Abstract
The lamellar body (LB) of the alveolar type II (ATII) cell is a lysosome-related organelle (LRO) that contains surfactant, a complex mix of mainly lipids and specific surfactant proteins. The major function of surfactant in the lung is the reduction of surface tension and stabilization of alveoli during respiration. Its lack or deficiency may cause various forms of respiratory distress syndrome (RDS). Surfactant is also part of the innate immune system in the lung, defending the organism against air-borne pathogens. The limiting (organelle) membrane that encloses the LB contains various transporters that are in part responsible for translocating lipids and other organic material into the LB. On the other hand, this membrane contains ion transporters and channels that maintain a specific internal ion composition including the acidic pH of about 5. Furthermore, P2X4 receptors, ligand gated ion channels of the danger signal ATP, are expressed in the limiting LB membrane. They play a role in boosting surfactant secretion and fluid clearance. In this review, we discuss the functions of these transporting pathways of the LB, including possible roles in disease and as therapeutic targets, including viral infections such as SARS-CoV-2.
1. Introduction
Lamellar bodies (LBs) are specialized lipid storage and secretory organelles found in various cells [1]. The most studied LBs are found in keratinocytes of the skin [2,3] and in alveolar type II (ATII) epithelial cells in the lung [4,5]. Next to lipids, LBs may also contain cell-type specific proteins and lytic enzymes. Due to their acidic pH and the shared compositional and physiological features with lysosomes, LBs have also been classified as lysosome-related organelles (LROs) [6,7].
LBs in ATII cells are storage organelles for pulmonary surfactant. Pulmonary surfactant is a complex mix of >90% lipids (mainly phospholipids) and specialized surfactant proteins. It is stored as densely packed multilamellar structures within LBs [8,9,10] and secreted into the alveolar lumen via exocytosis of LBs [5,11,12,13]. Secretion of pulmonary surfactant is essential for lung function. Defects in surfactant biogenesis or function are linked to a variety of severe diseases, some of which are directly related to perturbance of LB biogenesis and function [14,15,16,17].
The major function of pulmonary surfactant in the lung is the reduction of surface tension and stabilization of alveoli during respiration [18,19]. The surface tension in the lung creates a strong retractive force that tends to expel air and cause shrinkage of the organ. This was first discovered by von Neergaard in 1929 [20]. In the 1950s and early 1960s, a surface-active material (surfactant) that reduces the surface tension in the lungs was isolated and characterized by Clements [21,22]. By that time, LBs of the ATII cells were identified as the intracellular storage organelle for surfactant [4,23]. It was also shown that surfactant deficiency causes IRDS (infant respiratory distress syndrome), the first disease clearly related to immature LB biosynthesis in neonates that could be treated by surfactant replacement [24,25].
The first evidence in favor of an exocytotic release mechanism was provided by electron microscopy (EM) [26,27]. It has also been found that the phospholipid composition of LBs was similar to that of whole lung surfactant obtained from broncho–alveolar lavage (BAL) [28,29] and that LB exocytosis accounts for alveolar phospholipid composition [30].
Surfactant also contains four specific proteins (SP-A, SP-B, SP-C, and SP-D), which account for about 10% by weight [31]. They differ in their routes of secretion: The small hydrophobic SP-B and SP-C are localized within LBs and are co-secreted with phospholipids upon LB exocytosis. They are believed to play a role in enabling the formation of a highly organized, DPPC (dipalmitoyl phosphatidylcholine)-enriched, surface film [32]. The large, hydrophilic, surfactant proteins SP-A and SP-D, however, are secreted largely independently of LB contents. They are part of the collagenous family of proteins called collectins and appear to play an important role in host defense. SP-A and SP-D bind a wide spectrum of pathogens including viruses, bacteria, fungi, and pneumocystis (reviewed in [33]) fulfilling a crucial role in the innate pulmonary immune response, in addition to surfactant providing a physical barrier for pathogens, including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [34,35].
Many of the key features of alveolar LBs for surfactant maturation, including loading of lipids, maintenance of an acidic pH and intraorganellar ion homeostasis, as well as exocytosis and secretion of surfactant depend on functional ion channels and transport proteins localized on LBs. Despite this importance of the LB transportosome for LB homeostasis and function as well as for maintaining the vital surfactant system in the alveoli, our understanding thereof is far from completion. Within this review, we aim to summarize current knowledge on ion channel and transporter expression on LBs and their implication for LB function (physiology) and potential contribution to lung diseases (pathophysiology).
2. Ion Channels and Transporters on Lamellar Bodies
Historically, interest in ion channels and transporters expressed on LBs in ATII cells was linked to their main function, the storage of surfactant lipids and proteins. Surfactant proteins, either freshly synthesized surfactant proteins B (SP-B) and C (SP-C) or surfactant proteins that are recycled from the alveolar space, reach the LBs through intracellular vesicle trafficking via multivesicular bodies or the endosomal recycling route, respectively [10,36,37]. Surfactant lipids, however, reach the LBs via non vesicular pathways. This requires lipid transfer proteins for intracellular lipid trafficking in ATII cells [38] and subsequently transporters for lipid translocation into LBs [10]. To date, several lipid translocation proteins have been identified on LBs by either immunofluorescence, immunohistochemistry, or Western blot from isolated LBs (Figure 1). These include ATP-binding cassette sub-family A member 3 (ABCA3) [39,40,41], lysosomal integral membrane protein-2 (LIMP-2, also known as SCARB2) [42], Niemann–Pick C1 (NPC1) and Niemann–Pick C2 (NPC2) [43], and P4-type ATPase ATP8A1 [44].
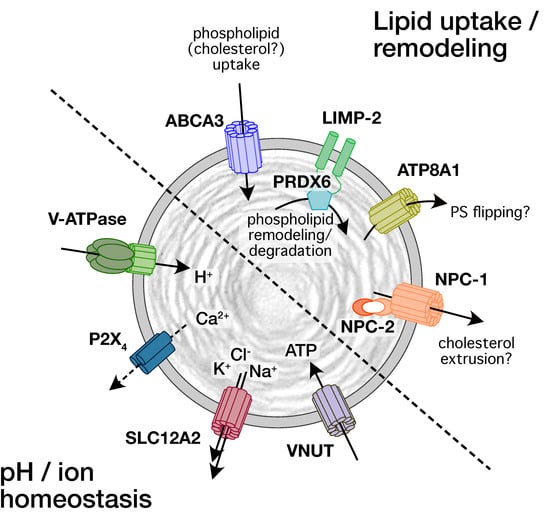
Figure 1.
Schematic representation of transporters and channels identified on LBs and their proposed function for lipid and pH/ion homeostasis.
Channels and transporters on LBs are required to maintain the intra-organellar proton and ion concentrations, but also facilitate surfactant secretion following LB exocytosis. Yet, apart from the identification of various subunits of the vacuolar V-ATPase [45,46,47] only an outwardly directed Na+-K+-2Cl- co-transporter (NKCC1 or SLC12A2, [45]), P2X4 purinergic receptors [41] and vesicular nucleotide transporter (VNUT or SLC17A9 [48]) have so far been identified on LBs and linked to a physiological function (Figure 1). Investigations of proton and electrical gradients also postulated the presence of Na+/H+ exchanger [45], Ca2+/H+ exchange(r), Ca2+-activated K+-channels, and possibly other K+-channels [49,50]. However, these have yet to be confirmed.
In addition, proteomic studies have expanded the repertoire of proteins expressed on LBs [51,52,53]. These include additional lipid transporters (ABCA8a), amino acid transporters (SLC3A2), ion transporters (SLC4A1), or ATPases including Na+/K+ (ATP1A1) or Ca2+-transporting (ATP2B1, ATP2B4) ATPases. Most of these were exclusively detected in the limiting membrane fraction of isolated LBs. However, the relevance of these proteins for LB homeostasis and function is yet to determined. It also needs to be considered that many of the ion channels and transporters expressed on LBs are trafficked from the LBs to the plasma membrane during LB exocytosis and surfactant secretion, and can subsequently be either recycled back to LBs or sent to lysosomes for degradation [54,55]. For example, we have recently shown that P2X4 receptors that have been delivered to the cell surface upon LB exocytosis are recycled from the plasma membered back to LBs. This recycling to acidic organelles is required for re-sensitization of the receptors [55].
Additional ion channels and transporters have also been identified on other species of LROs [56] and future studies are required to confirm whether these are also present on LBs. It is not unlikely that LROs share a similar décor of transporter to support common features amongst LROs, however, some ion channels and transporters may be unique on individual LROs serving their specific function [53].
3. Physiological Role for Ion Channels and Transporters on Lamellar Bodies
Pulmonary surfactant is predominately composed of lipids (approximately 90%) with phospholipids being the most abundant ones (80% in mass). Cholesterol is also present at significant levels (10% in mass) [57]. Lipid loading into LBs depends on specific lipid transporters expressed on the limiting membrane of LBs.
The best studied transporter is ABCA3 (reviewed in detail in [17]). Structure prediction and homology with other ABC transporters suggests that ABCA3 forms a lipid conduit channels in the membrane of LBs. The transfer of phospholipid species across the limiting membrane of LBs during their biogenesis is a highly energy-dependent process, catalyzed by binding and hydrolysis of ATP at two nucleotide-binding-domains [58,59,60]. It has been hypothesized that the energy accumulated during LB biogenesis confers an energy loaded state to the surfactant complexes, that converts LB particles (LBPs, i.e., the densely packed surfactant particles inside mature LBs) into a sort of pressurized particles, that readily transform into the surface-active interfacial film upon release from LBs [61,62] and contact with the air–liquid interface [63]. The detailed physiological substrates for ABCA3 are not yet fully characterized, but ample evidence suggests that ABCA3 facilitates loading of several phospholipid species into LBs. These include the main surfactant species phosphatidylcholine and phosphatidylglycerol, but also phosphatidylethanolamine and phosphatidylserine [60,64,65,66]. A role of ABCA3 in cholesterol loading is controversial [65,67].
