Clinical Utility of Optical Genome Mapping as an Additional Tool in a Standard Cytogenetic Workup in Hematological Malignancies

 ,
,  , , ,
, , ,  , ,
, ,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Chromosomal Analysis
2.3. Fluorescence In Situ Hybridization
2.4. OGM Analysis
2.5. Variants Interpretation
2.6. Assessment of OGM’s Additional Value
2.7. Statistical Analysis
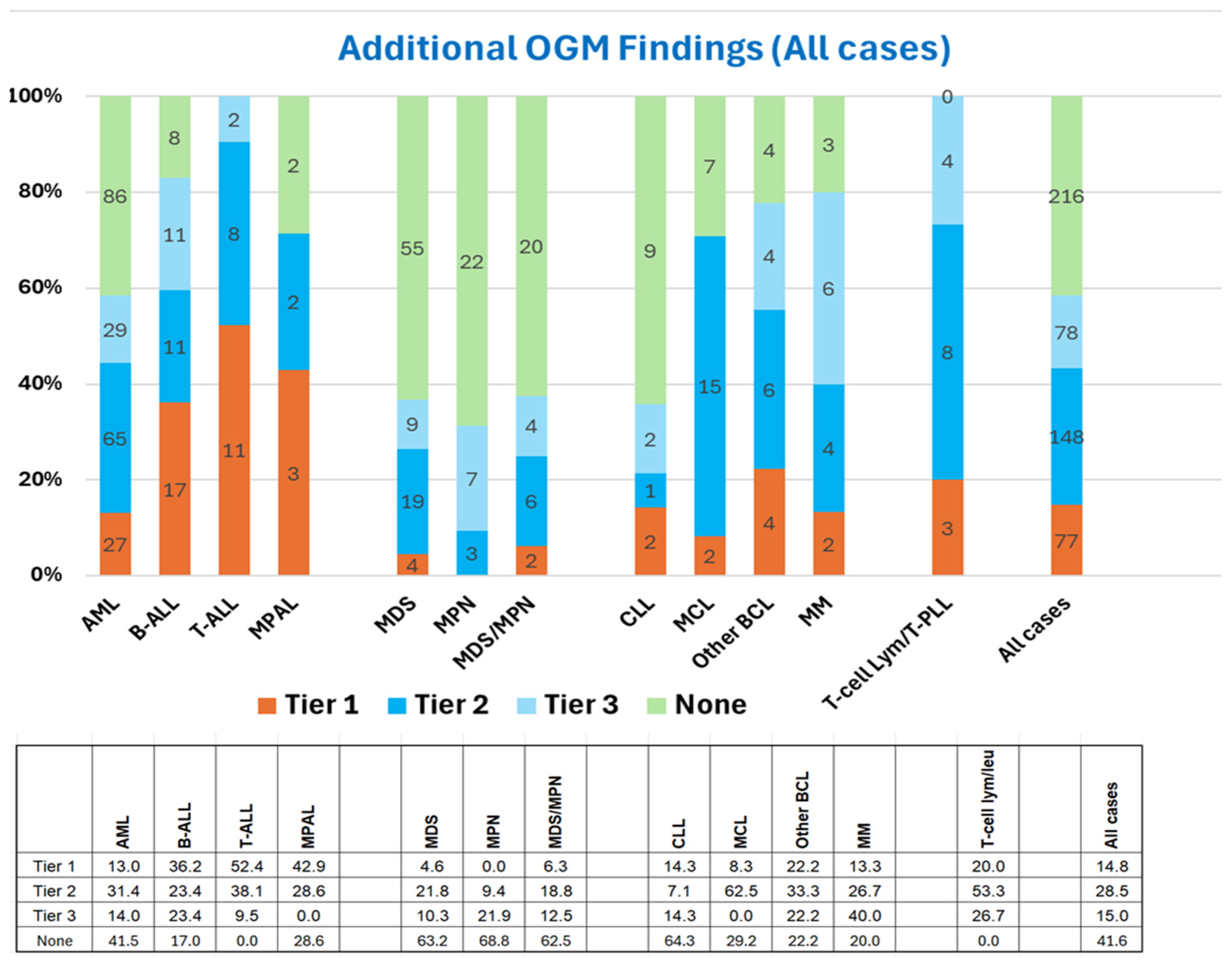
3. Results
3.1. OGM Enhances Analytical Sensitivity over SCGW
3.2. OGM Enhances the Clinical Utility of SCGW
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Tarantini, F.; Cumbo, C.; Tota, G.; Minervini, C.F.; Minervini, A.; Conserva, M.R.; Redavid, I.; et al. Feasibility of Optical Genome Mapping in Cytogenetic Diagnostics of Hematological Neoplasms: A New Way to Look at DNA. Diagnostics 2023, 13, 1841. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heras, J. Optical Genome Mapping: A Revolutionary Tool for “Next Generation Cytogenomics Analysis” with a Broad Range of Diagnostic Applications in Human Diseases. J. Assoc. Genet. Technol. 2021, 47, 191–200. [Google Scholar]
- Sahajpal, N.S.; Mondal, A.K.; Hastie, A.; Chaubey, A.; Kolhe, R. Optical Genome Mapping for Oncology Applications. Curr. Protoc. 2023, 3, e910. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Neveling, K.; Kanagal-Shamanna, R. Optical genome mapping for structural variation analysis in hematologic malignancies. Am. J. Hematol. 2022, 97, 975–982. [Google Scholar] [CrossRef]
- Balciuniene, J.; Ning, Y.; Lazarus, H.M.; Aikawa, V.; Sherpa, S.; Zhang, Y.; Morrissette, J.J. Cancer cytogenetics in a genomics world: Wedding the old with the new. Blood Rev. 2024, 66, 101209. [Google Scholar] [CrossRef]
- Balducci, E.; Kaltenbach, S.; Villarese, P.; Duroyon, E.; Zalmai, L.; Friedrich, C.; Suarez, F.; Marcais, A.; Bouscary, D.; Decroocq, J.; et al. Optical genome mapping refines cytogenetic diagnostics, prognostic stratification and provides new molecular insights in adult MDS/AML patients. Blood Cancer J. 2022, 12, 126. [Google Scholar] [CrossRef]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML/MDS patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef]
