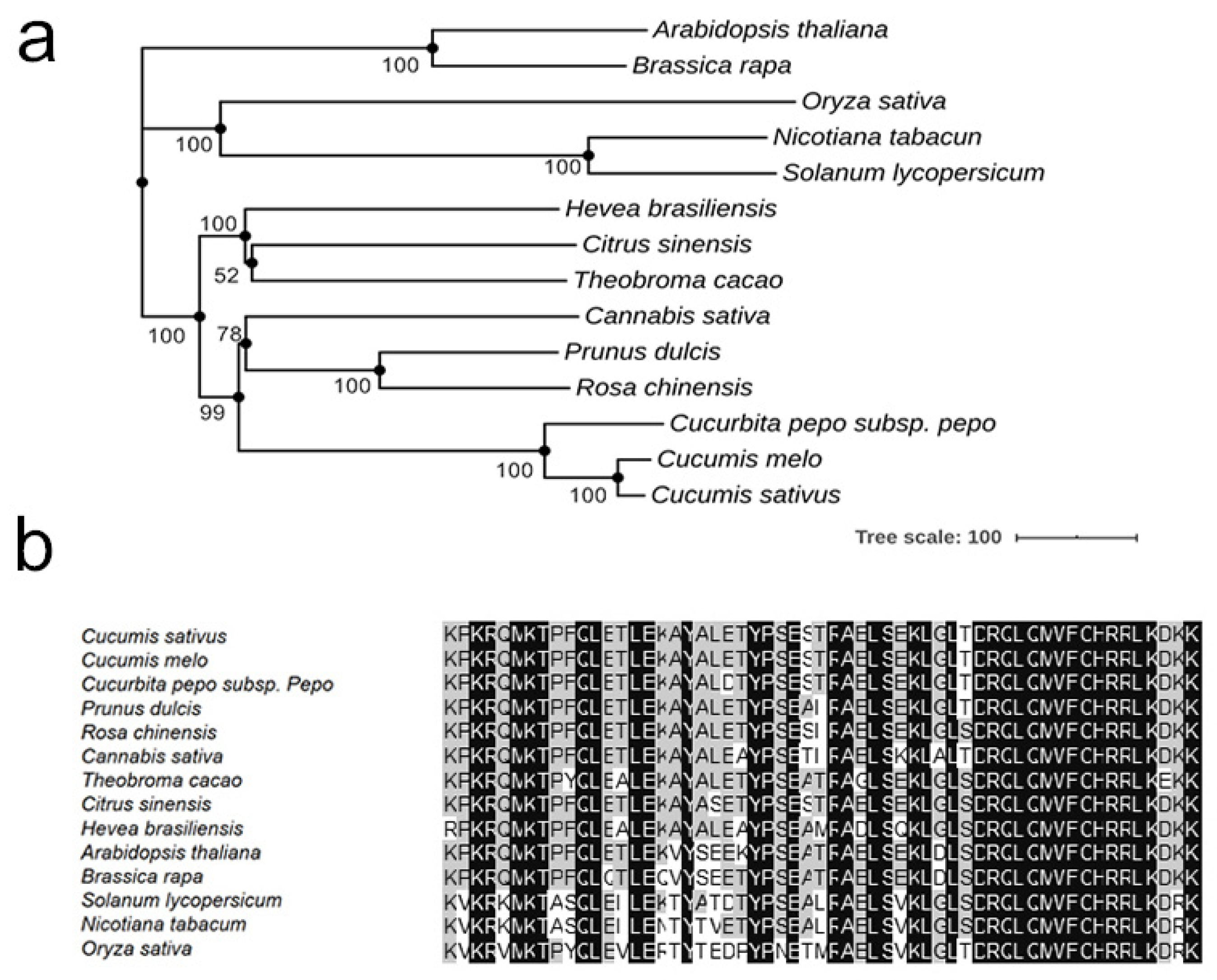
2.7. Sequence Alignments and Phylogenetic Tree Construction
2.8. Digital Gene Expression Analysis
3. Results
3.1. Morphological Characterization and Inheritance of a Multiple-Trichomes Mutant in Cucumber
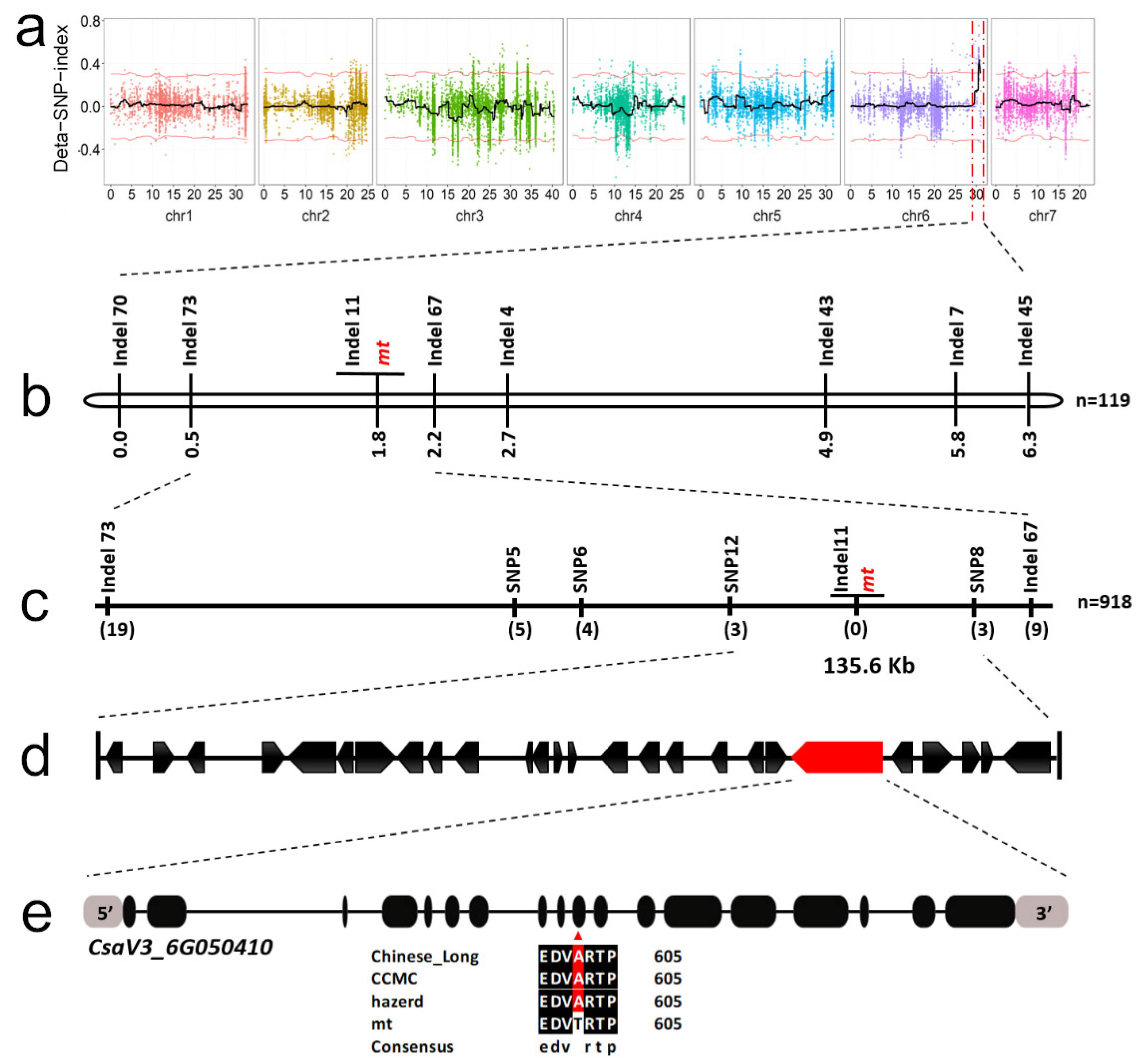
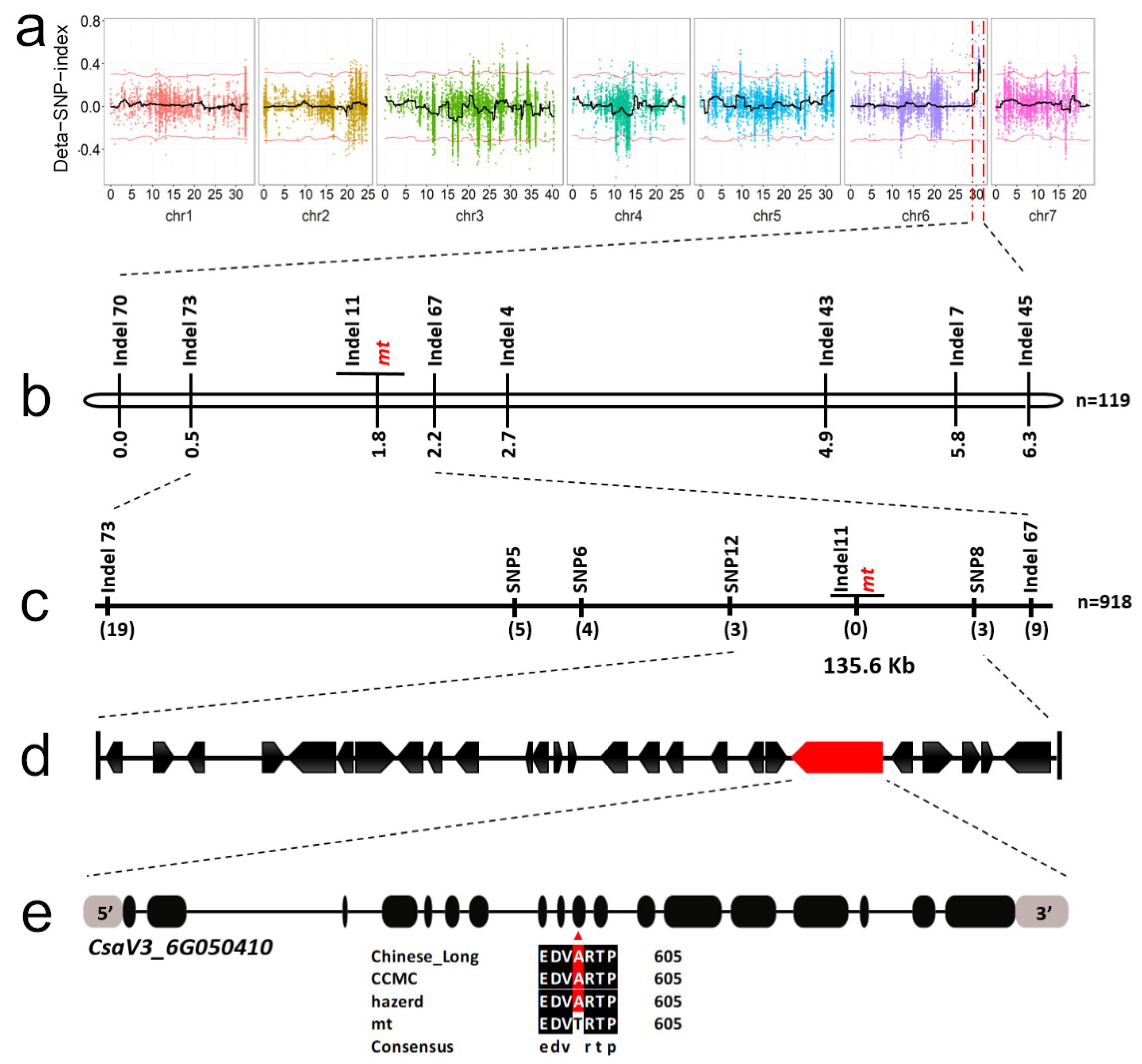
3.2. Linkage Analysis Identified a Candidate Mutation Site for mt Mutant
3.3. A Mutation in the HD-DDT Gene Resulted in the mt Mutant
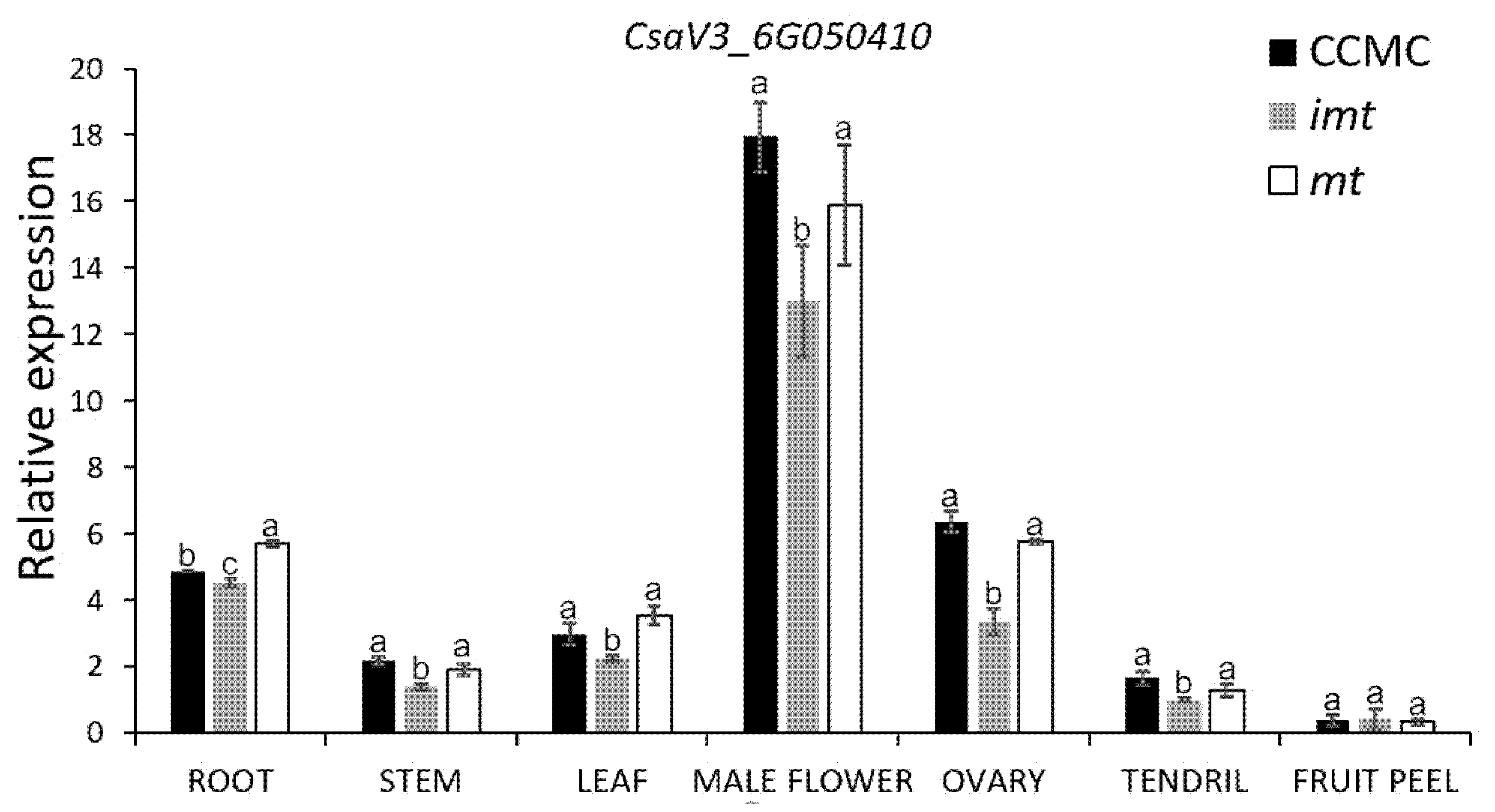
3.4. Expression Pattern of CsHD1 in Cucumber
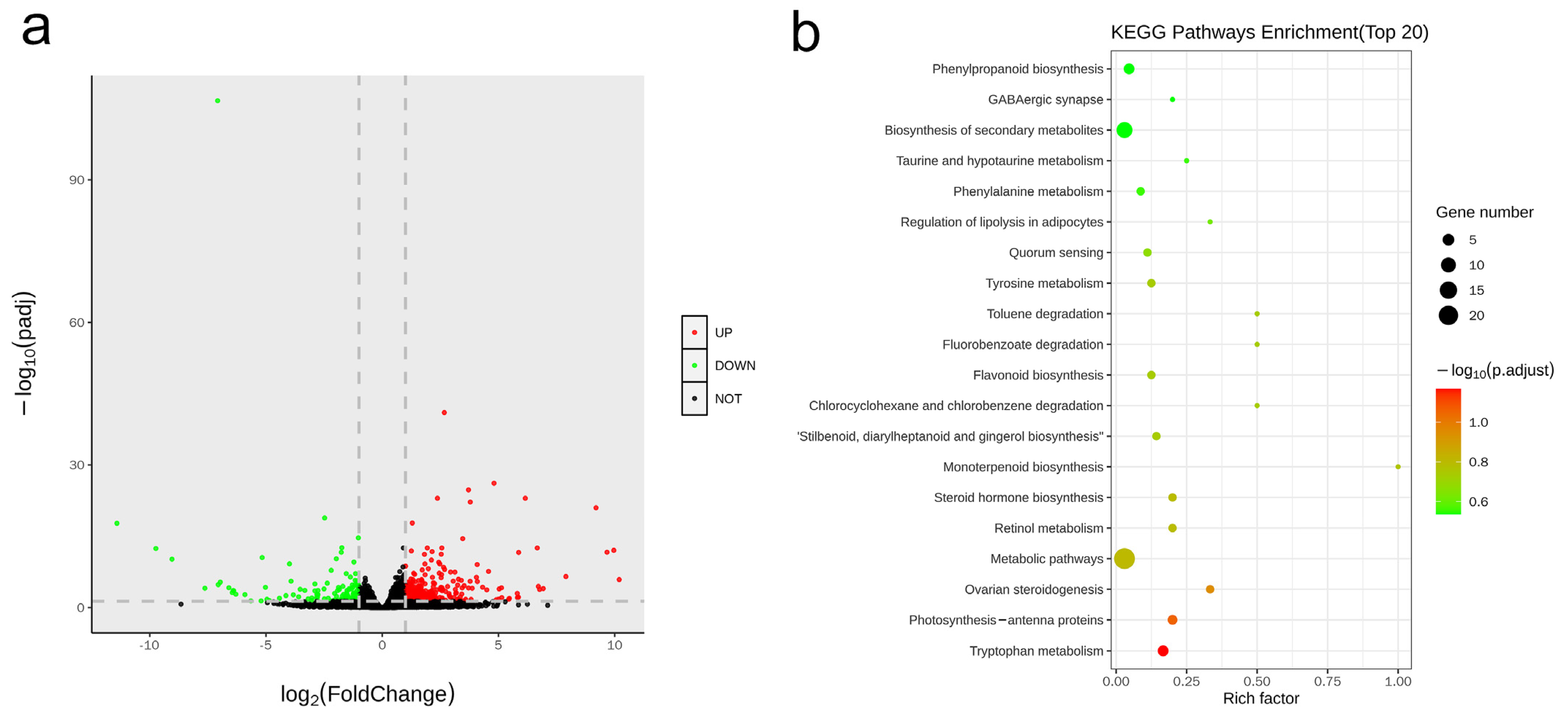
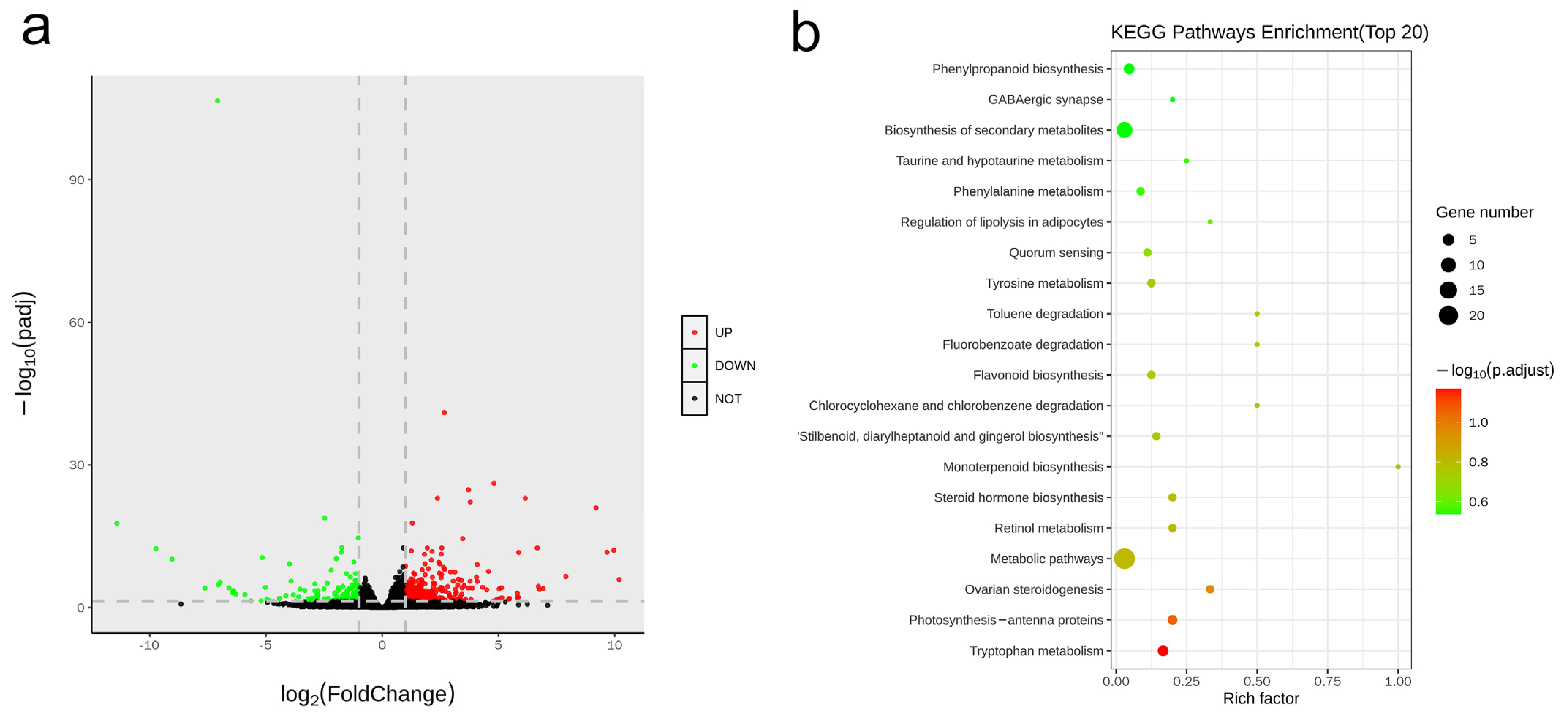
3.5. Transcriptome Profiling Reveals the Key Regulatory Molecules Involved in CsHD1-Dependent Trichome Development in Cucumber
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Balkunde, R.; Pesch, M.; Hulskamp, M. Trichome patterning in Arabidopsis thaliana: From genetic to molecular models. Plant Dev. 2010, 91, 299–321. [Google Scholar]
- Andrade, M.C.; Da Silva, A.A.; Neiva, I.P.; Oliveira, I.R.C.; De Castro, E.M.; Francis, D.M.; Maluf, W.R. Inheritance of type IV glandular trichome density and its association with whitefly resistance from Solanum galapagense accession LA1401. Euphytica 2017, 213, 52. [Google Scholar] [CrossRef]
- Schiefelbein, J. Cell-fate specification in the epidermis: A common patterning mechanism in the root and shoot. Curr. Opin. Plant Biol. 2003, 6, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; McRoberts, J.; Shi, F.; Moreno, J.E.; Jones, A.D.; Howe, G.A. The Flavonoid biosynthetic enzyme chalcone isomerase mod-ulates terpenoid production in glandular trichomes of tomato. Plant Physiol. 2014, 164, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Karabourniotis, G.; Papadopoulos, K.; Papamarkou, M.; Manetas, Y. Ultraviolet-bradiation absorbing capacity of leaf hairs. Physiol Plantarum. 1992, 86, 414–418. [Google Scholar] [CrossRef]