LBs express cholesterol transporting LIMP-2 and the NPC2/NPC1 complex [42,43]. LIMP-2 and NPC1 transport cholesterol across the limiting membrane of lysosomes [68,69,70]. However, LIMP-2 and NPC1 shuttle cholesterol from the inside of lysosomes into the cytosol to facilitate cholesterol uptake into the cell. Hence cholesterol loading into LBs would require reverse orientation of either LIMP-2 or NPC1 in the LB membrane. Evidence against such inverted orientation of LIPM-2 comes from a recent study that suggested that LIMP-2 ferries the soluble enzyme peroxiredoxin 6 (PRDX6) to the lumen of LBs. This study further suggested that PRDX6 remains bound to a helical loop of LIMP-2 inside LBs. This loop is also localized on the luminal side in lysosomes, arguing against a reverse orientation of LIMP-2 in LBs [42]. Interestingly, PRDX6 exhibits phospholipase A2 (PLA2) activity in the acidic environment of the LB and plays a key role in LB phospholipid homeostasis [71,72]. PRDX6 facilitates the remodeling of unsaturated phosphatidylcholine (PC) to enrich desaturated phosphatidylcholine (DSPC) in pulmonary surfactant [73] but may also contribute to degradation of phospholipids [74]. Lack of PRDX6 is associated with increased LB phospholipid content [74,75]. It has been suggested that trapping PRDX6 to LIMP-2 in LBs facilitate interaction of PRDX6 with phospholipids that are shuttled through a hydrophobic tunnel in LIMP-2 [42,76]. The main function of LIMP-2 on LBs may therefore be linked to regulating LB phospholipid content rather than cholesterol loading into LBs. Similar results, that is, increases in LB phospholipid content, was observed in NPC1 and NPC2 mutant mice [77]. Moreover, LB cholesterol content was increased in ATII cells from these mice, arguing against a role of NPC1/NPC2 in cholesterol loading into LBs. Rather NPC1 (and LIMP-2) may be involved in extrusion of excess cholesterol from LBs that has accumulated during LB biogenesis. Accumulation could result from fusion of cholesterol containing lysosomes with multivesicular bodies (during LB biogenesis) or from cholesterol that is released by lipases from cholesteryl esters to release fatty acids [78]. In such a model, cholesterol is enriched and needs to be eliminated from the LB lumen to adjust surfactant cholesterol levels. Recently, it has also been suggested that increased vesicular cholesterol levels reduces fusion pore expansion and hence could limit secretion [79].
Whether ATP8A1, which is expressed on LBs, contributes to lipid loading into LBs or modulating surfactant lipid composition needs to be confirmed. ATP8A1 is a member of the Class P4 ATPases that flip phospholipids from one side of a membrane to the other using ATP hydrolysis as an energy source [80,81]. ATP8A1 has a high specificity for flipping phosphatidyl-serine (PS) across membranes [82]. Again, ATP8A1 normally flips PS from the luminal to the cytoplasmic leaflet of intracellular organelles [83], arguing against an active loading mechanism. Extrusion of excess PS accumulated during PB morphogenesis could be an alternative function, but again, no evidence so far supports such a mechanism. Recently it’s been discussed that a more likely role of ATP8A1 flippase activity is in preparation of LBs for exocytosis [44] similar to the observation that PS facilitates fusion of insulin-containing granules with the PM in pancreatic beta cells β and silencing of flippases impairs insulin secretion [84,85].
The lumen of LBs is acidic (pH of 5–5.5) [50], contains high concentrations of ionized Ca2+ [86,87], and has been proposed to only contain very little free water [61,88]. Establishing and maintaining such conditions requires transmembrane transport of ions and water.
Acidification of isolated LBs depends on ATP and expression of the vacuolar V-ATPase on the LB membrane [45,46,50]. The acidic pH within LB is crucial for surfactant protein B and C processing [89,90,91] and the packaging of surfactant lipids [92]. In particular, the degradation and the remodeling of surfactant phospholipids by PRDX6, which expresses PLA2 and lysophosphatidylcholine acyl transferase (LPCAT) activities is strongly increased at acidic pH [71,93,94].
Besides the importance of intralumenal acidification for surfactant maturation, a shift in pH also affects intralumenal ion concentrations. It has been suggested that V-ATPase generates an electropositive LB lumen that ultimately affects the electrochemical gradient across the LB membrane via secondary active transport mechanisms. It´s been shown that the H+ influx is neutralized by a Cl- uptake which in turn increases V-ATPase activity in a positive feedback loop [45]. This probably accounts for the Cl- accumulation in LBs [86]. The entry of Cl- could also reduce the K+-dependent electrical gradient contributing to the trans LB membrane potential. It´s also been proposed that a Na+/H+ exchanger dissociates the electrical and chemical H+ gradient exchanging H+ for Na+. The V-ATPase could also serve as the driving force for the outwardly directed Na+-K+-2Cl- co-transporter [45].
An intimate link exists between acidic pH in LBs and luminal Ca2+ concentrations. Ca2+ uptake depends on the low pH in LBs [45]. Again, this is likely via secondary active transport of Ca2+ by a Ca2+/H+ exchanger [49]. Additional mechanism for Ca2+ uptake into LBs have been discussed [49] but may not be relevant at the very low cytoplasmic Ca2+ concentrations found in ATII cells [95,96]. Alkalinization of LBs, e.g., by inhibition of the vacuolar V-ATPase by bafilomycin A1 or H+ sequestration by ambroxol results in Ca2+ release from LBs and stimulates LB exocytosis and surfactant secretion from LBs [46,97]. The detailed pathways for this pH-dependent Ca2+ release are not yet fully understood. Two pore channels (TPCs), mucolipin TRP channels (TRPMLs), and P2X purinergic receptors have been linked to pH-dependent Ca2+ release from lysosomes [98,99,100]. We have recently reported that P2X4 receptors are expressed on LBs [41]. Alkalinization induced Ca2+ release from LBs through P2X4 receptors [48]. Activation of P2X4 receptors also depends on the presence of its natural ligand ATP. ATP is loaded into LBs by VNUT and present at high concentrations (∼1.9 mM) inside LBs [48]. The role for this alkalinization-induced, intracellular Ca2+ release is not yet clear. In lysosomes P2X4 forms channels activated by luminal ATP in a pH-dependent manner [101] and P2X4-mediated endolysosomal Ca2+ release promotes lysosome fusion [98]. Such fusion events have not been observed for mature LBs but may be relevant during LB biogenesis or in case of cell damage, when alkalization promotes a massive Ca2+ release from LBs to induce autophagy [102]. The most important function for P2X4 receptors on LBs, however, is to facilitate secretion and activation of pulmonary surfactant. Upon exocytosis of LBs and opening of the fusion pore, the intraluminal pH is rapidly neutralized and luminal ATP may activate the P2X4 receptors. This results in a fusion-activated cation entry (FACE) via P2X4 at the site of LB exocytosis. The resulting Ca2+ elevation around the fused LB accelerates the widening of the narrow fusion pore that restricts efficient release (secretion) of poorly soluble surfactant, i.e., LBPs [103,104,105,106]. This vital function is also supported by the observation that the Ca2+ concentration in exocytotic LBs is higher than in perinuclear LBs [86]. Moreover, FACE drives a net transepithelial fluid transport from the alveolar lumen. The resulting reduction of alveolar lining fluid thickness promotes direct contact between newly released surfactant and the air–liquid interphase, thus facilitating its adsorption and activation [107,108]. The multiple functions of FACE have been reviewed and represented in detail, recently [105,107,109].
Studies analyzing the structural organization of LBs propose a continuous intraorganellar dehydration during maturation of LBs as a result of the massive lipid accumulation by ABCA3 [61,88]. It´s been proposed that phospholipids adopt a particular high-energy structure as a result of the energy accumulated during continuous packing of lipids into the limited volume of LBs. The high level of dehydration might be promoted both by the high packing and as a consequence of osmotic stress induced by the segregation of large protein complexes out from the tightly packed multilamellar arrays [61]. Whether water is predominantly removed from tightly packed LBPs (but still residing inside the LB) or irrevocably extruded from the LB is yet to be determined. In the latter case water can either permeate passively across the LB membrane or via water conducting structures [110,111,112,113]. However, such entities (e.g., aquaporins) have not yet been described on LBs. A loss of free water would also affect intraorganellar pH and ion concentrations and raises the question if and how these are regulated. The observation that mature LBs contain high amounts of Ca2+ [86] is consistent with the general property of lysosomes as sites of intracellular Ca2+ storage [114] and in addition may result from concentration as a result of water loss.
4. Pathophysiology Linked to Ion Channels and Transporters on Lamellar Bodies
Defects in LB channel or transporter function are linked to a variety of diseases in the lung, including interstitial lung diseases (e.g., pulmonary fibrosis). Owing to its classification as a LRO, these channels and transporters are also intrinsically associated with functions required in host defense, including viral infections. Defects in lipid transporters affect surfactant maturation and LB homeostasis. This can result in changes in LB size and phospholipid content that leads to ATII cell injury and ultimately to development of pulmonary fibrosis. Damaged ATII cells secrete mediators that lead to fibroblast proliferation and differentiation to highly active myofibroblasts, which deposit excessive amounts of extracellular matrix (ECM) [115,116]. This results in overall remodeling of the alveolar structure, formation of scar tissue, thickening of the alveolar septae, and an increase in tissue stiffness [117,118].
