Extracellular Redox Regulation of α7β Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
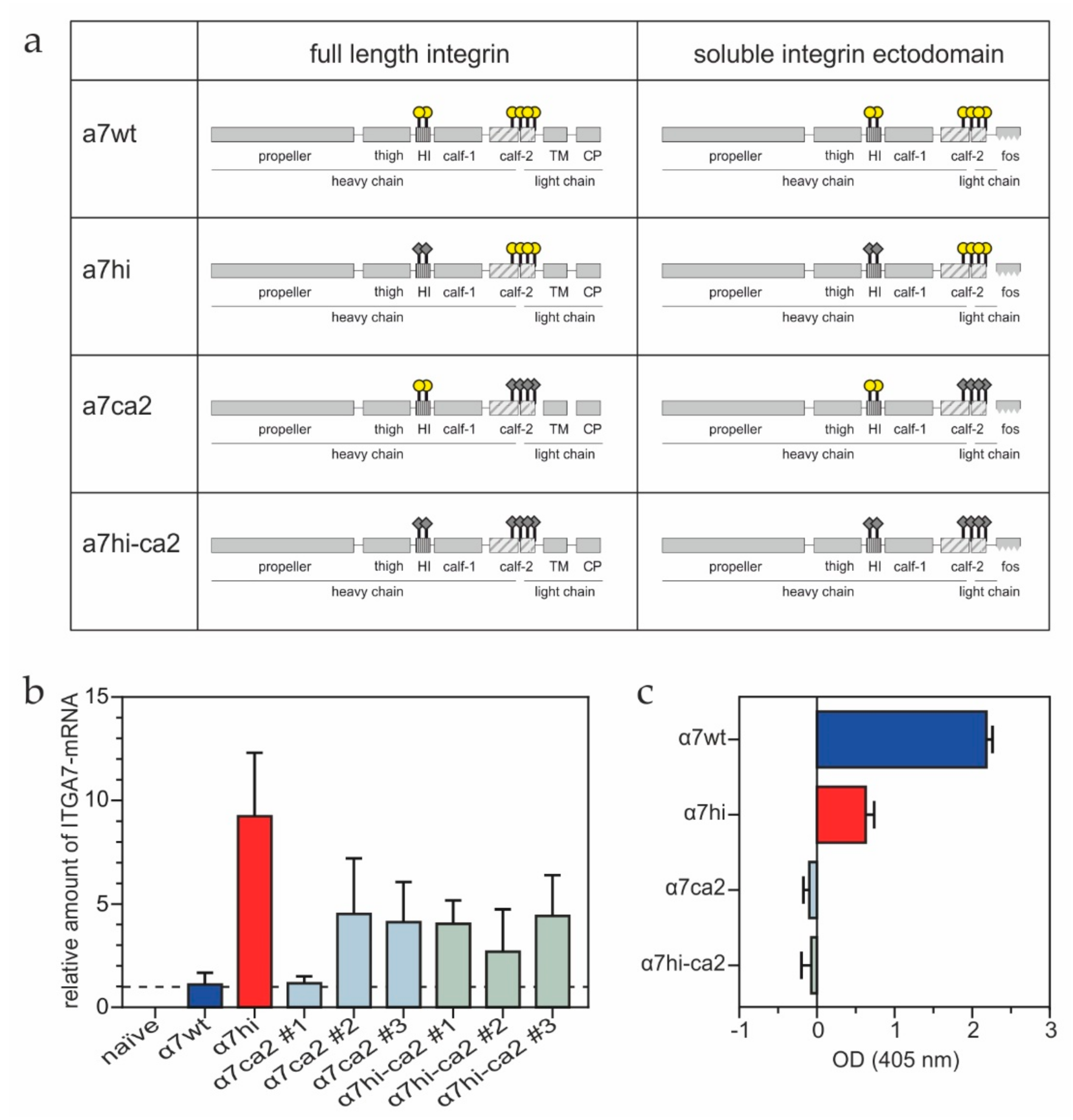
2.2. Mutagenesis of Redox-Modifiable Cysteine Residues within Integrin α7 Subunit
2.3. Construction of Expression Vectors pIRESneo3-a7 and pUC-hygMT-a7-Fos
2.4. Generation of Stably Transfected HT1080 Cells
2.5. Reverse Transcription and Real-Time PCR
2.6. Flow Cytometry
2.7. Immunofluorescence Staining of Transfected HT1080 Cells
2.8. Transduction of LifeAct GFP into HT1080 Cells and Life Cell Imaging
2.9. Production, Purification and Binding Test of Soluble Integrin α7β1
2.10. AFM Measurement of Intermolecular Forces between Soluble Integrin α7β1 and Laminin-111
2.11. Expression and Purification of Recombinant Redoxins
2.12. Intermediate Trapping of Trx1 to Integrins
2.13. Impedance-Based Measurement of Cell Adhesion and Migration
2.14. Negative Staining and Electron Microscopy of Recombinant Soluble α7β1 Integrin
3. Results
3.1. Cysteines of the Integrin α7 Subunit Calf-2 Domain Are Structurally Important
3.2. The Cysteine Pair within the α7 Integrin Hinge Region Determines the Morphology of Transfected HT1080 Cells
3.3. α7hi Supports Cell Migration Less Than Integrin α7wt, Despite a Similar Adhesion Reaction
3.4. Oxidation of the Cysteine Pair in the Integrin α7 Hinge Results in H2O2-Promoted Migration of α7wt-, but Not of α7hi-Expressing Cells
3.5. The Wild-Type α7β1 Integrin Occurs in a High Activity-Conformation on the Cell Surface at a Higher Frequency Than the α7hi-Mutant
3.6. Oxidation of the Cysteine Pair Within the Hinge Region of Integrin α7β1 Increases Binding Activity and Force to Laminin-111
3.7. The Integrin Ectodomain Is a Substrate for Thioredoxin-1, Which Reduces the Integrin α7wt Thiol-Switch
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


References
- Aumailley, M. The laminin family. Cell Adh. Migr. 2013, 7, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.L.; Graham, B.T.; Pabon, L.M.; Han, S.J.; Murry, C.E.; Sniadecki, N.J. Measuring the contractile forces of human induced pluripotent stem cell-derived cardiomyocytes with arrays of microposts. J. Biomech. Eng. 2014, 136, 051005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Xia, W.; Li, J.; Walz, T.; Humphries, M.J.; Vestweber, D.; Cabanas, C.; Lu, C.; Springer, T.A. Relating conformation to function in integrin alpha5beta1. Proc. Natl. Acad. Sci. USA 2016, 113, E3872–E3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askari, J.A.; Tynan, C.J.; Webb, S.E.; Martin-Fernandez, M.L.; Ballestrem, C.; Humphries, M.J. Focal adhesions are sites of integrin extension. J. Cell Biol. 2010, 188, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Lenter, M.; Uhlig, H.; Hamann, A.; Jeno, P.; Imhof, B.; Vestweber, D. A monoclonal antibody against an activation epitope on mouse integrin chain b1 blocks adhesion of lymphocytes to the endothelial integrin alpha 6 beta 1. Proc. Natl. Acad. Sci. USA 1993, 90, 9051–9055. [Google Scholar] [CrossRef] [Green Version]