- Wagner, G.J.; Wang, E.; Shepherd, R.W. New approaches for studying and exploiting an old protuberance, the plant trichome. Ann. Bot. 2004, 93, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champagne, A.; Boutry, M. A comprehensive proteome map of glandular trichomes of hop (Humulus lupulus L.) female cones: Identification of biosynthetic pathways of the major terpenoid-related compounds and possible transport proteins. Proteomics 2017, 17, 1600411. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Reddy, V.A.; Panicker, D.; Mao, H.Z.; Kumar, N.; Rajan, C.; Venkatesh, P.N.; Chua, N.H.; Sarojam, R. Metabolic engineering of terpene biosynthesis in plants using a trichome-specific transcription factor MsYABBY5 from spearmint (Mentha spicata). Plant Biotechnol. J. 2016, 14, 1619–1632. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.D.; Wright, R.J.; Chandnani, R.; Goff, V.H.; Ingles, J.; Paterson, A.H. EMS-mutated cotton populations suggest overlapping genetic control of trichome and lint fiber variation. Euphytica 2015, 208, 597–608. [Google Scholar] [CrossRef]
- Bryant, L.; Patole, C.; Cramer, R. Proteomic analysis of the medicinal plant Artemisia annua: Data from leaf and trichome ex-tracts. Data Brief 2016, 7, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Feller, A.; Machemer, K.; Braun, E.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef] [PubMed]
- Serna, L.; Martin, C. Trichomes: Different regulatory networks lead to convergent structures. Trends Plant Sci. 2006, 11, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, D.G.; Herman, P.L.; Sivakumaran, S.; Esch, J.; Marks, M.D. A myb gene required for leaf trichome differentiation in Arabidopsis is expressed in stipules. Cell 1991, 67, 483–493. [Google Scholar] [CrossRef]
- Payne, C.T.; Zhang, F.; Lloyd, A.M. GL3 encodes a bHLH protein that regulates trichome development in arabidopsis through interaction with gl1 and TTG1. Genetics 2000, 156, 1349–1362. [Google Scholar] [CrossRef]