The pathophysiology of the ABCA3 transporter is by far the best studied and has been outlined in extensive reviews elsewhere [17,119]. Over 200 distinct ABCA3 mutations have been identified. These constitute the most prevalent group of mutations among genes associated with surfactant-related lung disorders (excellently summarized in [17]). Mutations can either affect intracellular trafficking of ABCA3 (type I), ATP hydrolysis (type II) or both (i.e., heterozygote mutations, type III). Compound heterozygous variants appear to account for increasing disease severity. Mutations in the ABCA3 gene are associated with surfactant dysfunction [120,121], familial lung diseases ranging from respiratory failure in newborns or interstitial lung disease in children [122,123] to idiopathic pulmonary fibrosis (IPF) or diffuse parenchymal lung disease in adults [124,125,126]. Most ABCA3 mutations result in an ABCA3 null phenotype and are lethal within the first months following birth. Ultrastructural examination of lung tissues from these patients revealed a lack of mature LBs. Instead, numerous smaller vesicles with denser inclusion bodies were observed [10,65,127]. ABCA3 mutations that result in either partial loss-of-function or promote a toxic gain-of-function phenotype are less frequent and are associated with a more chronic disease phenotype.
LBs also contain transporters linked to lysosomal storage diseases [43,128]. Lysosomal storage diseases (LSDs) are inherited metabolic disorders characterized by the gradual accumulation of substrates inside lysosomes or LROs (that is, ‘storage’), which ultimately leads to cell dysfunction and cell death [129,130]. LSDs comprise a group of 70 monogenic disorders, several of which are associated with a pulmonary phenotype [130], ranging from the upper airways to the lung parenchyma (reviewed in detail in [131]). In this review, we consider diseases that are explicitly associated with a transporter found on LBs–Niemann–Pick disease (NPD) and Hermansky–Pudlak Syndrome (HPS).
NPD consist of autosomal recessive disorders associated with neurologic symptoms, splenomegaly, and the storage of lipids, including cholesterol. Three subtypes of the disease are described: Niemann-Pick disease type A, B and C. Niemann-Pick disease type C (NPC) is caused by mutations of the NPC1 and NPC2 genes that result in impaired cellular processing and transport of low-density lipoprotein (LDL)-cholesterol. NPC1 and NPC2 are found on LBs [132]. The majority (95%) of cases of NPC disease are caused by a mutation in NPC1, while only about 5% are due to mutations in NPC2 [43]. NPC1-mutant type II cells had uncharacteristically larger LB (mean area 2-fold larger), while NPC2-mutant cells had predominantly smaller LBs (mean area 50% of normal) than wild type [77]. The manifestation of NPD in the lung is an interstitial lung disease which is characterized by reduced diffusion capacity for carbon monoxide (DLCO) [130]. Pulmonary involvement in NPC1 was reported to be as well associated with an obstructive and restrictive lung disease [132]. However, to our knowledge, a causal link between a specific mutation and defective cholesterol transport in LBs to clinical manifestations (i.e., interstitial lung disease, fibrosis) has yet to be established. Recently, it has also been hypothesized that the inherent cellular and biochemical abnormalities of lysosomal storage diseases (LSDs) in general, and Niemann–Pick disease type C (NPC) in particular, create “unfavorable” (lysosomal) environments for SARS-CoV-2 infectivity in the host cells, that is ATII cells (see also below) [133].
HPS is a rare, genetic, multisystem disorder characterized by oculocutaneous albinism, bleeding diathesis, immunodeficiency, granulomatous colitis, and pulmonary fibrosis (reviewed in [16]). It represents a family of disorders in which the biogenesis of lysosome-related organelles (LROs) is compromised [134,135]. The underlying defects of HPS are mutations in genes that encode proteins which are essential for the synthesis of LROs, including the Biogenesis of LRO Complexes (BLOC)-1, -2, and -3 and the Adaptor Protein 3 complex (AP-3). Three subtypes of HPS, HPS1, -2, and -4, are associated with giant LBs, impaired surfactant secretion, ATII cell hyperplasia and fibrosis [136,137,138,139]. In ATII cells, it was recently shown that AP-3 is required to sort LIMP-2 and PRDX6 [42] but also the P4-type ATPase ATP8A1 from early endosomes to LBs [44]. Impaired trafficking of the luminal enzyme PRDX6 was associated with increased LB phospholipid content, that may lead to ATII cell injury and fibrosis [42]. ATP8A1 is a flippase that hydrolysis ATP to flip phospholipids from one side of a membrane to the other. Disruption of the AP-3/ATP8A1 interaction causes activation of Yes-activating protein, a transcriptional coactivator that augments cell migration and ATII cell numbers. It was suggested that this causes a toxic gain-of-function that results in activation of a repair process associated with severe, progressive pulmonary disease and fibrosis [44].
Much less is known about functional roles for ion channels or receptors on LBs in the pathogenesis of pulmonary diseases. Here we speculate whether LBs, and in particular P2X4 receptors expressed on LBs, may play a role in SARS-CoV-2 induced disease. Viruses such as SARS-CoV and SARS-CoV-2 are taken up by ATII cells after binding of the viral spike protein (S protein) to the SARS receptor, the angiotensin converting enzyme-2 (ACE2), on the host cell [140,141,142,143,144]. Entry of viral mRNA into the cytoplasm occurs by two possible mechanisms: First, by direct fusion of the viral membrane with the plasma membrane. This mechanism is mediated by transmembrane protease serine subtype 2 (TMPRSS2)-induced cleavage of the ACE2-spike protein (ACE2-S) complex [145,146,147]. ATII cells express both, ACE2 and TMPRSS2 [140,148,149,150,151]. Second, and more importantly, binding of S protein to ACE2 induces endocytosis of the virion. Cleavage of the viral proteins’ ‘S2′ site’ by cathepsin in the acidic endo-lysosomal compartment then induces fusion of the viral envelope with the late endosome/lysosome membrane to release the viral genome into the cytoplasm [152,153,154,155,156]. This is also referred to as “uncoating” [157,158].
As outlined above, LBs have a vesicular pH of about 5 [50,159,160] and also contain proteases such as cathepsins and others that belong to the class of lysosomal enzymes required for the viral uncoating [52,161,162]. During biogenesis LBs derive from late endosomes (multivesicular bodies) and receive cargo from the trans-Golgi network. In theory, LBs or fusion products of LBs with other acidic (i.e., endo-lysosomal) compartments could therefore constitute intracellular hubs for uncoating and processing of SARS-CoV-2 in the lung. In line, SARS-CoV-2 has been found in LBs of induced pluripotent stem cell-derived ATII (iATII) cells maintained at air–liquid culture conditions [163]. However, SARS-CoV and SARS-CoV-2 viral particles were not readily visible in LBs of infected primary ATII cells investigated by electron microscopy [143,164].
It is tempting to speculate whether LBs contribute to SARS-CoV-2 infection or clearance. There is ample evidence that lysosomotropic drugs and endosomal acidification inhibitors like (hydroxy)chloroquine, that are weak bases and trapped within acidic compartments, exert anti-viral activities. Through alkalinization of the vesicles, these compounds inhibit the activity of cathepsin, which is required for the viral uncoating process, [157,165,166]. We have recently demonstrated that ambroxol, another drug with anti-viral activities used to ameliorate respiratory infections, accumulates in LBs, and neutralizes LB pH by the same mechanism [97]. It has also been shown that ambroxol prevents SARS-CoV-2 entry into other epithelial cells by inhibition of acid sphingomyelinase [167]. As noted above, ambroxol also elicits Ca2+ release from LBs via alkalinization-dependent activation of P2X4 receptors on LBs. This results in LB fusion with the plasma membrane and surfactant release [97]. Thus, alkalinizing drugs are not only potential inhibitors of viral uncoating in LBs but could also contribute to viral clearance from ATII cells via stimulation of LB exocytosis and surfactant (content) secretion.
P2X4 receptors expressed on LB membrane could also play an important role for viral inactivation and protection of the alveolus from SARS-CoV-2-induced alveolar barrier damage and acute respiratory distress syndrome (ARDS). ARDS by any cause, including SARS-CoV-2 infection, is accompanied by a loss of alveolar barrier function, surfactant deficiency or dysfunction, changes of alveolar compliance, hypoxia, and mechanical stress of alveolar units (reviewed in [168,169,170]). ARDS-associated cell damage and/or the activation of the immune response can lead to the release of cytokines, including ATP, from various cell types. ATP is a danger signal in the lung [171,172]. In healthy conditions, extracellular ATP within the respiratory lining fluid is continuously hydrolyzed by the action of ectonucleotidases [172] and it is intrinsically difficult to estimate the concentration of ATP in the alveolar hypophase between the surface layer of secreted surfactant and the apical membrane of ATI and ATII cells, since this liquid layer has an estimated mean thickness of about 200 nm only. However, strong ATP release from epithelial cells has been demonstrated for various ways of cell stress, including hypoxia or mechanical stress [173,174] and epithelial cell lysis induced by viral infection will inevitably lead to a strong increase of the ATP concentration in the adjacent pulmonary lining fluid. Accordingly, significant elevations of ATP concentrations have been demonstrated in the bronchoalveolar lavage (BAL) fluid under various pathophysiological conditions including SARS-CoV-2 infection [171,175]. This led to ATP-driven purinergic-inflammasome signaling [175]. In addition, ATP is released from LBs upon LB exocytosis [48], which can also lead to a (temporal) increase in extracellular ATP levels. The physiological significance of this last mechanism is not entirely clear but most likely part of a positive feedback mechanism enabling or facilitating the release of surfactant by expansion of the fusion pore. Although it is not entirely clear at which stage of the infection the ATP concentration starts to rise in the alveolar space, there is overwhelming plausibility that it can reach levels sufficient for activation of P2X4 receptors. P2X4 receptors on LBs are integrated into the apical membrane of ATII cells upon LB exocytosis and the ATP binding site of the P2X4 receptor is suddenly exposed to the alveolar lumen [41]. In the absence of ATP, this added apical membrane is quite “tight”, because even when cell membrane capacitance increases by more than 10%, the cell membrane conductance is unaltered [160]. When ATP is on the apical side, however, it can readily bind to the receptor, enabling the entry of cations (mainly Ca2+ and Na+ ions) from the alveolar hypophase (or edema fluid) into the cytoplasm of type II cells [41]. This has two consequences:
First, a Ca2+ signal is generated in the ATII cell, which we termed “fusion-activated Ca2+ entry”, because it follows the fusion of an LB with the plasma membrane (see above). FACE facilitates surfactant secretion from the fused LB, and activates LB exocytosis. Second, Na+ entry into the cytoplasm through the P2X4 receptor removes osmotically active Na+ from the alveolar lumen, facilitating alveolar clearance from excess edema fluid by osmosis [107]. Both mechanisms potentially protect the alveolus by restoring surface tension and alveolar compliance on the one hand, and by reducing fluid accumulation and improving oxygenation on the other hand. Last but not least, the release of surfactant as part of the innate immune system facilitates viral clearance in many ways, as reviewed in detail [176].
