Translational Research in Osteogenesis Imperfecta and Cell Therapy †
by
Vrisha Madhuri
1,2,*, 
Sowmya Ramesh
1,2,
Renita Raymond
1,2,
Agnes Selina
1,2 and
Lakshmi Loganathan
1,2 1
Department of Paediatric Orthopaedics, Christian Medical College, Vellore 632 004, India
2
Center for Stem Cell Research, a Unit of inStem Bengaluru, Christian Medical College, Vellore 632 002, India
*
Author to whom correspondence should be addressed.
†
Presented at the Translational Research in Osteogenesis Imperfecta and Cell Therapy Symposium, Vellore, India, 6–7 February 2019.
Proceedings 2021, 72(1), 3; https://doi.org/10.3390/proceedings2021072003
Published: 10 May 2021
(This article belongs to the Proceedings of Stand Alone Papers 2021)
Abstract
:On 6 and 7 of February 2019, Center for Stem cell Research (CSCR) and Pediatric orthopedic Unit at Christian Medical College (CMC), Vellore, conducted a meet on Translational Research in Osteogenesis Imperfecta and Cell Therapy. Osteogenesis Imperfecta (OI) is a disease in which the individual has weak brittle bones which fracture easily, resulting in multiple fractures throughout their childhood. Children become deformed and often do not walk or grow normally. The meeting was conducted to highlight the newer advances and therapies for osteogenesis imperfecta and bring regulatory processes and challenges that need to be addressed.
1. Introduction
The meeting was held under the aegis of a project called “Boost to Brittle Bones” (BOOST2B) aimed at providing a new treatment for this condition and is funded by Department of Biotechnology, International division in India and VINNOVA in Sweden and is carried out jointly by CMC, Vellore and Karolinska Institutet, Sweden. The meeting was jointly organized by Professor Vrisha Madhuri from CMC Vellore and Cecilia Gotherström of Karolinska Institutet, Sweden, and comprises a clinical trial that looks at the safety and efficacy of transplanting mesenchymal stem cells in the affected children. There are only a handful of OI cases treated in this manner around the world, and a similar trial is about to begin in Europe.
The day 1 symposium focused on the current concepts, molecular genetics, clinical management, and newer treatment strategies of Osteogenesis Imperfecta. The speakers were renowned geneticists, Endocrinologists, Hemato-oncologists, radiologists, occupational and physiotherapists, Orthopedicians and scientists working in Osteogenesis Imperfecta and cell and gene therapy. The meeting brought out the most recent trends in the diagnosis and management of osteogenesis imperfecta, including newer management using bisphosphonates and cell therapies and the best practices in the molecular diagnosis, radiological and bone density assessment, surgery, and rehabilitation. The new project BOOST2B was presented by Dr. Vrisha Madhuri after the background information regarding the European project by Dr. Cecilia Gotherström.
Some of the notable speakers on Osteogenesis imperfecta were Prof. David Sillence from Australia, Dr. Eva Aström from Sweden, Dr. James Fernadez and Dr Claire Hill from Sheffield, United Kingdom, and Dr. Edwin M. Horwitz from Emory University, United States. Indian speakers included Dr. Hitesh Shah from Manipal, Dr. Suresh from Mediscan Chennai, Dr. Sridhar Gibikote, and Dr. Thomas Paul from Vellore.
The second-day workshop was on the “Current regulatory and Ethical requirements for conducting cell therapy-based clinical trials” where the best practices in this field in India, USA, and Europe were discussed along with the changing scenario in the regulatory field in India. The notable speakers included Lt. Gen (rtd) Dr. Velu Nair, Dr. Alok Srivastava, Dr. Pawan Gupta, Dr. Vikram Mathews, and Dr. Annie John from India. The European regulators included Vera Franzen, Dr. Gudmund Hedenskog, Annika Goos, and Dr. Lilian Jallow Walther. The participation by industry partners, clinical trialists and regulators in these deliberations allowed us to compare and learn from the best practices in this field in the world.
2. Day 1 Lectures
2.1. Prenatal Diagnosis and Intrauterine Assessment of Osteogenesis Imperfecta
Dr. Suresh, MediScan System, Chennai
Osteochondrodysplasias are the abnormalities of the bone or cartilage growth. Skeletal dysplasias are abnormalities of the skeleton. It continues throughout life. Fetal skeletal dysplasias are challenging to diagnose in utero due to several factors, which are mentioned below.