- Jean, J.; Kovach, A.E.; Doan, A.; Oberley, M.J.; Ji, J.; Schmidt, R.J.; Biegel, J.A.; Bhojwani, D.; Raca, G. Characterization of PAX5 intragenic tandem multiplication in pediatric B-lymphoblastic leukemia by optical genome mapping. Blood Adv. 2022, 6, 3343–3346. [Google Scholar] [CrossRef]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosom. Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Lühmann, J.L.; Stelter, M.; Wolter, M.; Kater, J.; Lentes, J.; Bergmann, A.K.; Schieck, M.; Göhring, G.; Möricke, A.; Cario, G.; et al. The Clinical Utility of Optical Genome Mapping for the Assessment of Genomic Aberrations in Acute Lymphoblastic Leukemia. Cancers 2021, 13, 4388. [Google Scholar] [CrossRef] [PubMed]
- Puiggros, A.; Ramos-Campoy, S.; Kamaso, J.; de la Rosa, M.; Salido, M.; Melero, C.; Rodríguez-Rivera, M.; Bougeon, S.; Collado, R.; Gimeno, E.; et al. Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL). Cancers 2022, 14, 3376. [Google Scholar] [CrossRef] [PubMed]
- Rack, K.; De Bie, J.; Ameye, G.; Gielen, O.; Demeyer, S.; Cools, J.; De Keersmaecker, K.; Vermeesch, J.R.; Maertens, J.; Segers, H.; et al. Optimizing the diagnostic workflow for acute lymphoblastic leukemia by optical genome mapping. Am. J. Hematol. 2022, 97, 548–561. [Google Scholar] [CrossRef]
- Ramos-Campoy, S.; Puiggros, A.; Kamaso, J.; Beà, S.; Bougeon, S.; Larráyoz, M.J.; Costa, D.; Parker, H.; Rigolin, G.M.; Blanco, M.L.; et al. TP53 Abnormalities Are Underlying the Poor Outcome Associated with Chromothripsis in Chronic Lymphocytic Leukemia Patients with Complex Karyotype. Cancers 2022, 14, 3715. [Google Scholar] [CrossRef]
- Suttorp, J.; Lühmann, J.L.; Behrens, Y.L.; Göhring, G.; Steinemann, D.; Reinhardt, D.; von Neuhoff, N.; Schneider, M. Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia. Cancers 2022, 14, 2058. [Google Scholar] [CrossRef]
- Giguère, A.; Raymond-Bouchard, I.; Collin, V.; Claveau, J.-S.; Hébert, J.; LeBlanc, R. Optical Genome Mapping Reveals the Complex Genetic Landscape of Myeloma. Cancers 2023, 15, 4687. [Google Scholar] [CrossRef]
- Díaz-González, Á.; Mora, E.; Avetisyan, G.; Furió, S.; De la Puerta, R.; Gil, J.V.; Liquori, A.; Villamón, E.; García-Hernández, C.; Santiago, M.; et al. Cytogenetic Assessment and Risk Stratification in Myelofibrosis with Optical Genome Mapping. Cancers 2023, 15, 3039. [Google Scholar] [CrossRef]
- Gao, H.; Xu, H.; Wang, C.; Cui, L.; Huang, X.; Li, W.; Yue, Z.; Tian, S.; Zhao, X.; Xue, T.; et al. Optical Genome Mapping for Comprehensive Assessment of Chromosomal Aberrations and Discovery of New Fusion Genes in Pediatric B-Acute Lymphoblastic Leukemia. Cancers 2022, 15, 35. [Google Scholar] [CrossRef]