- Walker, A.R.; Davison, P.A.; Bolognesi-Winfield, A.C.; James, C.M.; Srinivasan, N.; Blundell, T.L.; Esch, J.J.; Marks, M.D.; Gray, J.C. The Transparent Testa Glabra1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. Plant Cell 1999, 11, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Kirik, V.; Simon, M.; Wester, K.; Schiefelbein, J.; Hulskamp, M. Enhancer of TRY and CPC 2 (ETC2) reveals redundancy in the region-specific control of trichome development of Arabidopsis. Plant Mol. Biol. 2004, 55, 389–398. [Google Scholar] [CrossRef]
- Schellmann, S.; Schnittger, A.; Kirik, V.; Wada, T.; Okada, K.; Beermann, A.; Thumfahrt, J.; Jürgens, G.; Hülskamp, M. TRIPTYCHON and CAPRICE mediate lateral inhibition during trichome and root hair patterning in Arabidopsis. EMBO J. 2002, 21, 5036–5046. [Google Scholar] [CrossRef] [Green Version]
- Traw, M.B.; Bergelson, J. Interactive effects of jasmonic acid, salicylic acid, and gibberellin on induction of trichomes in Arabidopsis. Plant Physiol. 2003, 133, 1367–1375. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.-C.; Hu, Q.-Q.; Li, W.; Chen, Y.; Han, L.-H.; Tao, M.; Wu, W.-Y.; Li, X.-B.; Huang, G.-Q. Cotton (Gossypium hirsutum) JAZ3 and SLR1 function in jasmonate and gibberellin mediated epidermal cell differentiation and elongation. Plant Cell Tissue Organ. Cult. (PCTOC) 2018, 133, 1–14. [Google Scholar] [CrossRef]
- Zhou, Z.J.; Sun, L.L.; Zhao, Y.Q.; An, L.J.; Yan, A.; Meng, X.F.; Gan, Y.B. Zinc finger protein 6 (ZFP6) regulates trichome initiation by integrating gibberellin and cytokinin signaling in Arabidopsis thaliana. New Phytol. 2013, 198, 699–708. [Google Scholar] [CrossRef]