- Overlapping phenotypic features
- Lack of precise molecular diagnosis of many diseases
- Variability in the manifestation of skeletal findings
- Lack of a systematic approach
- The inability of the ultrasound to provide an integrated view.
The three-dimensional ultrasound is used for diagnostic accuracy and the parameters assessed are long bones, thorax, hands, feet, skull, spine, and pelvis. The prevalence of skeletal dysplasias is 2.4/10,000 births. The overall prevalence of skeletal dysplasias among perinatal deaths was 9.1 per thousand cases. Prenatal diagnosis is more straightforward in cases of positive family history and accurate phenotypic description. The individual parameters assessed during the prenatal period is explained as below.
In the case of long bones, its absence, any hypoplasias, its length, curvature, degree of mineralization, number of fractures, the lethality parameters like femur–foot length ratio (<1 suggests skeletal dysplasia; normal = 1) and femur–abdominal circumference ratio (<0.16 suggests lung hypoplasia) are measured. All long bones are measured, and any limb length shortening is noted.
In the thorax, chest circumference and the cardiothoracic ratio at the level of the heart is measured. Chest circumference < 5th centile of the gestational age is the indicator of pulmonary hypoplasia. Other parameters are chest–heart area ratio, chest area, a chest–trunk ratio less than 0.32, measurement of the clavicle, and the presence of scapula. Pulmonary hypoplasia occurs in many skeletal dysplasias and is the main reason for many perinatal deaths.
Hands and feet should be evaluated for the presence of features like polydactyly, syndactyly, clinodactyly, and other deformities.
In the skull, head circumference and biparietal diameter should be measured to exclude macrocephaly. Interorbital distance for hypertelorism and interocular diameter are also measured. Other features assessed are micrognathia, short upper lip, abnormally shaped ears, frontal bossing, cloverleaf skull, abnormal shape of the head also noted (brachycephaly, scaphocephaly, and craniosynostosis).
In the spine, its mineralization, curvature, neural arches, vertebral height should be noted to rule out scoliosis and platyspondyly.
In the pelvis, its shape is important in certain dysplasias and dysostosis, femoral hypoplasia, achondroplasia which has features of flat rounded iliac bones with lack of iliac flaring, broad horizontal superior horizontal margins, and small sacrosciatic notch.
In Ultrasonography, the echogenicity of the bone is measured. This assessment is done to diagnose limb shortening (severe, mild, or moderate) and agenesis. In severe shortening cases, depending on the region of shortening, three types of abnormal limb morphology, namely, rhizomelia, mesomelia, and rhizo-mesomelia, are diagnosed. Osteogenesis imperfect comes under mesomelia and rhizo-mesomelia. It also shows hypoechogenicity with the presence of fractures.
In osteogenesis imperfecta, the skull is thin, the cranial vault has a wavy outline and is easily compressible even with the US probe. The major radiological features are generalized osteoporosis, Wormian bones, collapsed vertebral bodies, rib fractures, thin cortex in tubular bones, and thin shaft with bowing deformities.
It is possible to make an in utero diagnosis for mutations in the following genes COMP, COL9A1, COL9A2, COL9A3, MATN3, EVC, COL1A1, COL1A2, COL2A1, FGFR3, DTDST, and RUNX2. Even when there is a termination or stillbirth, the postnatal workup is essential because diagnostic information is helpful for (1) counselling parents for future pregnancies, its recurrence and risk (2) designing strategies for prenatal monitoring and diagnosis in future pregnancies. In the postnatal skeletal survey, the following parameters are assessed, AP and lateral skull to include the atlas and axis, AP chest, AP pelvis, AP lumbar spine, and lateral thoracolumbar spine, AP of one lower limb and one upper limb, and PA of one hand for bone age assessment.
Prenatal CT can be used for the diagnosis of severe skeletal abnormalities. Most skeletal dysplasia die in utero, are stillborn, die as neonates, or be delivered by elective termination. The correct diagnosis requires a pathological workup. Minimal postmortem workup should include external examination with a photograph, postmortem whole body radiographs, skin, or other tissue biopsy specimens for chromosome analysis, preservation of fibroblast for possible later biochemical, enzymatic, or genetic studies, which should be sent to the concerned lab.