- Humphries, J.D.; Chastney, M.R.; Askari, J.A.; Humphries, M.J. Signal transduction via integrin adhesion complexes. Curr. Opin. Cell Biol. 2018, 56, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Horton, E.R.; Humphries, J.D.; James, J.; Jones, M.C.; Askari, J.A.; Humphries, M.J. The integrin adhesome network at a glance. J. Cell Sci. 2016, 129, 4159–4163. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.R.; Paul, N.R.; Morgan, M.R. Adhesion and growth factor receptor crosstalk mechanisms controlling cell migration. Essays Biochem. 2019, 63, 553–567. [Google Scholar] [CrossRef]
- Eble, J.A.; Wucherpfennig, K.W.; Gauthier, L.; Dersch, P.; Krukonis, E.; Isberg, R.R.; Hemler, M.E. Recombinant soluble human a3b1 integrin: Purification, processing, regulation, and specific binding to laminin-5 and invasin in a mutually exclusive manner. Biochemistry 1998, 37, 10945–10955. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A.; Bruckner, P.; Mayer, U. Vipera lebetina venom contains two disintegrins inhibiting laminin-binding b1 integrins. J. Biol. Chem. 2003, 278, 26488–26496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiuchi, R.; Takagi, J.; Hayashi, M.; Ido, H.; Yagi, Y.; Sanzen, N.; Tsuji, T.; Yamada, M.; Sekiguchi, K. Ligand-binding specificities of laminin-binding integrins: A comprehensive survey of laminin-integrin interactions using recombinant a3b1, a6b1, a7b1 and a6b4 integrins. Matrix Biol. 2006, 25, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Mayer, U.; Saher, G.; Fassler, R.; Bornemann, A.; Echtermeyer, F.; Von Der Mark, H.; Miosge, N.; Poschl, E.; von der Mark, K. Absence of integrin a7 causes a novel form of muscular dystrophy. Nat. Genet. 1997, 17, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.; Willem, M.; Jones, L.L.; Kreutzberg, G.W.; Mayer, U.; Raivich, G. Impaired axonal regeneration in a7 integrin-deficient mice. J. Neurosci. 2000, 20, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Haas, T.L.; Sciuto, M.R.; Brunetto, L.; Valvo, C.; Signore, M.; Fiori, M.E.; Di Martino, S.; Giannetti, S.; Morgante, L.; Boe, A.; et al. Integrin a7 Is a Functional Marker and Potential Therapeutic Target in Glioblastoma. Cell Stem Cell 2017, 21, 35–50.e39. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, Y.; Hansson, J.; Sasaki, T.; Rousselle, P.; Domogatskaya, A.; Rodin, S.; Tryggvason, K.; Patarroyo, M. Melanoma cells produce multiple laminin isoforms and strongly migrate on a5 laminin(s) via several integrin receptors. Exp. Cell Res. 2011, 317, 1119–1133. [Google Scholar] [CrossRef]
- Ziober, B.L.; Chen, Y.Q.; Ramos, D.M.; Waleh, N.; Kramer, R.H. Expression of the a7b1 laminin receptor suppresses melanoma growth and metastatic potential. Cell Growth Differ. 1999, 10, 479–490. [Google Scholar]
- Eble, J.A.; De Rezende, F.F. Redox-relevant aspects of the extracellular matrix and its cellular contacts via integrins. Antioxid. Redox Signal. 2014, 20, 1977–1993. [Google Scholar] [CrossRef] [Green Version]
- Grosche, J.; Meissner, J.; Eble, J.A. More than a syllable in fib-ROS-is: The role of ROS on the fibrotic extracellular matrix and on cellular contacts. Mol. Aspects Med. 2018, 63, 30–46. [Google Scholar] [CrossRef]
- Goitre, L.; Pergolizzi, B.; Ferro, E.; Trabalzini, L.; Retta, S.F. Molecular Crosstalk between Integrins and Cadherins: Do Reactive Oxygen Species Set the Talk? J. Signal Transduct. 2012, 2012, 807682. [Google Scholar] [CrossRef] [PubMed]