Interestingly, fusion-activated Ca2+ entry into type II pneumocytes is potentiated by ivermectin [41], an allosteric positive potentiator of P2X4 receptors [172,177,178]. Ivermectin, an antiparasitic drug used to treat persons or animals infested with helminths and insects, has gained substantial interest during the COVID-19 crisis, because it was used as an off-label drug to protect from COVID-19 infection. The issue is heavily debated and controversially discussed, as exemplified in a recent exchange of letters [179]. The general believes of a lack of plausibility that ivermectin may help to treat COVID-19 comes from its action on glutamate-gated chloride channels common to invertebrate nerve and muscle cells. These channels are not expressed in humans. Interestingly, the P2X4 receptor is never considered as a potential target of ivermectin in the COVID-19 literature. It should be considered that ivermectin is quite lipophilic, and lipophilic drugs can accumulate in the lipid environment of a LB [180]. For this reason, ivermectin may reach high local concentrations at the site of the P2X4 receptor when LBs are exocytosed. Given this experimental and theoretical background, ivermectin may be considered as an activator of the P2X4 receptor for a possible effect against COVID-19.
Apart from these speculative considerations regarding ivermectin, to date no specific treatment exists for disorders caused by defects in ion channels or transporters on LBs. This may be related to the fact that still little is known about the exact function of most of these proteins for LB biogenesis, homeostasis, a possible role in surfactant dysfunction or lung diseases related to ATII cell dysfunction. Even for ABCA3, which is clearly associated with development of various lung diseases, no specific treatment exists for disorders caused ABCA3 mutations [17]. One possible strategy to tackle this need could be a similar approach that has been very successful in the development of therapeutics for treatment of cystic fibrosis (CF). CF is caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) channel. Nearly 2000 cystic fibrosis-causing mutations have been described, many of which result in trafficking and / or channel conductance defects [181]. Development of small molecule CFTR modulators that either aid the trafficking (“correctors”) or improve the function (“potentiators”) of mutated CFTR or increase the amount of CFTR mRNA (and therefor protein, “amplifiers”) have been developed and are already approved drugs or entered clinical trials [182,183,184,185,186]. Such approaches (e.g., exploiting high-throughput screening technologies) may have the potential to identify novel drugs to correct mistrafficking and/or dysfunction of mutant ABCA3 isoforms.
Alternatively, the future may also bring novel therapeutic options based on gene therapy to treatment of genetic diseases that are beyond the reach of traditional approaches. The goal of gene therapy for genetic diseases is to achieve durable expression of the therapeutic gene or “transgene” at a level sufficient to ameliorate or cure disease symptoms with minimal adverse events. Hundreds of gene-therapy programs are in clinical development and several gene therapy products have already been approved [187].
Last but not least, repurposing of approved drugs could be a swift way to receive authority approval for novel applications. Several drugs have been developed and approved for clinical use that specifically target some of the channels and transporters expressed on LBs, e.g., clodronate, an anti-osteoporotic drug that inhibits VNUT [188]. Other approved drugs have been found to affect some of these proteins rather unexpectedly. Ambroxol, an over-the-counter mucolytic drug has been found to increase the levels of LIMP-2 in neurons of mice [189]. Whether this is related to the effects of ambroxol on intra-organellar pH levels needs to be seen.
Overall, although there are currently no specific treatments available for disorders related to ion channels and transporters on LBs, the future holds multiple strategic options for development of such therapeutics. However, in addition to drug development efforts, success will also depend on a better understanding of the exact physiological function of the individual transport proteins on LBs and how misfunction contributes to disease development.
5. Summary and Outlook
The pulmonary LB was originally discovered as the storage organelle for surfactant, the vital multi-molecular substance needed for breathing and preventing respiratory distress of the neonate [4]. Since then, our knowledge about the LB has been continuously expanding (Table 1), and its equipment as a LRO with a growing number of transporters, channels, and receptors prompts questions about its involvement in multiple additional functions/dysfunctions, including its exact role in the pathogenesis of chronic pulmonary disease such as fibrosis of various origin, and of its possible involvement in the processing and transmission of viral disease.

Table 1.
Channels and transporters that have been identified on LBs and for which a function and/or a possible relevance in lung disease has been reported. The table does not list channels and transporters identified on LBs for which no physiological function has been described so far.
Author Contributions
Writing—original draft preparation, review, and editing, P.D. and M.F.; visualization, P.D. and M.F.; funding acquisition, P.D. and M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummern: 175083951 and 251293561.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schmitz, G.; Muller, G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J. Lipid Res. 1991, 32, 1539–1570. [Google Scholar] [CrossRef]
- Wertz, P. Epidermal Lamellar Granules. Skin Pharmacol. Physiol. 2018, 31, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Matoltsy, A.G.; Parakkal, P.F. Membrane-coating granules of keratinizing epithelia. J. Cell Biol. 1965, 24, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Campiche, M. Les inclusions lamellaires des cellules alvéolaires dans le poumon du Raton. J. Ultrastruct. Res. 1960, 3, 302–312. [Google Scholar] [CrossRef]
- Dietl, P.; Haller, T. Exocytosis of lung surfactant: From the secretory vesicle to the air-liquid interface. Annu. Rev. Physiol. 2005, 67, 595–621. [Google Scholar] [CrossRef]
- Raposo, G.; Marks, M.S.; Cutler, D.F. Lysosome-related organelles: Driving post-Golgi compartments into specialisation. Curr. Opin. Cell Biol. 2007, 19, 394–401. [Google Scholar] [CrossRef]
- Weaver, T.E.; Na, C.L.; Stahlman, M. Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin. Cell Dev. Biol. 2002, 13, 263–270. [Google Scholar] [CrossRef]
- Castillo-Sánchez, J.C.; Cruz, A.; Pérez-Gil, J. Structural hallmarks of lung surfactant: Lipid-protein interactions, membrane structure and future challenges. Arch. Biochem. Biophys. 2021, 703, 108850. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Wimmer, B.H.; Winter, S.L.; Kolovou, A.; Laketa, V.; Chlanda, P. Post-correlation on-lamella cryo-CLEM reveals the membrane architecture of lamellar bodies. Commun. Biol. 2021, 4, 137. [Google Scholar] [CrossRef]
- Olmeda, B.; Martínez-Calle, M.; Pérez-Gil, J. Pulmonary surfactant metabolism in the alveolar airspace: Biogenesis, extracellular conversions, recycling. Ann. Anat. 2017, 209, 78–92. [Google Scholar] [CrossRef]
- Mason, R.J.; Voelker, D.R. Regulatory mechanisms of surfactant secretion. Biochim. Biophys. Acta 1998, 1408, 226–240. [Google Scholar] [CrossRef]
- Frick, M.; Eschertzhuber, S.; Haller, T.; Mair, N.; Dietl, P. Secretion in alveolar type II cells at the interface of constitutive and regulated exocytosis. Am. J. Respir. Cell Mol. Biol. 2001, 25, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Chander, A.; Fisher, A.B. Regulation of lung surfactant secretion. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1990, 258, L241–L253. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Diseases of pulmonary surfactant homeostasis. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 371–393. [Google Scholar] [CrossRef] [PubMed]
- Vicary, G.W.; Vergne, Y.; Santiago-Cornier, A.; Young, L.R.; Roman, J. Pulmonary fibrosis in Hermansky-Pudlak syndrome. Ann. Am. Thorac. Soc. 2016, 13, 1839–1846. [Google Scholar] [CrossRef]
- Velázquez-Díaz, P.; Nakajima, E.; Sorkhdini, P.; Hernandez-Gutierrez, A.; Eberle, A.; Yang, D.; Zhou, Y. Hermansky-Pudlak Syndrome and Lung Disease: Pathogenesis and Therapeutics. Front. Pharmacol. 2021, 12, 644671. [Google Scholar] [CrossRef]