2.2. Radiological Assessment of Osteogenesis Imperfecta and Basilar Impression
Dr Sridhar Gibikote, Professor and Head, Department of Radiology Christian Medical College, Vellore
Brittle bone disease is a genetic, qualitative or quantitative abnormality in type 1 collagen. Old classifications include OI congenita which is diagnosed prenatally, and OI Tarda, which is diagnosed in the postnatal period. The newer classification includes OI TYPE I to TYPE VII depending on the phenotype.
Radiographic findings in OI are described with (i) Osteopenia which is radiologically identified as cortical bone thinning, trabecular bone transparency. To exclude the other causes of osteopenia, DEXA helps establish the diagnosis of OI; (ii) Bone fractures are few or numerous throughout their lives. The most common fractures occur in long bone diaphyses, the spine, and the apophyses; (iii) bone deformities as lower limbs/upper limbs bowing, mainly due to excessive bone malleability and plasticity.
In the skull, radiography shows a prominent occipital region called the “Darth Vader” appearance or a flattening of the cranial vault with transverse infolding of the cranial base called “Tam O’Shanter skull”, which is rare. More frequently, multiple wormian bones leading to a “mosaic” or “paving” appearance to the cranial vault is seen.
Some radiological features display the phenotype/type of OI; in OI type III, they show a lack of bone modelling with “bamboo cane” appearance with multiple healed fractures, severe deformities of the femur called “shepherd’s crook” and in tibia called the “sabre shin’s deformity”. Hyperplastic callus formation is rarely reported in type V, in the femur bones of males. Ossification of the interosseous membrane is seen in type V forearm or leg. “Popcorn” calcification is usually seen in type III, in the metaphyseal and epiphyseal region of the knee, leading to limb length discrepancy.
After bisphosphonate administration, radiological changes in bones include increased bone density in the spine and long bones. Dense metaphyseal lines called “Zebra lines” are seen parallel to growth plate in the long bones; each line corresponds to one treatment course. Multiple lines show a “ladder-rung” appearance, and in the spine or the flat bones, a “bone-within-a bone” pattern is observed.
Three radiologic categories of OI:
- Ø Category I—Thin and gracile bones
- Ø Category II—Short and thick limbs
- Ø Category III—Cystic changes
Craniovertebral junction deformities
The three major abnormalities noted in osteogenesis imperfecta are basilar invagination, basilar impression, and platybasia. Basilar invagination is the protrusion of the odontoid process into the foramen magnum. The radiographic criterion is a McRae measure at or above zero. Basilar impression is a condition in which the odontoid process is positioned far above the skull’s caudal borders. The radiographic criteria are the chamberlain measure, the Mcgregor measure, or the DM distance (perpendicular distance from D-point to a line drawn through the most caudal point of the occipital curve(M) parallel to nasion-sella line) is elevated by more than 3 standard deviations above the average of age-matched healthy controls. Platybasia is a flat anterior cranial base angle (nasion-sella-basion angle). Radiologically it is diagnosed when the anterior cranial base angle is more than 3 SDs above the age-matched healthy controls. The most sensitive indicator of basilar impression is the DM line measure, as this index changes less with age than Chamberlain and McGregor lines.
2.3. Osteogenesis Imperfecta—A Combined Phenomic and Genomic Approach
Dr. David Sillence, Genetic Medicine, Children’s Hospital, Westmead, Australia
Genetic heterogeneity in the heritable disorders of bone fragility (osteoporosis and fractures) has been well established for over 40 years. Ekman, 1788 wrote his doctorate on “A fragile boned family”. There was no mention of blue sclerae. Later, when families were described with an autosomal dominant inheritance of bone fragility and distinctly blue sclerae, the debate commenced about whether Osteogenesis Imperfecta (OI) was a single disorder or multiple disorders with varying severities of bone fragility.
Over the last three decades, progress has been made through classical genetic and clinical studies to delineate five major phenotype groupings. Diverse studies, firstly in connective tissue proteins, then molecular genetics and more recently genomics, demonstrate that these five phenotypes and several special syndromes with phenotypic features overlapping with those in OI result from mutations in at least 24 distinct gene loci. In the last five years, genetic and functional studies have resulted in even more insights into the pathogenic mechanism responsible for bone fragility. In particular, GWAS studies (Rivadeneira and Makitie) suggest that there are many more loci that have a quantitative trait locus (QTL) modifying effect on bone density and bone strength.