- de Rezende, F.F.; Martins Lima, A.; Niland, S.; Wittig, I.; Heide, H.; Schroder, K.; Eble, J.A. Integrin a7b1 is a redox-regulated target of hydrogen peroxide in vascular smooth muscle cell adhesion. Free Radic. Biol. Med. 2012, 53, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Boronat, S.; Domenech, A.; Hidalgo, E. Proteomic Characterization of Reversible Thiol Oxidations in Proteomes and Proteins. Antioxid. Redox Signal. 2017, 26, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Pan, Y.; Qi, J.; Yue, J.; Zhang, M.; Xu, C.; Li, G.; Chen, J. Disruption of disulfide restriction at integrin knees induces activation and ligand-independent signaling of a4b7. J. Cell Sci. 2013, 126, 5030–5041. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; Pan, Y.; Sun, L.; Zhang, K.; Liu, J.; Lu, L.; Chen, J. The unique disulfide bond-stabilized W1 b4-b1 loop in the a4 b-propeller domain regulates integrin a4b7 affinity and signaling. J. Biol. Chem. 2013, 288, 14228–14237. [Google Scholar] [CrossRef] [Green Version]
- Laragione, T.; Bonetto, V.; Casoni, F.; Massignan, T.; Bianchi, G.; Gianazza, E.; Ghezzi, P. Redox regulation of surface protein thiols: Identification of integrin a4 as a molecular target by using redox proteomics. Proc. Natl. Acad. Sci. USA 2003, 100, 14737–14741. [Google Scholar] [CrossRef] [Green Version]
- Passam, F.; Chiu, J.; Ju, L.; Pijning, A.; Jahan, Z.; Mor-Cohen, R.; Yeheskel, A.; Kolsek, K.; Tharichen, L.; Aponte-Santamaria, C.; et al. Mechano-redox control of integrin de-adhesion. Elife 2018, 7. [Google Scholar] [CrossRef]
- Mor-Cohen, R. Disulfide Bonds as Regulators of Integrin Function in Thrombosis and Hemostasis. Antioxid. Redox Signal. 2016, 24, 16–31. [Google Scholar] [CrossRef]
- Leader, A.; Mor-Cohen, R.; Ram, R.; Sheptovitsky, V.; Seligsohn, U.; Rosenberg, N.; Lahav, J. The role of protein disulfide isomerase in the post-ligation phase of b3 integrin-dependent cell adhesion. Thromb. Res. 2015, 136, 1259–1265. [Google Scholar] [CrossRef]
- Levin, L.; Zelzion, E.; Nachliel, E.; Gutman, M.; Tsfadia, Y.; Einav, Y. A single disulfide bond disruption in the b3 integrin subunit promotes thiol/disulfide exchange, a molecular dynamics study. PLoS ONE 2013, 8, e59175. [Google Scholar] [CrossRef]
- Sun, Q.H.; Liu, C.Y.; Wang, R.; Paddock, C.; Newman, P.J. Disruption of the long-range GPIIIa Cys(5)-Cys(435) disulfide bond results in the production of constitutively active GPIIb-IIIa (aIIbb3) integrin complexes. Blood 2002, 100, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.C.; Khosla, C. Thiol-Disulfide Exchange Reactions in the Mammalian Extracellular Environment. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 197–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-DDC(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Eble, J.A. Titration ELISA as a Method to Determine the Dissociation Constant of Receptor Ligand Interaction. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Urbainsky, C.; Nolker, R.; Imber, M.; Lubken, A.; Mostertz, J.; Hochgrafe, F.; Godoy, J.R.; Hanschmann, E.M.; Lillig, C.H. Nucleoredoxin-Dependent Targets and Processes in Neuronal Cells. Oxid. Med. Cell. Longev. 2018, 2018, 4829872. [Google Scholar] [CrossRef]