- Beers, M.F.; Mulugeta, S. The biology of the ABCA3 lipid transporter in lung health and disease. Cell Tissue Res. 2017, 367, 481–493. [Google Scholar] [CrossRef]
- Daniels, C.B.; Orgeig, S. Pulmonary surfactant: The key to the evolution of air breathing. News Physiol. Sci. 2003, 18, 151–157. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, E.; Pérez-Gil, J. Structure-function relationships in pulmonary surfactant membranes: From biophysics to therapy. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1568–1585. [Google Scholar] [CrossRef]
- Neergaard, K. Neue Auffassungen über einen Grundbegriff der Atemmechanik. Z. Gesamte Exp. Med. 1929, 66, 373–394. [Google Scholar] [CrossRef]
- Pattle, R.E. Surface Lining of Lung Alveoli. Physiol. Rev. 1965, 45, 48–79. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.A. Lung surfactant: A personal perspective. Annu. Rev. Physiol. 1997, 59, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.; Avery, M.E. Time of appearance of lung surfactant in the foetal mouse. Nature 1962, 193, 688–689. [Google Scholar] [CrossRef]
- Avery, M.E.; Mead, J. Surface Properties in Relation to Atelectasis and Hyaline Membrane Disease. AMA. J. Dis. Child. 1959, 97, 517–523. [Google Scholar] [CrossRef]
- Avery, M.E.; Taeusch, H.; Floros, J. Surfactant replacement. N. Engl. J. Med. 1986, 315, 825–826. [Google Scholar] [CrossRef]
- Ryan, U.S.; Ryan, J.W.; Smith, D.S. Alveolar type II cells: Studies on the mode of release of lamellar bodies. Tissue Cell 1975, 7, 587–599. [Google Scholar] [CrossRef]
- Balis, J.U.; Conen, P.E. The role of alveolar inclusion bodies in the developing lung. Lab. Investig. 1964, 13, 1215–1229. [Google Scholar] [PubMed]
- Kikkawa, Y.; Smith, F. Cellular and biochemical aspects of pulmonary surfactant in health and disease. Lab. Investig. 1983, 49, 122–139. [Google Scholar]
- Veldhuizen, R.; Nag, K.; Orgeig, S.; Possmayer, F. The role of lipids in pulmonary surfactant. Biochim. Biophys. Acta-Mol. Basis Dis. 1998, 1408, 90–108. [Google Scholar] [CrossRef]
- Askin, F.B.; Kuhn, C. The cellular origin of pulmonary surfactant. Lab. Investig. 1971, 25, 260–268. [Google Scholar]
- Veldhuizen, E.J.A.; Haagsman, H.P. Role of pulmonary surfactant components in surface film formation and dynamics. Biochim. Biophys. Acta-Biomembr. 2000, 1467, 255–270. [Google Scholar] [CrossRef]
- Schürch, S.; Green, F.H.Y.; Bachofen, H. Formation and structure of surface films: Captive bubble surfactometry. Biochim. Biophys. Act-Mol. Basis Dis. 1998, 1408, 180–202. [Google Scholar] [CrossRef]
- Mason, R.J.; Greene, K.; Voelker, D.R. Surfactant protein A and surfactant protein D in health and disease. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1998, 275, L1–L13. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.H.; Beirag, N.; Murugaiah, V.; Chou, Y.C.; Kuo, W.S.; Kao, H.F.; Madan, T.; Kishore, U.; Wang, J.Y. Human Surfactant Protein D Binds Spike Protein and Acts as an Entry Inhibitor of SARS-CoV-2 Pseudotyped Viral Particles. Front. Immunol. 2021, 12, 641360. [Google Scholar] [CrossRef]
- Watson, A.; Madsen, J.; Clark, H.W. SP-A and SP-D: Dual Functioning Immune Molecules with Antiviral and Immunomodulatory Properties. Front. Immunol. 2021, 11, 622598. [Google Scholar] [CrossRef]
- Osanai, K.; Mason, R.J.; Voelker, D.R. Trafficking of newly synthesized surfactant protein A in isolated rat alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 1998, 19, 929–935. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C.; Ruckert, P.; Tao, J.Q.; Bates, S.R. Pathway to lamellar bodies for surfactant protein A. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2010, 299, L51–L58. [Google Scholar] [CrossRef]
- Lin, S.; Ikegami, M.; Moon, C.; Naren, A.P.; Shannon, J.M. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) specifically interacts with phospholipid transfer protein StarD10 to facilitate surfactant phospholipid trafficking in alveolar type II Cells. J. Biol. Chem. 2015, 290, 18559–18574. [Google Scholar] [CrossRef]
- Yamano, G.; Funahashi, H.; Kawanami, O.; Zhao, L.X.; Ban, N.; Uchida, Y.; Morohoshi, T.; Ogawa, J.; Shioda, S.; Inagaki, N. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett. 2001, 508, 221–225. [Google Scholar] [CrossRef]
- Mulugeta, S.; Gray, J.M.; Notarfrancesco, K.L.; Gonzales, L.W.; Koval, M.; Feinstein, S.I.; Ballard, P.L.; Fisher, A.B.; Shuman, H. Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J. Biol. Chem. 2002, 277, 22147–22155. [Google Scholar] [CrossRef]
- Miklavc, P.; Mair, N.; Wittekindt, O.H.; Haller, T.; Dietl, P.; Felder, E.; Timmler, M.; Frick, M. Fusion-activated Ca 2+ entry via vesicular P2X 4 receptors promotes fusion pore opening and exocytotic content release in pneumocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 14503–14508. [Google Scholar] [CrossRef]
- Kook, S.; Wang, P.; Young, L.R.; Schwake, M.; Saftig, P.; Weng, X.; Meng, Y.; Neculai, D.; Marks, M.S.; Gonzales, L.; et al. Impaired lysosomal integral membrane protein 2-dependent peroxiredoxin 6 delivery to lamellar bodies accounts for altered alveolar phospholipid content in adaptor protein-3-deficient pearl mice. J. Biol. Chem. 2016, 291, 8414–8427. [Google Scholar] [CrossRef]
- Roszell, B.R.; Tao, J.Q.; Yu, K.J.; Huang, S.; Bates, S.R. Characterization of the Niemann-Pick C pathway in alveolar type II cells and lamellar bodies of the lung. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 302, L919–L932. [Google Scholar] [CrossRef]
- Kook, S.; Wang, P.; Meng, S.; Jetter, C.S.; Sucre, J.M.S.; Benjamin, J.T.; Gokey, J.J.; Hanby, H.A.; Jaume, A.; Goetzl, L.; et al. AP-3-dependent targeting of flippase ATP8A1 to lamellar bodies suppresses activation of YAP in alveolar epithelial type 2 cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2025208118. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, S.J.; Spitzer, A.R.; Chander, A. Ionic regulation of proton chemical (pH) and electrical gradients in lung lamellar bodies. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1997, 273, L427–L436. [Google Scholar] [CrossRef] [PubMed]
- Chintagari, N.R.; Mishra, A.; Su, L.; Wang, Y.; Ayalew, S.; Hartson, S.D.; Liu, L. Vacuolar ATPase regulates surfactant secretion in rat alveolar type II cells by modulating lamellar body calcium. PLoS ONE 2010, 5, e9228. [Google Scholar] [CrossRef]
- Sun-Wada, G.H.; Murata, Y.; Namba, M.; Yamamoto, A.; Wada, Y.; Futai, M. Mouse Proton Pump ATPase C Subunit Isoforms (C2-a and C2-b) Specifically Expressed in Kidney and Lung. J. Biol. Chem. 2003, 278, 44843–44851. [Google Scholar] [CrossRef] [PubMed]
- Fois, G.; Winkelmann, V.E.; Bareis, L.; Staudenmaier, L.; Hecht, E.; Ziller, C.; Ehinger, K.; Schymeinsky, J.; Kranz, C.; Frick, M. ATP is stored in lamellar bodies to activate vesicular P2X4 in an autocrine fashion upon exocytosis. J. Gen. Physiol. 2018, 150, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, S.J.; Chander, A. H+- and K+-dependence of Ca2+ uptake in lung lamellar bodies. J. Membr. Biol. 2000, 174, 41–51. [Google Scholar] [CrossRef]
- Chander, A.; Johnson, R.G.; Reicherter, J.; Fisher, B. Lung lamellar bodies maintain an acidic internal pH. J. Biol. Chem. 1986, 261, 6126–6131. [Google Scholar] [CrossRef]
- Ridsdale, R.; Lewis, D.F.; Weaver, T.E.; Akinbi, H.T. Proteomic analysis of lamellar bodies isolated from amniotic fluid: Implications for function. Am. J. Perinatol. 2012, 29, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chintagari, N.R.; Narayanaperumal, J.; Ayalew, S.; Hartson, S.; Liu, L. Proteomic analysis of lamellar bodies isolated from rat lungs. BMC Cell Biol. 2008, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Ridsdale, R.; Na, C.L.; Xu, Y.; Greis, K.D.; Weaver, T. Comparative proteomic analysis of lung lamellar bodies and lysosome-related organelles. PLoS ONE 2011, 6, e16482. [Google Scholar] [CrossRef] [PubMed]
- Schaller-Bals, S.; Bates, S.R.; Notarfrancesco, K.; Tao, J.Q.; Fisher, A.B.; Shuman, H. Surface-expressed lamellar body membrane is recycled to lamellar bodies. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2000, 279, L631–L640. [Google Scholar] [CrossRef]
- Fois, G.; Föhr, K.J.; Kling, C.; Fauler, M.; Wittekindt, O.H.; Dietl, P.; Frick, M. P2X4 receptor re-sensitization depends on a protonation/deprotonation cycle mediated by receptor internalization and recycling. J. Physiol. 2018, 596, 4893–4907. [Google Scholar] [CrossRef]
- Hu, Z.Z.; Valencia, J.C.; Huang, H.; Chi, A.; Shabanowitz, J.; Hearing, V.J.; Appella, E.; Wu, C. Comparative bioinformatics analyses and profiling of lysosome-related organelle proteomes. Int. J. Mass Spectrom. 2007, 259, 147–160. [Google Scholar] [CrossRef][Green Version]