The current international nosology (naming) and classification have been maintained as a descriptive grouping of syndromes. In the tables below, the descriptive grouping is compared with the numerical nomenclature. One of the concerns expressed from time to time is that the numerical nomenclature (I–V) is confused with the severity scale. This breaks down immediately in that the numerical nomenclature ordered the syndromes in terms of the order they were described, not in the order of severity. The grouping is compared with the historic numerical nomenclature in the tables below, but note that the International Nosology committee recommends Arabic numerals rather than Roman numerals.
The genome database, OMIM, now lists 18 types of Osteogenesis Imperfecta, OI Types I–XVIII. This numerical listing is a synthesis of the original five clinical types with 13 further genotypes resulting from dominant, autosomal recessive or sex-linked inheritance, but the gene types will again increase in the upcoming years (Table 1, Table 2, Table 3, Table 4, Table 5, Table 6, Table 7, Table 8, Table 9, Table 10 and Table 11).
Further genes in which mutations have been discovered have been reported since the last Nosology meeting 2017, and several other gene loci are still being actively researched.
In a Whole Exome Sequencing report of 411 patients from 288 families with Skeletal dysplasia, Maddirevula and colleagues 2018 reported several newly discovered genes in which mutations result in a predisposition to low impact fractures. Homozygous nucleotide sequence variations in SUCO (SUN domain ossification factor) results in an extremely severe OI with broad femurs and abundant callus, which has an appearance reminiscent of OI type 2. In addition, there are at least nine named syndromes and multiple premature ageing disorders with overlap in phenotypic features with Osteogenesis Imperfecta.
2.4. Molecular Diagnosis and Genotype–Phenotype Correlation of Osteogenesis Imperfecta
Dr. Hitesh Shah, Pediatric Orthopedics, Kasturba Medical College, Manipal
Osteogenesis Imperfecta (OI) is a clinically and genetically heterogeneous disorder with low bone mineral density leading to bone fragility and fractures. Clinical features include orthopedics and non-orthopedics manifestations. Initially, only autosomal dominant forms caused by COL1A1 and COL1A2 genes were recognized. However, with advancement in molecular technologies, new genes have been identified, leading to the recognition of autosomal recessive OI. Currently, mutation of around 20 genes is responsible for OI. The classification of OI thus is now shifted on genetic analysis from clinical types. Pathogenic variants in approximately twenty genes (COL1A1, COL1A2, CRTAP, LEPRE1, FKBP10, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, WNT1, BMP1, TMEM38B, IFITM5, PLS3, CREB3L1, SEC24D, SPARC, P4HB, and MBTPS2) are known to cause OI. Molecular diagnostics of genetically heterogeneous disorders is greatly facilitated by parallel testing of disease genes through next-generation sequencing. However, different approaches are practiced, from small gene panels for specific disease entities to broader panels containing all disease genes to exome or even genome sequencing. We made an effort to assess the severity of the condition in patients using the severity grading scale proposed by Van Dijk and Sillence based on the clinical features. An NGS-based bone mass panel containing 70 genes known or suggested to cause alterations in bone mineral density was developed, and the enrichment of the coding region of these genes was performed. We correlated genotype with phenotype.
A total of 50 patients were evaluated for the clinical and molecular spectrum of OI in Indian patients. The molecular defects were identified with gene panel and exome sequencing in all but one family. We observed 24 novel mutations and 24 known mutations in ten genes known to be associated with OI. Autosomal dominant (COL1A1, COL1A2, and IFITM5) and autosomal recessive (WNT1, LEPRE1, FKBP10, SERPINF1, and BMP1) inheritance patterns are observed in almost equal proportions. Severe scoliosis, hyperplastic callus formation, severe osteopenia, and long bones’ bowing were associated with IFITM5 mutation. Extensive bowing of long bones, dense metaphyseal bands, fractures, and osteopenia was associated with WNT1 mutation. Joint contractures with osteogenesis imperfecta were associated with FKBP10.
Ours is one of the largest cohorts of patients with OI in the literature and demonstrates panel testing’s clinical utility for establishing a molecular diagnosis in genetically heterogeneous conditions like OI. In 48 of the 50 unselected OI patients recruited by a single Indian clinical center, mutation was identified by two next-generation sequencing approaches. The percentage of autosomal recessive forms was unusually high (48%) due to a high degree of consanguinity in the local population. Exome sequencing provided no higher diagnostic yield than specific testing by a small gene panel. Based on their characteristic clinical and radiological phenotype, a targeted screening of the genes FKBP10, IFITM5, and WNT1 is feasible.