- Godoy, J.R.; Funke, M.; Ackermann, W.; Haunhorst, P.; Oesteritz, S.; Capani, F.; Elsasser, H.P.; Lillig, C.H. Redox atlas of the mouse. Immunohistochemical detection of glutaredoxin-, peroxiredoxin-, and thioredoxin-family proteins in various tissues of the laboratory mouse. Biochim. Biophys. Acta 2011, 1810, 2–92. [Google Scholar] [CrossRef]
- Grube, L.; Dellen, R.; Kruse, F.; Schwender, H.; Stuhler, K.; Poschmann, G. Mining the Secretome of C2C12 Muscle Cells: Data Dependent Experimental Approach To Analyze Protein Secretion Using Label-Free Quantification and Peptide Based Analysis. J. Proteome Res. 2018, 17, 879–890. [Google Scholar] [CrossRef]
- Navdaev, A.; Lochnit, G.; Eble, J.A. The rhodocetin alphabeta subunit targets GPIb and inhibits von Willebrand factor induced platelet activation. Toxicon Off. J. Int. Soc. Toxinology 2011, 57, 1041–1048. [Google Scholar] [CrossRef]
- Krokhin, O.V.; Cheng, K.; Sousa, S.L.; Ens, W.; Standing, K.G.; Wilkins, J.A. Mass spectrometric based mapping of the disulfide bonding patterns of integrin a chains. Biochemistry 2003, 42, 12950–12959. [Google Scholar] [CrossRef]
- Calvete, J.J. Structures of integrin domains and concerted conformational changes in the bidirectional signaling mechanism of aIIbb3. Exp. Biol. Med. 2004, 229, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.P.; Stehle, T.; Diefenbach, B.; Zhang, R.; Dunker, R.; Scott, D.L.; Joachimiak, A.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin aVb3. Science 2001, 294, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahav, J.; Wijnen, E.M.; Hess, O.; Hamaia, S.W.; Griffiths, D.; Makris, M.; Knight, C.G.; Essex, D.W.; Farndale, R.W. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin a2b1. Blood 2003, 102, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Blouin, E.; Halbwachs-Mecarelli, L.; Rieu, P. Redox regulation of b2-integrin CD11b/CD18 activation. Eur. J. Immunol. 1999, 29, 3419–3431. [Google Scholar] [CrossRef]
- Verhaar, R.; Dekkers, D.W.; De Cuyper, I.M.; Ginsberg, M.H.; De Korte, D.; Verhoeven, A.J. UV-C irradiation disrupts platelet surface disulfide bonds and activates the platelet integrin aIIbb3. Blood 2008, 112, 4935–4939. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Y.; Tsai, M.Y.; Chuang, K.P.; Huang, Y.F.; Shieh, C.C. Ligand binding of leukocyte integrin very late antigen-4 involves exposure of sulfhydryl groups and is subject to redox modulation. Eur. J. Immunol. 2008, 38, 410–423. [Google Scholar] [CrossRef]
- Hemler, M.E.; Huang, C.; Takada, Y.; Schwarz, L.; Strominger, J.L.; Clabby, M.L. Characterization of the cell surface heterodimer VLA-4 and related peptides. J. Biol. Chem. 1987, 262, 11478–11485. [Google Scholar]
- Mor-Cohen, R.; Rosenberg, N.; Peretz, H.; Landau, M.; Coller, B.S.; Awidi, A.; Seligsohn, U. Disulfide bond disruption by a b3-Cys549Arg mutation in six Jordanian families with Glanzmann thrombasthenia causes diminished production of constitutively active aIIbb3. Thromb. Haemost. 2007, 98, 1257–1265. [Google Scholar]
- Arjonen, A.; Alanko, J.; Veltel, S.; Ivaska, J. Distinct recycling of active and inactive b1 integrins. Traffic 2012, 13, 610–625. [Google Scholar] [CrossRef] [Green Version]