- Kriegova, E.; Fillerova, R.; Minarik, J.; Savara, J.; Manakova, J.; Petrackova, A.; Dihel, M.; Balcarkova, J.; Krhovska, P.; Pika, T.; et al. Whole-genome optical mapping of bone-marrow myeloma cells reveals association of extramedullary multiple myeloma with chromosome 1 abnormalities. Sci. Rep. 2021, 11, 14671. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Soler, G.; Ouedraogo, Z.G.; Goumy, C.; Lebecque, B.; Requena, G.A.; Ravinet, A.; Kanold, J.; Véronèse, L.; Tchirkov, A. Optical Genome Mapping in Routine Cytogenetic Diagnosis of Acute Leukemia. Cancers 2023, 15, 2131. [Google Scholar] [CrossRef]
- Valkama, A.; Vorimo, S.; Kumpula, T.A.; Räsänen, H.; Savolainen, E.-R.; Pylkäs, K.; Mantere, T. Optical Genome Mapping as an Alternative to FISH-Based Cytogenetic Assessment in Chronic Lymphocytic Leukemia. Cancers 2023, 15, 1294. [Google Scholar] [CrossRef] [PubMed]
- Vangala, D.B.; Nilius-Eliliwi, V.; Gerding, W.M.; Schroers, R.; Nguyen, H.P. Optical Genome Mapping in MDS and AML as tool for structural variant profiling—Comment and data update on Yang et al.: “High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance”. Leukemia 2022, 37, 248–249. [Google Scholar] [CrossRef]
- Vieler, L.-M.; Nilius-Eliliwi, V.; Schroers, R.; Ben Vangala, D.; Nguyen, H.P.; Gerding, W.M. Optical Genome Mapping Reveals and Characterizes Recurrent Aberrations and New Fusion Genes in Adult ALL. Genes 2023, 14, 686. [Google Scholar] [CrossRef] [PubMed]
- Valkama, A.; Vorimo, S.; Tervasmäki, A.; Räsänen, H.; Savolainen, E.; Pylkäs, K.; Mantere, T. Structural Variant Analysis of Complex Karyotype Myelodysplastic Neoplasia Through Optical Genome Mapping. Genes Chromosom. Cancer 2025, 64, e70024. [Google Scholar] [CrossRef]
- Xu, H.; Gao, H.; Wang, C.; Cheng, X.; Li, Z.; Lei, C.; Huang, X.; Li, W.; Yue, Z.; Tian, S.; et al. Optical Genome Mapping Reveals Novel Structural Variants in Lymphoblastic Lymphoma. J. Pediatr. Hematol. 2024, 46, e71–e82. [Google Scholar] [CrossRef]
- Seto, A.; Downs, G.; King, O.; Salehi-Rad, S.; Baptista, A.; Chin, K.; Grenier, S.; Nwachukwu, B.; Tierens, A.; Minden, M.D.; et al. Genomic Characterization of Partial Tandem Duplication Involving the KMT2A Gene in Adult Acute Myeloid Leukemia. Cancers 2024, 16, 1693. [Google Scholar] [CrossRef]
- Budurlean, L.; Tukaramrao, D.B.; Zhang, L.; Dovat, S.; Broach, J. Integrating Optical Genome Mapping and Whole Genome Sequencing in Somatic Structural Variant Detection. J. Pers. Med. 2024, 14, 291. [Google Scholar] [CrossRef]