- Goerke, J. Pulmonary surfactant: Functions and molecular composition. Biochim. Biophys. Acta 1998, 1408, 79–89. [Google Scholar] [CrossRef]
- Kos, V.; Ford, R.C. The ATP-binding cassette family: A structural perspective. Cell. Mol. Life Sci. 2009, 66, 3111–3126. [Google Scholar] [CrossRef]
- Tusnády, G.E.; Sarkadi, B.; Simon, I.; Váradi, A. Membrane topology of human ABC proteins. FEBS Lett. 2006, 580, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Matsumura, Y.; Sakai, H.; Takanezawa, Y.; Sasaki, M.; Arai, H.; Inagaki, N. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J. Biol. Chem. 2007, 282, 9628–9634. [Google Scholar] [CrossRef]
- Cerrada, A.; Haller, T.; Cruz, A.; Pérez-Gil, J. Pneumocytes Assemble Lung Surfactant as Highly Packed/Dehydrated States with Optimal Surface Activity. Biophys. J. 2015, 109, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Ravasio, A.; Olmeda, B.; Bertocchi, C.; Haller, T.; Pérez-Gil, J. Lamellar bodies form solid three-dimensional films at the respiratory air-liquid interface. J. Biol. Chem. 2010, 285, 28174–28182. [Google Scholar] [CrossRef] [PubMed]
- Haller, T.; Dietl, P.; Stockner, H.; Frick, M.; Mair, N.; Tinhofer, I.; Ritsch, A.; Enhorning, G.; Putz, G. Tracing surfactant transformation from cellular release to insertion into an air-liquid interface. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 286, L1009–L1015. [Google Scholar] [CrossRef]
- Cheong, N.; Zhang, H.; Madesh, M.; Zhao, M.; Yu, K.; Dodia, C.; Fisher, A.B.; Savani, R.C.; Shuman, H. ABCA3 is critical for lamellar body biogenesis in vivo. J. Biol. Chem. 2007, 282, 23811–23817. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Xavier, R.; Haley, K.J.; Welti, R.; Goss, J.L.; Brown, C.E.; Zhuang, D.Z.; Bell, S.A.; Lu, N.; Mckee, M.; et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J. Lipid Res. 2007, 48, 621–632. [Google Scholar] [CrossRef]
- Matsumura, Y.; Sakai, H.; Sasaki, M.; Ban, N.; Inagaki, N. ABCA3-mediated choline-phospholipids uptake into intracellular vesicles in A549 cells. FEBS Lett. 2007, 581, 3139–3144. [Google Scholar] [CrossRef]
- Cheong, N.; Madesh, M.; Gonzales, L.W.; Zhao, M.; Yu, K.; Ballard, P.L.; Shuman, H. Functional and trafficking defects in ATP binding cassette A3 mutants associated with respiratory distress syndrome. J. Biol. Chem. 2006, 281, 9791–9800. [Google Scholar] [CrossRef]
- Rosenbaum, A.I.; Maxfield, F.R. Niemann-Pick type C disease: Molecular mechanisms and potential therapeutic approaches. J. Neurochem. 2011, 116, 789–795. [Google Scholar] [CrossRef]
- Li, X.; Saha, P.; Lib, J.; Blobel, G.; Pfeffer, S.R. Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc. Natl. Acad. Sci. USA 2016, 113, 10079–10084. [Google Scholar] [CrossRef]
- Heybrock, S.; Kanerva, K.; Meng, Y.; Ing, C.; Liang, A.; Xiong, Z.J.; Weng, X.; Ah Kim, Y.; Collins, R.; Trimble, W.; et al. Lysosomal integral membrane protein-2 (LIMP-2/SCARB2) is involved in lysosomal cholesterol export. Nat. Commun. 2019, 10, 3521. [Google Scholar] [CrossRef]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef]
- Manevich, Y.; Shuvaeva, T.; Dodia, C.; Kazi, A.; Feinstein, S.I.; Fisher, A.B. Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase A2 activities. Arch. Biochem. Biophys. 2009, 485, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Rooney, S.A.; Young, S.L.; Mendelson, C.R. Molecular and cellular processing of lung surfactant. FASEB J. 1994, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C.; Feinstein, S.I.; Ho, Y.S. Altered lung phospholipid metabolism in mice with targeted deletion of lysosomal-type phospholipase A2. J. Lipid Res. 2005, 46, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C.; Yu, K.; Manevich, Y.; Feinstein, S.I. Lung phospholipid metabolism in transgenic mice overexpressing peroxiredoxin 6. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2006, 1761, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Conrad, K.S.; Cheng, T.W.; Ysselstein, D.; Heybrock, S.; Hoth, L.R.; Chrunyk, B.A.; Am Ende, C.W.; Krainc, D.; Schwake, M.; Saftig, P.; et al. Lysosomal integral membrane protein-2 as a phospholipid receptor revealed by biophysical and cellular studies. Nat. Commun. 2017, 8, 1908. [Google Scholar] [CrossRef]
- Roszell, B.R.; Tao, J.Q.; Yu, K.J.; Gao, L.; Huang, S.; Ning, Y.; Feinstein, S.I.; Vite, C.H.; Bates, S.R. Pulmonary Abnormalities in Animal Models Due to Niemann-Pick Type C1 (NPC1) or C2 (NPC2) Disease. PLoS ONE 2013, 8, e67084. [Google Scholar] [CrossRef]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-Terminal Domain of NPC1 Reveals Distinct Subdomains for Binding and Transfer of Cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef]
- Rituper, B.; Guček, A.; Lisjak, M.; Gorska, U.; Šakanović, A.; Bobnar, S.T.; Lasič, E.; Božić, M.; Abbineni, P.S.; Jorgačevski, J.; et al. Vesicle cholesterol controls exocytotic fusion pore. Cell Calcium 2021, 101, 102503. [Google Scholar] [CrossRef]
- Palmgren, M.; Østerberg, J.T.; Nintemann, S.J.; Poulsen, L.R.; López-Marqués, R.L. Evolution and a revised nomenclature of P4 ATPases, a eukaryotic family of lipid flippases. Biochim. Biophys. Acta-Biomembr. 2019, 1861, 1135–1151. [Google Scholar] [CrossRef]
- Best, J.T.; Xu, P.; Graham, T.R. Phospholipid flippases in membrane remodeling and transport carrier biogenesis. Curr. Opin. Cell Biol. 2019, 59, 8–15. [Google Scholar] [CrossRef]
- Soupene, E. ATP8A1 activity and phosphatidylserine transbilayer movement. J. Receptor. Ligand Channel Res. 2008, 1, 1–10. [Google Scholar] [CrossRef]
- Fairn, G.D.; Schieber, N.L.; Ariotti, N.; Murphy, S.; Kuerschner, L.; Webb, R.I.; Grinstein, S.; Parton, R.G. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J. Cell Biol. 2011, 194, 257–275. [Google Scholar] [CrossRef]
- Ansari, I.U.H.; Longacre, M.J.; Paulusma, C.C.; Stoker, S.W.; Kendrick, M.A.; MacDonald, M.J. Characterization of P4 ATPase phospholipid translocases (flippases) in human and rat pancreatic beta cells: Their gene silencing inhibits insulin secretion. J. Biol. Chem. 2015, 290, 23110–23123. [Google Scholar] [CrossRef]
- MacDonald, M.J.; Ade, L.; Ntambi, J.M.; Ansari, I.U.H.; Stoker, S.W. Characterization of phospholipids in insulin secretory granules and mitochondria in pancreatic beta cells and their changes with glucose stimulation. J. Biol. Chem. 2015, 290, 11075–11092. [Google Scholar] [CrossRef] [PubMed]
- Eckenhoff, R.C.; Somlyo, A.P. Rat lung type II cell and lamellar body: Elemental composition in situ. Am. J. Physiol.-Cell Physiol. 1988, 254, C614-20. [Google Scholar] [CrossRef]
- Eckenhoff, R.G.; Rannels, S.R.; Fisher, A.B. Secretory granule calcium loss after isolation of rat alveolar type II cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1991, 260, L129–L135. [Google Scholar] [CrossRef]
- Malacrida, L.; Astrada, S.; Briva, A.; Bollati-Fogolín, M.; Gratton, E.; Bagatolli, L.A. Spectral phasor analysis of LAURDAN fluorescence in live A549 lung cells to study the hydration and time evolution of intracellular lamellar body-like structures. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2625–2635. [Google Scholar] [CrossRef]
- Beers, M.F. Inhibition of cellular processing of surfactant protein C by drugs affecting intracellular pH gradients. J. Biol. Chem. 1996, 271, 14361–14370. [Google Scholar] [CrossRef]
- Serrano, A.G.; Cabré, E.J.; Pérez-Gil, J. Identification of a segment in the precursor of pulmonary surfactant protein SP-B, potentially involved in pH-dependent membrane assembly of the protein. Biochim. Biophys. Acta-Biomembr. 2007, 1768, 1059–1069. [Google Scholar] [CrossRef][Green Version]
- Bañares-Hidalgo, A.; Pérez-Gil, J.; Estrada, P. Acidic pH triggers conformational changes at the NH2-terminal propeptide of the precursor of pulmonary surfactant protein B to form a coiled coil structure. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1738–1751. [Google Scholar] [CrossRef][Green Version]
- Chander, A.; Sen, N.; Wu, A.M.; Higgins, S.; Wadsworth, S.; Spitzer, A.R. Methylamine decreases trafficking and packaging of newly synthesized phosphatidylcholine in lamellar bodies in alveolar type II cells. Biochem. J. 1996, 318, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C. Lysosomal-type PLA2 and turnover of alveolar DPPC. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2001, 280, L748-54. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C. Role of acidic Ca2+-independent phospholipase A2 in synthesis of lung dipalmitoyl phosphatidylcholine. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1997, 272, L238-43. [Google Scholar] [CrossRef]
- Rice, W.R.; Dorn, C.C.; Singleton, F.M. P2-purinoceptor regulation of surfactant phosphatidylcholine secretion. Relative roles of calcium and protein kinase C. Biochem. J. 1990, 266, 407–413. [Google Scholar] [CrossRef]