- Boye, K.; Ligezowska, A.; Eble, J.A.; Hoffmann, B.; Klosgen, B.; Merkel, R. Two barriers or not? Dynamic force spectroscopy on the integrin a7b1 invasin complex. Biophys. J. 2013, 105, 2771–2780. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.; Garcia, A.J.; Mould, A.P.; Humphries, M.J.; Zhu, C. Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 2009, 185, 1275–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprecht, V.; Wieser, S.; Callan-Jones, A.; Smutny, M.; Morita, H.; Sako, K.; Barone, V.; Ritsch-Marte, M.; Sixt, M.; Voituriez, R.; et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 2015, 160, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amanso, A.; Lyle, A.N.; Griendling, K.K. NADPH Oxidases and Measurement of Reactive Oxygen Species. Methods Mol. Biol. 2017, 1527, 219–232. [Google Scholar] [CrossRef]
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target. 2015, 23, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Bajetto, A.; Allavena, G.; Wollman, E.; Sitia, R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J. Biol. Chem. 1992, 267, 24161–24164. [Google Scholar] [PubMed]
- Soderberg, A.; Sahaf, B.; Rosen, A. Thioredoxin reductase, a redox-active selenoprotein, is secreted by normal and neoplastic cells: Presence in human plasma. Cancer Res. 2000, 60, 2281–2289. [Google Scholar]
- Leveillard, T.; Ait-Ali, N. Cell Signaling with Extracellular Thioredoxin and Thioredoxin-Like Proteins: Insight into Their Mechanisms of Action. Oxid. Med. Cell. Longev. 2017, 2017, 8475125. [Google Scholar] [CrossRef] [Green Version]
- Son, A.; Kato, N.; Horibe, T.; Matsuo, Y.; Mochizuki, M.; Mitsui, A.; Kawakami, K.; Nakamura, H.; Yodoi, J. Direct association of thioredoxin-1 (TRX) with macrophage migration inhibitory factor (MIF): Regulatory role of TRX on MIF internalization and signaling. Antioxid. Redox Signal. 2009, 11, 2595–2605. [Google Scholar] [CrossRef] [Green Version]
- Schwertassek, U.; Balmer, Y.; Gutscher, M.; Weingarten, L.; Preuss, M.; Engelhard, J.; Winkler, M.; Dick, T.P. Selective redox regulation of cytokine receptor signaling by extracellular thioredoxin-1. EMBO J. 2007, 26, 3086–3097. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergerhausen, L.; Grosche, J.; Meißner, J.; Hecker, C.; Caliandro, M.F.; Westerhausen, C.; Kamenac, A.; Rezaei, M.; Mörgelin, M.; Poschmann, G.; et al. Extracellular Redox Regulation of α7β Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch. Antioxidants 2020, 9, 227. https://doi.org/10.3390/antiox9030227
Bergerhausen L, Grosche J, Meißner J, Hecker C, Caliandro MF, Westerhausen C, Kamenac A, Rezaei M, Mörgelin M, Poschmann G, et al. Extracellular Redox Regulation of α7β Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch. Antioxidants. 2020; 9(3):227. https://doi.org/10.3390/antiox9030227
Chicago/Turabian StyleBergerhausen, Lukas, Julius Grosche, Juliane Meißner, Christina Hecker, Michele F. Caliandro, Christoph Westerhausen, Andrej Kamenac, Maryam Rezaei, Matthias Mörgelin, Gereon Poschmann, and et al. 2020. "Extracellular Redox Regulation of α7β Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch" Antioxidants 9, no. 3: 227. https://doi.org/10.3390/antiox9030227