- Zou, Y.S.; Klausner, M.; Ghabrial, J.; Stinnett, V.; Long, P.; Morsberger, L.; Murry, J.B.; Beierl, K.; Gocke, C.D.; Xian, R.R.; et al. A comprehensive approach to evaluate genetic abnormalities in multiple myeloma using optical genome mapping. Blood Cancer J. 2024, 14, 78. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Akkari, Y.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical genome mapping in acute myeloid leukemia: A multicenter evaluation. Blood Adv. 2023, 7, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Tvrdik, T.; Hauenstein, J.; Shi, H.; Deeb, K.K.; Saxe, D.; Hastie, A.R.; Chaubey, A.; Savage, N.M.; et al. Clinical Validation and Diagnostic Utility of Optical Genome Mapping for Enhanced Cytogenomic Analysis of Hematological Neoplasms. J. Mol. Diagn. 2022, 24, 1279–1291. [Google Scholar] [CrossRef]
- Wei, Q.; Hu, S.; Loghavi, S.; Toruner, G.A.; Ravandi-Kashani, F.; Tang, Z.; Li, S.; Xu, J.; Daver, N.; Medeiros, L.J.; et al. Chromoanagenesis Is Frequently Associated with Highly Complex Karyotypes, Extensive Clonal Heterogeneity, and Treatment Refractoriness in Acute Myeloid Leukemia. Am. J. Hematol. 2025, 100, 417–426. [Google Scholar] [CrossRef]
- Wei, Q.; Hu, S.; Xu, J.; Loghavi, S.; Daver, N.; Toruner, G.A.; Wang, W.; Medeiros, L.J.; Tang, G. Detection of KMT2A Partial Tandem Duplication by Optical Genome Mapping in Myeloid Neoplasms: Associated Cytogenetics, Gene Mutations, Treatment Responses, and Patient Outcomes. Cancers 2024, 16, 4193. [Google Scholar] [CrossRef]
- Hidalgo-Gómez, G.; Tazón-Vega, B.; Palacio, C.; Saumell, S.; Martínez-Morgado, N.; Navarro, V.; Murillo, L.; Velasco, P.; Murciano, T.; de Heredia, C.D.; et al. How to combine multiple tools for the genetic diagnosis work-up of pediatric B-cell acute lymphoblastic leukemia. Ann. Hematol. 2025, 1–16. [Google Scholar] [CrossRef]
- Loghavi, S.; Wei, Q.; Ravandi, F.; Quesada, A.E.; Routbort, M.J.; Hu, S.; Toruner, G.A.; Wang, S.A.; Wang, W.; Miranda, R.N.; et al. Optical genome mapping improves the accuracy of classification, risk stratification, and personalized treatment strategies for patients with acute myeloid leukemia. Am. J. Hematol. 2024, 99, 1959–1968. [Google Scholar] [CrossRef]
- Levy, B.; Kanagal-Shamanna, R.; Sahajpal, N.S.; Neveling, K.; Rack, K.; Dewaele, B.; Weghuis, D.O.; Stevens-Kroef, M.; Puiggros, A.; Mallo, M.; et al. A framework for the clinical implementation of optical genome mapping in hematologic malignancies. Am. J. Hematol. 2024, 99, 642–661. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, S.A.; Gagnon, V.; Béliveau, F.; Lavallée, S.; Collin, V.; Hébert, J. Unveiling the Complexity of KMT2A Rearrangements in Acute Myeloid Leukemias with Optical Genome Mapping. Cancers 2024, 16, 4171. [Google Scholar] [CrossRef]