2.5. Current Surgical Management of Osteogenesis Imperfecta
Dr. James Fernandez, Sheffield Children’s Hospital, United Kingdom
Though medical management of Osteogenesis imperfecta has vast benefits by altering the fracture patterns, surgical management has a significant effect on the walking milestones, limb length equalization for the ambulatory and social integration. Primary goals are to maximize function and minimize disability by reducing fracture patterns and improve relative independence to carry out their activities. Orthopedic management needs some internal support for the long bones as the plates in isolation is contraindicated, and hence rodding is preferred. Initial days stabilization is done by fixed length rods, in which the complication rate is much higher by 72% as rods stick out easily. Extensible rods (Bailey–Dubow) Sofield Miller technique reduced the complication rate of fixed rods by 14% out of total 24 children evaluated after rodding; fracture incidence reduced markedly for eight children with no fractures and 1 to 4 fractures only in 13 children, mobilization improved from wheelchair dependent to walker from 3 to 12 (quadrupled), milestones improved from 12 to 17 in standing and 9 to 16 in walking, educational, and social development by 21 attending regular school, 13 independent, and 5 with one person dependent, 5 out of 8 driving a standard car. Their survival rate is also increased as compared with the survival rate before rodding.
The use of bisphosphonates has influenced surgical management in many ways by changing the pattern of fracture. Fractures are located all along the long bones’ shaft in pre bisphosphonate era, whereas only along the subtrochanteric region of femur bone after bisphosphonate therapy. Intracapsular fractures are more common after rodding because of the lengthening of long bones after rodding and coxa vera’s possible development. Hence, to prevent neck fractures, reconstructive locking rods are made in practice. Management of coxa vara is by initial valgus osteotomy and by double fixation technique, which is a combination of tension band wiring with k wire fixation. Management of non-union is aided by placing BMP (bone matrix protein), guided growth in selective patients by stainless steel eight plates. Limb length discrepancy, if any, is managed by different techniques such as by Ilizarov, distal femur osteotomy, bifocal tibial corticotomy, hinged knee, femoral nail left in situ, and PRECICE intramedullary lengthening nail.
The long term outcomes of using Sheffield telescopic intramedullary rod system in the management of 22 (13 females + 9 Males) OI patients with an average of 24.7 y follow up were evaluated. Most of the patients had Sillence type III OI. Fracture incidence reduced markedly after rodding. Indications for re-operation are majorly for patients who are growing and due to bent rod due to trauma. Complications were contributed mainly by rod migration during surgical treatment and symptomatic non-union, probably by the bisphosphonate treatment. After surgery, the patient’s mobility shows significant improvement in mobility with walking aids and independent mobility. Buttock and hip symptoms such as ache and painful clicking with femoral rods were much less. With tibial rods, most of them had no symptoms, while some had minimal protruding rods. Compared to the UK adult normative data for physical and mental health, the OI cohort’s mean scores have poorer scores. The retrograde rodding technique managed the upper limb as the most common fracture site was the distal humerus because of the forearm’s deformity. Analysis of the surgical management of the OI showed improvement in mobility and functional status as well.
2.6. Clinical Assessment and Management of Osteogenesis Imperfecta
Dr. Eva Åström Astrid Lindgren Children’s Hospital at Karolinska University Hospital and Karolinska Institutet Stockholm, Sweden
Assessment and management of osteogenesis imperfecta (OI) depend on the clinical severity, family history, and child’s habitat.
In the delivery room, the APGAR Appearance, Pulse, Grimace, Activity, and Respiration GAR screening is used to quickly summarize the newborn’s health. Inspection of body posture, size, limb curvatures, and movement asymmetries is essential.
Mainly the severe forms of OI are detected before birth or on the first day of life. Early monitoring of head circumference, weight and height is essential. Within the first day of life, a whole-body X-ray (babygram) can show fractures and help in OI’s phenotyping. A lab screening with p-calcium, phosphate, albumin, creatinine, ALP, PTH, and 25OH vitamin D is important for further management and treatment. Handling with gentle care is recommended. Neonatal fractures should be fixated with soft material (padding).
Milder forms of OI are often detected later in life, and in case of suspected OI, parental handling information and routine care is provided.
On the whole, every child with OI needs to be assessed individually. Fracture tendency often varies during life and often increases when the child becomes more physically active, especially before learning motor skills. For children, deformities may decrease or increase during childhood.