- Chang, J.; Yu, T.; Yang, Q.; Li, C.; Xiong, C.; Gao, S.; Xie, Q.; Zheng, F.; Li, H.; Tian, Z.; et al. Hair, encoding a single C2H2 zinc-finger protein, regulates multicellular trichome formation in tomato. Plant J. 2018, 96, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, L.; Zheng, S.; Liu, Z.; Wu, X.; Gao, Z.; Cao, C.; Lina, W.; Ren, Z. A fragment substitution in the promoter of CsHDZIV11/CsGL3 is responsible for fruit spine density in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2016, 129, 1289–1301. [Google Scholar] [CrossRef]
- Pan, Y.; Bo, K.; Cheng, Z.; Weng, Y. The loss-of-function GLABROUS 3 mutation in cucumber is due to LTR-retrotransposon in-sertion in a class IV HD-ZIP transcription factor gene CsGL3 that is epistatic over CsGLBMC. Plant Biol. 2015, 15, 302. [Google Scholar]
- Chen, C.; Liu, M.; Jiang, L.; Liu, X.; Zhao, J.; Yan, S.; Yang, S.; Ren, H.; Liu, R.; Zhang, X. Transcriptome profiling reveals roles of meristem regulators and polarity genes during fruit trichome development in cucumber (Cucumis sativus L.). J. Exp. Bot. 2014, 65, 4943–4958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Cao, C.; Zhang, C.; Zheng, S.; Wang, Z.; Ren, Z. The identification of Cucumis sativus Glabrous 1 (CsGL1) required for the formation of trichomes uncovers a novel function for the homeodomain-leucine zipper I gene. J. Exp. Bot. 2015, 66, 2515–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.L.; Pan, J.S.; Guan, Y.; Zhang, W.W.; Bie, B.B.; Wang, Y.L.; He, H.L.; Lian, H.L.; Cai, R. Micro-trichome as a class I homeodo-main-leucine zipper gene regulates multicellular trichome development in Cucumis sativus. J. Integr. Plant Biol. 2015, 57, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lv, D.; Pan, J.; Zhang, K.; Wen, H.; Chen, Y.; Du, H.; He, H.; Cai, R.; Pan, J.; et al. A SNP of HD-ZIP I transcription factor leads to distortion of trichome morphology in cucumber (Cucumis sativus L.). BMC Plant Biol. 2021, 21, 182. [Google Scholar] [CrossRef]