- Haller, T.; Auktor, K.; Frick, M.; Mair, N.; Dietl, A.P. Threshold calcium levels for lamellar body exocytosis in type II pneumocytes. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1999, 277, L893–L900. [Google Scholar] [CrossRef]
- Fois, G.; Hobi, N.; Felder, E.; Ziegler, A.; Miklavc, P.; Walther, P.; Radermacher, P.; Haller, T.; Dietl, P. A new role for an old drug: Ambroxol triggers lysosomal exocytosis via pH-dependent Ca2+ release from acidic Ca2+ stores. Cell Calcium 2015, 58, 628–637. [Google Scholar] [CrossRef]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Murrell-Lagnado, R.; Zhu, M.X.; Dong, X.P. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J. Cell Biol. 2015, 209, 879–894. [Google Scholar] [CrossRef]
- Cang, C.; Bekele, B.; Ren, D. The voltage-gated sodium channel TPC1 confers endolysosomal excitability. Nat. Chem. Biol. 2014, 10, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Li, G.; Zhang, X.; Xu, H.; Abraham, S.N. A TRP channel senses lysosome neutralization by pathogens to trigger their expulsion. Cell 2015, 161, 1306–1319. [Google Scholar] [CrossRef]
- Huang, P.; Zou, Y.; Zhong, X.Z.; Cao, Q.; Zhao, K.; Zhu, M.X.; Murrell-Lagnado, R.; Dong, X.P. P2X4 forms functional ATP-activated cation channels on lysosomal membranes regulated by luminal pH. J. Biol. Chem. 2014, 289, 17658–17667. [Google Scholar] [CrossRef] [PubMed]
- La Rovere, R.M.L.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87. [Google Scholar] [CrossRef]
- Neuland, K.; Sharma, N.; Frick, M. Synaptotagmin-7 links fusion-activated Ca2+ entry and fusion pore dilation. J. Cell Sci. 2014, 127, 5218–5227. [Google Scholar] [CrossRef] [PubMed]
- Miklavc, P.; Albrecht, S.; Wittekindt, O.H.; Schullian, P.; Haller, T.; Dietl, P. Existence of exocytotic hemifusion intermediates with a lifetime of up to seconds in type II pneumocytes. Biochem. J. 2009, 424, 7–14. [Google Scholar] [CrossRef]
- Dietl, P.; Haller, T.; Frick, M. Spatio-temporal aspects, pathways and actions of Ca2+ in surfactant secreting pulmonary alveolar type II pneumocytes. Cell Calcium 2012, 52, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Haller, T.; Dietl, P.; Pfaller, K.; Frick, M.; Mair, N.; Paulmichl, M.; Hess, M.W.; Fürst, J.; Maly, K. Fusion pore expansion is a slow, discontinuous, and Ca2+-dependent process regulating secretion from alveolar type II cells. J. Cell Biol. 2001, 155, 279–289. [Google Scholar] [CrossRef]
- Thompson, K.E.; Korbmacher, J.P.; Hecht, E.; Hobi, N.; Wittekindt, O.H.; Dietl, P.; Kranz, C.; Frick, M. Fusion-activated cation entry (FACE) via P2X4 couples surfactant secretion and alveolar fluid transport. FASEB J. 2013, 27, 1772–1783. [Google Scholar] [CrossRef]
- Miklavc, P.; Thompson, K.E.; Frick, M. A new role for P2X4 receptors as modulators of lung surfactant secretion. Front. Cell. Neurosci. 2013, 7, 171. [Google Scholar] [CrossRef]
- Murrell-Lagnado, R.D.; Frick, M. P2X4 and lysosome fusion. Curr. Opin. Pharmacol. 2019, 47, 126–132. [Google Scholar] [CrossRef]
- Verkman, A.S. Water permeability measurement in living cells and complex tissues. J. Membr. Biol. 2000, 173, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Vitkova, V.; Genova, J.; Bivas, I. Permeability and the hidden area of lipid bilayers. Eur. Biophys. J. 2004, 33, 706–714. [Google Scholar] [CrossRef]
- Olbrich, K.; Rawicz, W.; Needham, D.; Evans, E. Water permeability and mechanical strength of polyunsaturated lipid bilayers. Biophys. J. 2000, 79, 321–327. [Google Scholar] [CrossRef]
- Borgnia, M.; Nielsen, S.; Engel, A.; Agre, P. Cellular and molecular biology of the aquaporin water channels. Annu. Rev. Biochem. 1999, 68, 425–458. [Google Scholar] [CrossRef] [PubMed]
- Haller, T.; Dietl, P.; Deetjen, P.; Völkl, H. The lysosomal compartment as intracellular calcium store in MDCK cells: A possible involvement in InsP3-mediated Ca2+ release. Cell Calcium 1996, 19, 157–165. [Google Scholar] [CrossRef]
- Wolters, P.J.; Blackwell, T.S.; Eickelberg, O.; Loyd, J.E.; Kaminski, N.; Jenkins, G.; Maher, T.M.; Molina-Molina, M.; Noble, P.W.; Raghu, G.; et al. Time for a change: Is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir. Med. 2018, 6, 154–160. [Google Scholar] [CrossRef]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. 2015, 24, 102–114. [Google Scholar] [CrossRef]
- Tscherny, V.V.; Markin, V.S.; Decoursey, T.E. The Voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the pH gradient. J. Gen. Physiol. 1995, 105, 861–896. [Google Scholar] [CrossRef]
- Bullard, J.E.; Wert, S.E.; Nogee, L.M. ABCA3 Deficiency: Neonatal Respiratory Failure and Interstitial Lung Disease. Semin. Perinatol. 2006, 30, 327–334. [Google Scholar] [CrossRef]
- Garmany, T.H.; Moxley, M.A.; White, F.V.; Dean, M.; Hull, W.M.; Whitsett, J.A.; Nogee, L.M.; Hamvas, A. Surfactant composition and function in patients with ABCA3 mutations. Pediatr. Res. 2006, 59, 801–805. [Google Scholar] [CrossRef]
- Shulenin, S.; Nogee, L.M.; Annilo, T.; Wert, S.E.; Whitsett, J.A.; Dean, M. ABCA3 Gene Mutations in Newborns with Fatal Surfactant Deficiency. N. Engl. J. Med. 2004, 350, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.P.; Pinheiro, L.; Costa, M.; Silva, A.; Gonçalves, A.; Pereira, A. Novel ABCA3 mutations as a cause of respiratory distress in a term newborn. Gene 2014, 534, 417–420. [Google Scholar] [CrossRef]
- Bullard, J.E.; Wert, S.E.; Whitsett, J.A.; Dean, M.; Nogee, L.M. ABCA3 mutations associated with pediatric interstitial lung disease. Am. J. Respir. Crit. Care Med. 2005, 172, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Crossno, P.F.; Polosukhin, V.V.; Blackwell, T.S.; Johnson, J.E.; Markin, C.; Moore, P.E.; Worrell, J.A.; Stahlman, M.T.; Phillips, J.A.; Loyd, J.E.; et al. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest 2010, 137, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Epaud, R.; Delestrain, C.; Louha, M.; Simon, S.; Fanen, P.; Tazi, A. Combined pulmonary fibrosis and emphysema syndrome associated with ABCA3 mutations. Eur. Respir. J. 2014, 43, 638–641. [Google Scholar] [CrossRef]
- Ota, C.; Kimura, M.; Kure, S. ABCA3 mutations led to pulmonary fibrosis and emphysema with pulmonary hypertension in an 8-year-old girl. Pediatr. Pulmonol. 2016, 51, E21–E23. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, R.; Kaltenborn, E.; Frixel, S.; Wittmann, T.; Liebisch, G.; Schmitz, G.; Griese, M. ABCA3 protects alveolar epithelial cells against free cholesterol induced cell death. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2015, 1851, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Bowman, S.L.; Bi-Karchin, J.; Le, L.; Marks, M.S. The road to lysosome-related organelles: Insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic 2019, 20, 404–435. [Google Scholar] [CrossRef]
- Huizing, M.; Helip-Wooley, A.; Westbroek, W.; Gunay-Aygun, M.; Gahl, W.A. Disorders of lysosome-related organelle biogenesis: Clinical and molecular genetics. Annu. Rev. Genomics Hum. Genet. 2008, 9, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef]
- Faverio, P.; Stainer, A.; De Giacomi, F.; Gasperini, S.; Motta, S.; Canonico, F.; Pieruzzi, F.; Monzani, A.; Pesci, A.; Biondi, A. Molecular pathways and respiratory involvement in lysosomal storage diseases. Int. J. Mol. Sci. 2019, 20, 327. [Google Scholar] [CrossRef] [PubMed]
- Staretz-Chacham, O.; Aviram, M.; Morag, I.; Goldbart, A.; Hershkovitz, E. Pulmonary involvement in Niemann-Pick C type 1. Eur. J. Pediatr. 2018, 177, 1609–1615. [Google Scholar] [CrossRef]
- Ballout, R.A.; Sviridov, D.; Bukrinsky, M.I.; Remaley, A.T. The lysosome: A potential juncture between SARS-CoV-2 infectivity and Niemann-Pick disease type C, with therapeutic implications. FASEB J. 2020, 34, 7253–7264. [Google Scholar] [CrossRef] [PubMed]
- Dell’Angelica, E.C. Lysosome-related organelles. FASEB J. 2000, 14, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.G.; Griffiths, G.M. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef]
- Guttentag, S.H.; Akhtar, A.; Tao, J.Q.; Atochina, E.; Rusiniak, M.E.; Swank, R.T.; Bates, S.R. Defective surfactant secretion in a mouse model of Hermansky-Pudlak syndrome. Am. J. Respir. Cell Mol. Biol. 2005, 33, 14–21. [Google Scholar] [CrossRef]
- Atochina-Vasserman, E.N.; Bates, S.R.; Zhang, P.; Abramova, H.; Zhang, Z.; Gonzales, L.; Tao, J.Q.; Gochuico, B.R.; Gahl, W.; Guo, C.J.; et al. Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am. J. Respir. Crit. Care Med. 2011, 184, 449–458. [Google Scholar] [CrossRef]