In severe OI, parents often learn to know when their child has a fracture, and not all undislocated fractures/fissures should be X-rayed.
Multidisciplinary pediatric OI-team assessments are important during childhood to prevent deformities and minimize pain and fracture rates and decide if and when an intervention is needed for each individual.
While caring for a newborn, the team has to have more intensive and focused care. Gentle care while handling, fracture fixation with soft padding, analgesic administration timed to facilitate nutrition and later the use of a foam rubber cushion under the infants bottom area when learning to walk (to minimize the risk of vertebral compressions) are some of the points to be considered.
During childhood, all clinical features associated with OI should be addressed: bleeding- and coagulation disorders, calcium and D-vitamin, growth, motor development, cardiology, respiration, and skeletal: curvatures, fractures, scoliosis, basilar impression, radiographs, lateral radiograph of the spine to assess vertebral compression fractures, lateral radiograph of the skull, fractures and curvatures of long bones, bone mineral density (dxa), dental examination of teeth and jaws, hearing, vision, pain/fatigue, and quality of life.
Anesthetic considerations include risk from hyperlaxity of the neck, risk for basilar impression, risk for bleeding/coagulopathy, be prepared for excess bleeding even if laboratory tests are normal, be prepared for hyperthermia.
The main treatment options are physiotherapy, orthopedic operations and medication, often in combination (see later chapter). A consensus statement of physical rehabilitation in children and adolescents with OI was published in 2018 (Mueller B, Semler O). Fassier–Duval telescopic rod is the modern surgical treatment to prevent long bones’ bowing and fractures (if needed). Intravenous bisphosphonate treatment is the medication of choice (if needed). Extensive research studies are made to collect more data for the creation of monitoring and treatment recommendations. In a longitudinal study of OI conducted by OI Linked Clinical Research Centers (LCRC) in North America, the large sample size of the cohort allows examination in various subtypes of OI and assess potential correlations which may provide important knowledge on growth characteristics and pulmonary function in OI:
The growth characteristics involve the height over the years of age, and in the study cohort of 552 individuals, there was an increasing age correlation with reduced height.
The spirometry analyses for pulmonary evaluation in correlation with the growth assessment (using either measured height or arm span calculated height) can underestimate the pulmonary involvement in severe OI forms (who have multiple bone deformities). So serial monitoring of observed FCV and FEV1 is probably the most reasonable way to assess pulmonary functions. Spirometry should be considered in all individuals with OI, especially in OI types III and IV. Like adults with FEV1 less than 1.5 L, pneumococcal vaccination and annual influenza vaccination should be considered in individuals with decreased pulmonary reserve.
According to Osteogenesis Imperfecta Federation Europe (2018), the needs of the OI community are management of pain related to fracture, medication to improve bone strength, orthopedic emergency care, regular follow-up to monitor medical issues like pulmonary or cardiac issues and provide assistance by occupation therapy, and dental care and audiology assessment. At a Brittle Bone Society meeting, important issues were pain/fatigue and quality of life at all ages.
2.7. Medical Management of OI: Action of Bisphosphonate Treatment
Dr. Eva Åström, Astrid Lindgren Children’s Hospital at Karolinska University Hospital and Karolinska Institutet Stockholm, Sweden
Bisphosphonates are stable analogues of pyrophosphate that inhibit osteoclastic function and have long half live in bone. The mechanism of action of the nitrogen-containing bisphosphonates is due to the mevalonate pathway’s disruption, primarily in osteoclastogenesis.
Recently Australian consensus guidelines on the use of bisphosphonate therapy in children and adolescents has been published (J Pediatrics & Child Health: Simm PJ 2018) that states:
Intravenous bisphosphonates should be used in children with severe osteogenesis imperfecta (OI) (e.g., type III), children with vertebral compression fractures and considered in children who have had two or more long-bone fractures per year.
Oral bisphosphonates should only be considered for those with mild to moderate OI in the absence of vertebral compression fractures.
Do not recommend to children with severe OI to cease therapy once bone mineral density (BMD) improves; they should be continued on a long term lower dose of bisphosphonate to preserve bone strength during growth.
The annual dose of intravenous bisphosphonate can be halved once the height-adjusted BMD z-score falls within the range of −2 to 0. Once the BMD z-score > 0, the dose can be reduced further, and treatment continued at this lower dose until the cessation of growth.