- Joseph, L.; Cankovic, M.; Caughron, S.; Chandra, P.; Emmadi, R.; Hagenkord, J.; Hallam, S.; Jewell, K.E.; Klein, R.D.; Pratt, V.M.; et al. The Spectrum of Clinical Utilities in Molecular Pathology Testing Procedures for Inherited Conditions and Cancer. J. Mol. Diagn. 2016, 18, 605–619. [Google Scholar] [CrossRef]
- Liu, W.; Burger, J.A.; Xu, J.; Tang, Z.; Toruner, G.; Khanlari, M.; Medeiros, L.J.; Tang, G. LPL deletion is associated with poorer response to ibrutinib-based treatments and overall survival in TP53-deleted chronic lymphocytic leukemia. Ann. Hematol. 2020, 99, 2343–2349. [Google Scholar] [CrossRef] [PubMed]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. (Eds.) ISCN (2020): An International System for Human Cytogenetic Nomenclature (2020); S. Kager AG: Basel, Switzerland, 2020. [Google Scholar]
- Mascarello, J.T.; Hirsch, B.; Kearney, H.M.; Ketterling, R.P.; Olson, S.B.; Quigley, D.I.; Rao, K.W.; Tepperberg, J.H.; Tsuchiya, K.D.; Wiktor, A.E. Section E9 of the American College of Medical Genetics technical standards and guidelines: Fluorescence in situ hybridization. Anesth. Analg. 2011, 13, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.A.; Shah, B.; Advani, A.; Aoun, P.; Boyer, M.W.; Burke, P.W.; DeAngelo, D.J.; Dinner, S.; Fathi, A.T.; Gauthier, J.; et al. Acute Lymphoblastic Leukemia, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 1079–1109. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Altman, J.K.; Assi, R.; Bixby, D.; Fathi, A.T.; Foran, J.M.; Gojo, I.; Hall, A.C.; Jonas, B.A.; Kishtagari, A.; et al. Acute Myeloid Leukemia, Version 3.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2023, 21, 503–513. [Google Scholar] [CrossRef]
- Wierda, W.G.; Brown, J.; Abramson, J.S.; Awan, F.; Bilgrami, S.F.; Bociek, G.; Brander, D.; Chanan-Khan, A.A.; Coutre, S.E.; Davis, R.S.; et al. NCCN Guidelines® Insights: Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma, Version 3. J. Natl. Compr. Cancer Netw. 2022, 20, 622–634. [Google Scholar] [CrossRef]
- Zelenetz, A.D.; Gordon, L.I.; Abramson, J.S.; Advani, R.H.; Andreadis, B.; Bartlett, N.L.; Budde, L.E.; Caimi, P.F.; Chang, J.E.; Christian, B.; et al. NCCN Guidelines® Insights: B-Cell Lymphomas, Version 6. J. Natl. Compr. Cancer Netw. 2023, 21, 1118–1131. [Google Scholar] [CrossRef]