- Cui, J.-Y.; Miao, H.; Ding, L.-H.; Wehner, T.C.; Liu, P.-N.; Wang, Y.; Zhang, S.-P.; Gu, X.-F. A new glabrous gene (csgl3) identified in trichome development in cucumber (Cucumis sativus L.). PLoS ONE 2016, 11, e0148422. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-L.; Nie, J.-T.; Chen, H.-M.; Guo, C.-L.; Pan, J.; He, H.-L.; Pan, J.-S.; Cai, R. Identification and mapping of Tril, a homeodomain-leucine zipper gene involved in multicellular trichome initiation in Cucumis sativus. Theor. Appl. Genet. 2016, 129, 305–316. [Google Scholar] [CrossRef]
- Du, H.; Wang, G.; Pan, J.; Chen, Y.; Xiao, T.; Zhang, L.; Zhang, K.; Wen, H.; Xiong, L.; Yu, Y.; et al. The HD-ZIP IV transcription factor Tril regulates fruit spine density through gene dosage effects in cucumber. J. Exp. Bot. 2020, 71, 6297–6310. [Google Scholar] [CrossRef]
- Chen, C.H.; Yin, S.; Liu, X.W.; Liu, B.; Yang, S.; Xue, S.D.; Cai, Y.L.; Black, K.; Liu, H.L.; Dong, M.M.; et al. The WD-repeat protein CsTTG1 regulates fruit wart formation through interaction with the homeodomain-heucine zipper I protein mict. Plant Physiol. 2016, 171, 1156–1168. [Google Scholar]
- Yang, S.; Cai, Y.; Liu, X.; Dong, M.; Zhang, Y.; Chen, S.; Zhang, W.; Li, Y.; Tang, M.; Zhai, X.; et al. A CsMYB6-CsTRY module regulates fruit trichome initiation in cucumber. J. Exp. Bot. 2018, 69, 1887–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Yang, X.; Wang, Y.; Nie, J.; Yang, Y.; Sun, J.; Du, H.; Zhu, W.; Pan, J.; Chen, Y.; et al. Identification and mapping of ts (tender spines), a gene involved in soft spine development in Cucumis sativus. Theor. Appl. Genet. 2017, 131, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Liu, P.; Shi, L.; Miao, H.; Bo, K.; Wang, Y.; Gu, X.; Zhang, S. Combined fine mapping, genetic diversity, and transcriptome profiling reveals that the auxin transporter gene ns plays an important role in cucumber fruit spine development. Theor. Appl. Genet. 