In children with less severe OI, it may be possible to stop bisphosphonate therapy during childhood without deterioration in clinical status or BMD.
Once a child with OI stops growing, it is recommended that therapy is suspended, and the child is monitored.
Intravenous BP treatment is associated with improvement in the number of vertebral fractures in the growing skeleton and modelling, and some studies have shown a significant reduction in the incidence of long-bone fractures.
In mild OI, oral BP has been shown to reduce fracture rates to a degree similar to that of intravenous agents but have neither been associated with improvements in spinal morphology nor reduction in bone pain.
Not all children with OI require intravenous BP.
Treatment should be instigated in children with severe OI (e.g., type III) and strongly considered in children with two or more long-bone fractures per year or children with vertebral compression fractures.
Most OI data pertains to the use of pamidronate, with increasing data accumulating on the use of other bisphosphonates, primarily zoledronate.
The best agent, dose or frequency is yet to be determined.
Treatment approaches vary according to the resources available and the experience of the treating clinician.
Pamidronate is often used in children younger than two years of age, followed by switching to zoledronate in older children with moderate to severe OI. Pamidronate doses vary from 9 to 12 mg/kg/year, and zoledronate is commenced at 0.1 mg/kg/year in two divided doses. Many centers reduce the first-ever dose of pamidronate (0.5 mg/kg) or zoledronate (0.0125 mg/kg or 0.025 mg/kg) in bisphosphonate naïve patients to minimize acute phase reactions and hypocalcemia. Once the height-adjusted z score is >0, therapy should be reduced to 0.025 mg/kg/year of zoledronic acid and 1.5 mg/kg every six months of pamidronate until the end of growth.
It may be possible to cease treatment in children with OI type 1, but there remains no clear evidence to assist in these decisions, which remains controversial.
The Swedish treatment schedule:
Bisphosphonate is used to treat type III and severe type IV phenotypes from one-month of age (usually from 2 to 4 months of age, when op intravenous port). Milder form with several progressive vertebral compressions is also started with bisphosphonates at that point of time; they are treated until growth arrest.
First required normal D-vitamin level. Oral D-vitamin is given to all Swedish young children. Pamidronate (APD) given as monthly infusions 10–30 mg/m2. More specifically, dose 1–2: 10 mg/m2, dose 3–5: 20 mg/m2, and from sixth dose: 30 mg/m2. When normalized, BMD (bone mineral density) individualized increased interval between infusions. In a Swedish study of 79 children with osteogenesis imperfecta, the researcher administered pamidronate infusions at regular intervals and found decreased fracture rate (Lindahl K et al., 2016). Assessment is done every six months during the 1–2 years, then yearly. Diary of pain, well-being, mobility, BMD (DXA whole body and lumbar spine), growth and mobility. Bone markers such as serum ALP, Osteocalcin, Ca, PO4, PTH, and urine deoxypyridinoline/creatinine are done. Some of the risks of BP-treatment include a short episode of flu-like symptoms, secondary hyperparathyroidism, osteonecrosis of the jaw (in adults), stress fractures after the treatment of duration > 5 years (in adults). Low calcium and D-vitamin should be normalized before treatment start due to the risk of severe hypocalcemia.
2.8. Update on Therapy of Osteogenesis Imperfecta 2019
Dr. David Sillence, Genetic Medicine, Children’s Hospital, Westmead, Australia
Treatment in OI requires multidisciplinary care encompassing orthopedic care with attention to fractures and correction of deformities. Rehabilitation includes physiotherapy, occupational therapy, and orthotics. Medical therapy includes cyclic intravenous or intermittent oral bisphosphonates. Finally, the genetic diagnosis has done for understanding prognosis and counselling.
Commonly, five types of bisphosphonates are available. Cyclic intravenous Pamidronate, Neridronate, and Zoledronate are extensively used in clinical trials showing unequivocal safety and benefit in OI patients. Then the oral bisphosphonates include Alendronate and Risedronate.
All other medical therapies are still experimental.
Denosumab is a monoclonal antibody that is given once in six months. It binds to the RANK ligand and blocks the signal transduction through the RANK receptor. It, therefore, reduces osteoclast activity and increases bone density. This infusion also causes severe suppression of bone turnover. It has the concern of atypical hip, pelvis, and rib fractures. It is also mainly used for aseptic necrosis of the jaw.