- Mikhail, F.M.; Biegel, J.A.; Cooley, L.D.; Dubuc, A.M.; Hirsch, B.; Horner, V.L.; Newman, S.; Shao, L.; Wolff, D.J.; Raca, G. Technical laboratory standards for interpretation and reporting of acquired copy-number abnormalities and copy-neutral loss of heterozygosity in neoplastic disorders: A joint consensus recommendation from the American College of Medical Genetics and Genomics (ACMG) and the Cancer Genomics Consortium (CGC). Anesth. Analg. 2019, 21, 1903–1916. [Google Scholar] [CrossRef]
- Bochtler, T.; Granzow, M.; Stölzel, F.; Kunz, C.; Mohr, B.; Kartal-Kaess, M.; Hinderhofer, K.; Heilig, C.E.; Kramer, M.; Thiede, C.; et al. Marker chromosomes can arise from chromothripsis and predict adverse prognosis in acute myeloid leukemia. Blood 2017, 129, 1333–1342. [Google Scholar] [CrossRef]
- Fontana, M.C.; Marconi, G.; Feenstra, J.D.M.; Fonzi, E.; Papayannidis, C.; Di Rorá, A.G.L.; Padella, A.; Solli, V.; Franchini, E.; Ottaviani, E.; et al. Chromothripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia 2018, 32, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.E.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Ma, M.; Wu, X.; Deng, J.; Liu, X.; Zheng, X.; Gong, Y. Prognostic significance of KMT2A-PTD in patients with acute myeloid leukaemia: A systematic review and meta-analysis. BMJ Open 2023, 13, e062376. [Google Scholar] [CrossRef] [PubMed]
- Nilius-Eliliwi, V.; Gerding, W.M.; Schroers, R.; Nguyen, H.P.; Vangala, D.B. Optical Genome Mapping for Cytogenetic Diagnostics in AML. Cancers 2023, 15, 1684. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | AML | B-ALL | T-ALL | MPAL | MDS | MPN | MDS/MPN | CLL | MCL | Other BCL | MM | T-Cell lym | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aberrations | ||||||||||||||
| MECOM-R | 11 | 3 | 14 | |||||||||||
| KMT2A-R | 5 | 1 | 6 | |||||||||||
| NUP98-R | 5 | 1 | 6 | |||||||||||
| DEK::NUP214 | 1 | 1 | ||||||||||||
| NPM1::MLF1 | 1 | 1 | ||||||||||||
| CBFA2T3::GLIS2 | 1 | 1 | ||||||||||||
| JAK2-R | 2 | 1 | 3 | |||||||||||
| FGFR1-R | 1 | 1 | ||||||||||||
| del(5q) | 1 | 1 | ||||||||||||
| LYN-R | 2 | 2 | ||||||||||||
| SYK | 1 | 1 | ||||||||||||
| IGH::IL3 | 1 | 1 | ||||||||||||
| ETV6::RUNX1 | 1 | 1 | ||||||||||||
| PAX5alt | 2 | 2 | ||||||||||||
| MEF2D-R | 1 | 1 | ||||||||||||
| ZNF384-R | 2 | 2 | ||||||||||||
| IKZF1loss | 6 | 6 | ||||||||||||
| MLLT10::PICALM | 1 | 1 | 1 | 3 | ||||||||||
| BCL11B-R | 3 | 1 | 4 | |||||||||||
| TLX3-R | 3 | 3 | ||||||||||||
| HOXA3::TCRB | 1 | 1 | ||||||||||||
| NUP214::ABL1 | 1 | 1 | 2 | |||||||||||