2018, 131, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Q.; Zhang, W.W.; He, H.L.; Nie, J.T.; Bie, B.B.; Zhao, J.L.; Ren, G.L.; Li, Y.; Zhang, D.B.; Pan, J.S.; et al. Tuberculate fruit gene Tu encodes a C2H2 zinc finger protein that is required for the warty fruit phenotype in cucumber (Cucumis sativus L.). Plant J. 2014, 78, 1034–1046. [Google Scholar] [CrossRef]
- Jain, M.; Tyagi, A.K.; Khurana, J.P. Genome-wide identification, classification, evolutionary expansion and expression analyses of homeobox genes in rice. FEBS J. 2008, 275, 2845–2861. [Google Scholar] [CrossRef]
- Mukherjee, K.; Brocchieri, L.; Bürglin, T.R. A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol. Biol. Evol. 2009, 26, 2775–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouannic, S.; Collin, M.; Vidal, B.; Verdeil, J.L.; Tregear, J.W. A class IKNOX gene from the palm species Elaeis guineensis (Arecaceae) is associated with meristem function and a distinct mode of leaf dissection. New Phytol. 2007, 174, 551–568. [Google Scholar] [CrossRef]
- Castellano, M.; Sablowski, R. Intercellular signalling in the transition from stem cells to organogenesis in meristems. Curr. Opin. Plant Biol. 2005, 8, 26–31. [Google Scholar] [CrossRef]
- Long, J.A.; Moan, E.I.; Medford, J.I.; Barton, M.K. A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature 1996, 379, 66–69. [Google Scholar] [CrossRef]
- Sato, Y.; Hong, S.K.; Tagiri, A.; Kitano, H.; Yamamoto, N.; Nagato, Y.; Matsuoka, M. A rice homeobox gene, OSH1, is expressed before organ differentiation in a specific region during early embryogenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 8117–8122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Wu, Q.F.; Xie, Y.H.; Ru, H.; Xie, F.; Wang, X.Y.; Wang, C.Y. Ectopic expression of the Pttkn1 gene induces alterations in the morphology of the leaves and flowers in Petunia hybrida Vilm. J. Integr. Plant Biol. 2005, 47, 1153–1158. [Google Scholar] [CrossRef]