- Young, L.R.; Gulleman, P.M.; Bridges, J.P.; Weaver, T.E.; Deutsch, G.H.; Blackwell, T.S.; McCormack, F.X. The alveolar epithelium determines susceptibility to lung fibrosis in Hrmansky-Pdlak syndrome. Am. J. Respir. Crit. Care Med. 2012, 186, 1014–1024. [Google Scholar] [CrossRef]
- Young, L.R.; Pasula, R.; Gulleman, P.M.; Deutsch, G.H.; McCormack, F.X. Susceptibility of hermansky-pudlak mice to bleomycin-induced type II cell apoptosis and fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 67–74. [Google Scholar] [CrossRef]
- Peng, R.; Wu, L.A.; Wang, Q.; Qi, J.; Gao, G.F. Cell entry by SARS-CoV-2. Trends Biochem. Sci. 2021, 46, 848–860. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446. [Google Scholar] [CrossRef]
- Qian, Z.; Travanty, E.A.; Oko, L.; Edeen, K.; Berglund, A.; Wang, J.; Ito, Y.; Holmes, K.V.; Mason, R.J. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am. J. Respir. Cell Mol. Biol. 2013, 48, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A Transmembrane Serine Protease Is Linked to the Severe Acute Respiratory Syndrome Coronavirus Receptor and Activates Virus Entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Schuler, B.A.; Habermann, A.C.; Plosa, E.J.; Taylor, C.J.; Jetter, C.; Negretti, N.M.; Kapp, M.E.; Benjamin, J.T.; Gulleman, P.; Nichols, D.S.; et al. Age-determined expression of priming protease TMPRSS2 and localization of SARS-CoV-2 in lung epithelium. J. Clin. Investig. 2021, 131, e140766. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.C.; Bosch, B.J.; Li, F.; Li, W.; Kyoung, H.L.; Ghiran, S.; Vasilieva, N.; Dermody, T.S.; Harrison, S.C.; Dormitzer, P.R.; et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 2006, 281, 3189–3203. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, S.; Wu, Y.; Hou, W.; Yuan, L.; Shen, C.; Wang, J.; Ye, J.; Zheng, Q.; Ma, J.; et al. Virus-Free and Live-Cell Visualizing SARS-CoV-2 Cell Entry for Studies of Neutralizing Antibodies and Compound Inhibitors. Small Methods 2021, 5, 2001031. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef] [PubMed]
- Blaess, M.; Kaiser, L.; Sauer, M.; Csuk, R.; Deigner, H.P. COVID-19/SARS-CoV-2 infection: Lysosomes and lysosomotropism implicate new treatment strategies and personal risks. Int. J. Mol. Sci. 2020, 21, 4953. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Qin, P.; Huang, Y.W. Lysosomal ion channels involved in cellular entry and uncoating of enveloped viruses: Implications for therapeutic strategies against SARS-CoV-2. Cell Calcium 2021, 94, 102360. [Google Scholar] [CrossRef]
- Haller, T.; Ortmayr, J.; Friedrich, F.; Völkl, H.; Dietl, P. Dynamics of surfactant release in alveolar type II cells. Proc. Natl. Acad. Sci. USA 1998, 95, 1579–1584. [Google Scholar] [CrossRef]
- Mair, N.; Haller, T.; Dietl, P.; Shaul, P.W. Exocytosis in alveolar type II cells revealed by cell capacitance and fluorescence measurements. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1999, 276, L376–L382. [Google Scholar] [CrossRef]
- Yayoi, Y.; Ohsawa, Y.; Koike, M.; Zhang, G.; Kominami, E.; Uchiyama, Y. Specific localization of lysosomal aminopeptidases in type II alveolar epithelial cells of the rat lung. Arch. Histol. Cytol. 2001, 64, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.E.R.; Gilmore, L.B. Hydrolases of pulmonary lysosomes and lamellar bodies. J. Biol. Chem. 1982, 257, 9211–9220. [Google Scholar] [CrossRef]
- Huang, J.; Hume, A.J.; Abo, K.M.; Werder, R.B.; Villacorta-Martin, C.; Alysandratos, K.D.; Beermann, M.L.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Olejnik, J.; et al. SARS-CoV-2 Infection of Pluripotent Stem Cell-Derived Human Lung Alveolar Type 2 Cells Elicits a Rapid Epithelial-Intrinsic Inflammatory Response. Cell Stem Cell 2020, 27, 962–973. [Google Scholar] [CrossRef]
- Nardacci, R.; Colavita, F.; Castilletti, C.; Lapa, D.; Matusali, G.; Meschi, S.; Del Nonno, F.; Colombo, D.; Capobianchi, M.R.; Zumla, A.; et al. Evidences for lipid involvement in SARS-CoV-2 cytopathogenesis. Cell Death Dis. 2021, 12, 263. [Google Scholar] [CrossRef]
- Ou, T.; Mou, H.; Zhang, L.; Ojha, A.; Choe, H.; Farzan, M. Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog. 2021, 17, e1009212. [Google Scholar] [CrossRef]
- Gorshkov, K.; Chen, C.Z.; Bostwick, R.; Rasmussen, L.; Tran, B.N.; Cheng, Y.S.; Xu, M.; Pradhan, M.; Henderson, M.; Zhu, W.; et al. The SARS-CoV-2 Cytopathic Effect Is Blocked by Lysosome Alkalizing Small Molecules. ACS Infect. Dis. 2021, 7, 1389–1408. [Google Scholar] [CrossRef]
- Carpinteiro, A.; Gripp, B.; Hoffmann, M.; Pöhlmann, S.; Hoertel, N.; Edwards, M.J.; Kamler, M.; Kornhuber, J.; Becker, K.A.; Gulbins, E. Inhibition of acid sphingomyelinase by ambroxol prevents SARS-CoV-2 entry into epithelial cells. J. Biol. Chem. 2021, 296, 100701. [Google Scholar] [CrossRef]
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 31–39. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 2019, 5, 18. [Google Scholar] [CrossRef]
- Swenson, K.E.; Swenson, E.R. Pathophysiology of Acute Respiratory Distress Syndrome and COVID-19 Lung Injury. Crit. Care Clin. 2021, 37, 749–776. [Google Scholar] [CrossRef]
- Riteau, N.; Gasse, P.; Fauconnier, L.; Gombault, A.; Couegnat, M.; Fick, L.; Kanellopoulos, J.; Quesniaux, V.F.J.; Marchand-Adam, S.; Crestani, B.; et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 774–783. [Google Scholar] [CrossRef]
- Kanellopoulos, J.M.; Almeida-da-Silva, C.L.C.; Rüütel Boudinot, S.; Ojcius, D.M. Structural and Functional Features of the P2X4 Receptor: An Immunological Perspective. Front. Immunol. 2021, 12, 645834. [Google Scholar] [CrossRef]
- Wirsching, E.; Fauler, M.; Fois, G.; Frick, M. P2 purinergic signaling in the distal lung in health and disease. Int. J. Mol. Sci. 2020, 21, 4973. [Google Scholar] [CrossRef]
- Diem, K.; Fauler, M.; Fois, G.; Hellmann, A.; Winokurow, N.; Schumacher, S.; Kranz, C.; Frick, M. Mechanical stretch activates piezo1 in caveolae of alveolar type I cells to trigger ATP release and paracrine stimulation of surfactant secretion from alveolar type II cells. FASEB J. 2020, 34, 12785–12804. [Google Scholar] [CrossRef]
- Wauters, E.; Van Mol, P.; Garg, A.D.; Jansen, S.; Van Herck, Y.; Vanderbeke, L.; Bassez, A.; Boeckx, B.; Malengier-Devlies, B.; Timmerman, A.; et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021, 31, 272–290. [Google Scholar] [CrossRef]
- Ji, J.; Sun, L.; Luo, Z.; Zhang, Y.; Xianzheng, W.; Liao, Y.; Tong, X.; Shan, J. Potential Therapeutic Applications of Pulmonary Surfactant Lipids in the Host Defence Against Respiratory Viral Infections. Front. Immunol. 2021, 12, 730022. [Google Scholar] [CrossRef]
- Kaczmarek-Hájek, K.; Lörinczi, É.; Hausmann, R.; Nicke, A. Molecular and functional properties of P2X receptors-recent progress and persisting challenges. Purinergic Signal. 2012, 8, 375–417. [Google Scholar] [CrossRef]
- North, R.A. Molecular physiology of P2X receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef]
- Bryant, A.; Lawrie, T.A.; Dowswell, T.; Fordham, E.J.; Mitchell, S.; Hill, S.R.; Tham, T.C. Ivermectin for Prevention and Treatment of COVID-19 Infection: A Systematic Review, Meta-analysis, and Trial Sequential Analysis to Inform Clinical Guidelines. Am. J. Ther. 2021, 28, e434–e460. [Google Scholar] [CrossRef]
- Haller, T.; Cerrada, A.; Pfaller, K.; Braubach, P.; Felder, E. Polarized light microscopy reveals physiological and drug-induced changes in surfactant membrane assembly in alveolar type II pneumocytes. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 1152–1161. [Google Scholar] [CrossRef]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 5–11. [Google Scholar] [CrossRef]
- Patel, S.D.; Bono, T.R.; Rowe, S.M.; Solomon, G.M. CFTR targeted therapies: Recent advances in cystic fibrosis and possibilities in other diseases of the airways. Eur. Respir. Rev. 2020, 29, 190068. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 1783–1784. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Dukovski, D.; Villella, A.; Bastos, C.; King, R.; Finley, D.; Kelly, J.W.; Morimoto, R.I.; Hartl, F.U.; Munoz, B.; Lee, P.S.; et al. Amplifiers co-translationally enhance CFTR biosynthesis via PCBP1-mediated regulation of CFTR mRNA. J. Cyst. Fibros. 2020, 19, 733–741. [Google Scholar] [CrossRef]
- High, K.D.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef]
- Moriyama, Y.; Nomura, M. Clodronate: A Vesicular ATP Release Blocker. Trends Pharmacol. Sci. 2018, 39, 13–23. [Google Scholar] [CrossRef]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef]
- Sheth, J.; Joseph, J.J.; Shah, K.; Muranjan, M.; Mistri, M.; Sheth, F. Pulmonary manifestations in Niemann-Pick type C disease with mutations in NPC2 gene: Case report and review of literature. BMC Med. Genet. 2017, 18, 5. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).