| Complex karyotype | 2 | 2 | ||||||||||||
| CCND1-R | 1 | 1 | ||||||||||||
| CCND2-R | 1 | 1 | 2 | |||||||||||
| BCL2-R | 1 | 1 | ||||||||||||
| del(7q) | 1 | 1 | ||||||||||||
| MYC-R | 1 | 2 | 3 | |||||||||||
| MTCP1-R | 2 | 2 | ||||||||||||
| Total | 27 | 17 | 10 | 3 | 4 | 0 | 2 | 2 | 2 | 4 | 2 | 2 | 75 | |
| Disease | AML | B-ALL | T-ALL | MPAL | MDS | MPN | MDS/MPN | CLL | MCL | Other BCL | MM | T-Cell lym | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aberrations | ||||||||||||||
| Chromoanagenesis | 32 | 5 | 5 | 12 | 5 | 3 | 2 | 3 | 67 | |||||
| KMT2A PTD | 14 | 1 | 2 | 2 | 19 | |||||||||
| CNVs >=5Mb | 7 | 2 | 2 | 1 | 2 | 2 | 1 | 10 | 3 | 5 | 35 | |||
| RUNX1-R | 4 | 1 | 2 | 7 | ||||||||||
| ETV6-R | 2 | 1 | 3 | |||||||||||
| KMT2A amp | 2 | 1 | 3 | |||||||||||
| TET2 loss | 1 | 1 | 1 | 1 | 4 | |||||||||
| VDR::CBF2AT3 | 1 | 1 | ||||||||||||
| MECOM amp | 1 | 1 | ||||||||||||
| CDKN2A/B del | 1 | 4 | 5 | |||||||||||
| MYB-R | 1 | 1 | ||||||||||||
| TRA-R | 1 | 1 | ||||||||||||
| Hyperdiploidy | 1 | 1 | ||||||||||||
| MYC amp | 1 | 1 | ||||||||||||
| TYK2-R | 1 | 1 | ||||||||||||
| Total | 65 | 11 | 9 | 2 | 19 | 3 | 6 | 1 | 15 | 6 | 4 | 9 | 150 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toruner, G.A.; Hu, S.; Loghavi, S.; OK, C.Y.; Tang, Z.; Wei, Q.; Kanagal-Shamanna, R.; Medeiros, L.J.; Tang, G. Clinical Utility of Optical Genome Mapping as an Additional Tool in a Standard Cytogenetic Workup in Hematological Malignancies. Cancers 2025, 17, 1436. https://doi.org/10.3390/cancers17091436
Toruner GA, Hu S, Loghavi S, OK CY, Tang Z, Wei Q, Kanagal-Shamanna R, Medeiros LJ, Tang G. Clinical Utility of Optical Genome Mapping as an Additional Tool in a Standard Cytogenetic Workup in Hematological Malignancies. Cancers. 2025; 17(9):1436. https://doi.org/10.3390/cancers17091436
Chicago/Turabian StyleToruner, Gokce A., Shimin Hu, Sanam Loghavi, Chi Young OK, Zhenya Tang, Qing Wei, Rashmi Kanagal-Shamanna, L. Jeffrey Medeiros, and Guilin Tang. 2025. "Clinical Utility of Optical Genome Mapping as an Additional Tool in a Standard Cytogenetic Workup in Hematological Malignancies" Cancers 17, no. 9: 1436. https://doi.org/10.3390/cancers17091436
APA StyleToruner, G. A., Hu, S., Loghavi, S., OK, C. Y., Tang, Z., Wei, Q., Kanagal-Shamanna, R., Medeiros, L. J., & Tang, G. (2025). Clinical Utility of Optical Genome Mapping as an Additional Tool in a Standard Cytogenetic Workup in Hematological Malignancies. Cancers, 17(9), 1436. https://doi.org/10.3390/cancers17091436