- Xu, Q.; Gao, N.; Ruan, M.; Ding, W.; Hu, X.; Wang, C.; Wang, X. Ectopic expression of the PttKN1 gene induced altered leaf morphology and hormonal levels in transgenic tobacco. J. Plant Biochem. Biotechnol. 2015, 24, 197–203. [Google Scholar] [CrossRef]
- Song, M.; Wei, Q.; Wang, J.; Fu, W.; Qin, X.; Lu, X.; Cheng, F.; Yang, K.; Zhang, L.; Yu, X.; et al. Fine mapping of CsVYL, conferring virescent leaf through the regulation of chloroplast development in cucumber. Front. Plant Sci. 2018, 9, 432. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, H.; Huang, W.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience 2019, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Vernoud, V.; Laigle, G.; Rozier, F.; Meeley, R.B.; Perez, P.; Rogowsky, P. The HD-ZIP IV transcription factor OCL4 is necessary for trichome patterning and anther development in maize. Plant J. 2009, 59, 883–894. [Google Scholar] [CrossRef]
- Yang, C.; Li, H.; Zhang, J.; Luo, Z.; Gong, P.; Zhang, C.; Li, J.; Wang, T.; Zhang, Y.; Lu, Y.; et al. A regulatory gene induces trichome formation and embryo lethality in tomato. Proc. Natl. Acad. Sci. USA 2011, 108, 11836–11841. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Zhang, L.; Chen, G.; Wen, H.; Chen, Y.; Du, H.; Zhao, J.; He, H.; Lian, H.; Chen, H.; et al. Study of micro-trichome (mict) reveals novel connections between transcriptional regulation of multicellular trichome development and specific metabolism in cucumber. Hortic. Res. 2021, 8, 21. [Google Scholar] [CrossRef]
- Zhu, H.; Sun, X.; Zhang, Q.; Song, P.; Hu, Q.; Zhang, X.; Li, X.; Hu, J.; Pan, J.; Sun, S.; et al. GLABROUS (CmGL) encodes a HD-ZIP IV transcription factor playing roles in multicellular trichome initiation in melon. Theor. Appl. Genet. 2018, 131, 569–579. [Google Scholar] [CrossRef]
- Kazama, H.; Dan, H.; Imaseki, H.; Wasteneys, G. Transient exposure to ethylene stimulates cell division and alters the fate and polarity of hypocotyl epidermal cells. Plant Physiol. 2004, 134, 1614–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, T.; Bartholomew, E.; Black, K.; Dong, M.; Zhang, Y.; Yang, S.; Cai, Y.; Xue, S.; Weng, Y.; et al. Comprehensive analysis of NAC transcription factors and their expression during fruit spine development in cucumber (Cucumis sativus L.). Hortic. Res. 2018, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Wang, X.; Guo, H.; Cheng, Y.; Hou, C.; Chen, J.-G.; Wang, S. NTL8 regulates trichome formation in arabidopsis by directly activating R3 MYB genes TRY and TCL1. Plant Physiol. 2017, 174, 2363–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Song, M.; Cheng, F.; Zhang, M.; Davoudi, M.; Chen, J.; Lou, Q. A SNP Mutation in Homeodomain-DDT (HD-DDT) Transcription Factor Results in Multiple Trichomes (mt) in Cucumber (Cucumis sativus L.). Genes 2021, 12, 1478. https://doi.org/10.3390/genes12101478
Yang Z, Song M, Cheng F, Zhang M, Davoudi M, Chen J, Lou Q. A SNP Mutation in Homeodomain-DDT (HD-DDT) Transcription Factor Results in Multiple Trichomes (mt) in Cucumber (Cucumis sativus L.). Genes. 2021; 12(10):1478. https://doi.org/10.3390/genes12101478
Chicago/Turabian StyleYang, Zhige, Mengfei Song, Feng Cheng, Mengru Zhang, Marzieh Davoudi, Jinfeng Chen, and Qunfeng Lou. 2021. "A SNP Mutation in Homeodomain-DDT (HD-DDT) Transcription Factor Results in Multiple Trichomes (mt) in Cucumber (Cucumis sativus L.)" Genes 12, no. 10: 1478. https://doi.org/10.3390/genes12101478
APA StyleYang, Z., Song, M., Cheng, F., Zhang, M., Davoudi, M., Chen, J., & Lou, Q. (2021). A SNP Mutation in Homeodomain-DDT (HD-DDT) Transcription Factor Results in Multiple Trichomes (mt) in Cucumber (Cucumis sativus L.). Genes, 12(10), 1478. https://doi.org/10.3390/genes12101478