Anti-Sclerostin monoclonal antibody is another potential therapeutic agent commonly known as Romosozumab, which causes severe suppression of bone turnover. There is the potential risk of atypical hip, pelvis, and rib fractures, and in phase II trials, a 30% increase in cardiovascular events were observed.
Sequential use of bisphosphonate therapy followed by antiresorptive therapy with Denosumab has a high risk of causing low bone turnover induced fractures.
2.9. Quality of Life in Osteogenesis Imperfecta
Dr Claire Hill, Physiotherapist, Sheffield Children’s Hospital, United Kingdom
Individuals with Osteogenesis Imperfecta (OI) have low bone mass, recurrent fractures, varying degrees of short stature and deformity, and reduced functional ability. The disease affects the physical, social and emotional well-being of the individual and their family.
Patients undergo X-rays, blood tests, CT, MRI, and bone density scans, physical and occupational therapy assessments. However, disease-specific Quality of life (QoL) is not regularly recorded.
QoL has both subjective and objective components; health-related quality of life (HRQoL) is described as the patient’s perception of an illness’s impact and its treatment. The key to measuring QoL appears to be the idea that all individuals have their perspective on QoL. It is dependent on their current lifestyle, past experiences, hopes, dreams, and ambition for the future (Eiser and Morse, 2001).
Previous attempts at measuring QoL in OI have used several generic instruments. However, it is hypothesized that the development of disease-specific QoL questionnaires for individuals with OI would improve outcome measurement following medical, surgical, and physical therapy intervention.
Semi-structured interviews took place in the UK with ten children, ten parents, and five health professionals, attempting to uncover concepts relevant to children’s quality of life. Similar interviews were undertaken with 15 adults from both the US and UK to highlight concepts relevant to adult quality of life.
These interviews uncovered six main themes relevant to children with OI and seven concepts that were important to adults. The Six Main Themes are being safe and careful, reduced function, fear, pain, being different/Isolated, and independence and the main concepts are fractures, pain, social and relationships, hearing loss, sleep disturbance, activities—household chores and fatigue.
Together with the vocabulary used by individuals with OI, these concepts were utilized to develop pediatric and adult QoL questionnaires. These questionnaires were piloted, adjusted and then tested for validity and reliability. The pediatric Osteogeneis Imperfecta Quality of Life (OIQoL) questionnaire OIQoL-P is now available for use; further research is needed to translate and further validate the OIQoL in an international setting.
2.10. Importance of Physiotherapy in the Treatment of Osteogenesis Imperfecta
Dr Claire Hill Physiotherapist, Sheffield Children’s Hospital, United Kingdom
Osteogenesis Imperfecta (OI) is a disease with varying severity affecting the child and their family’s physical, social and emotional well-being (Hill, 2014). It is a hereditary condition affecting approximately 1 in 20,000 births with eleven recognized phenotypes (Glorieux, 2008; Forlino, 2011), and according to the international nosology committee 2019, there are 18 genes identified for osteogenesis imperfecta and will be more in the upcoming years. Children with Osteogenesis Imperfecta (OI) have low bone mass, recurrent fractures, often with minimal trauma, varying degrees of short stature and long bone deformity, scoliosis, kyphosis, pain, some hearing loss, and respiratory failure in the severest types, which can be lethal.
Osteogenesis Imperfecta (OI) cannot be cured currently, so it is managed rather than healed (Rauch, 2014). Non-surgical management of children with OI aims to prevent and treat fractures; enhance motor development, muscle strength, range of movement; reduce contractures and deformity; improve functional ability and ambulation (Monti et al., 2010).
Therapy intervention for children with OI is best placed within a multidisciplinary team, including clinical nurse specialist, occupational therapist, physiotherapist, psychologist, social worker, dietician, and speech and language specialist, but this is not essential. Early treatment involves educating the parents and carers in handling, positioning and caring for their newborn. Prevention of increased deformity and facilitation of normal development is an important management approach from the onset. The motor development and functional ability of children with OI can be delayed, often related to the severity of the disease (Engelbert et al., 2004). Children with severe disease may be encouraged to remain reclined within the first year of life, with gradual progression into supported vertical sitting. This is done to prevent increased crush fractured vertebrae and poor spinal alignment.
Many children with mild and moderate OI achieve independent walking with or without equipment; some may use wheelchairs for longer distances. Some severely affected children may achieve household walking or therapeutic walking (Bleck, 1981). However, fatigue, reduced exercise capacity, and exercise intolerance are frequently reported to limit daily living activities (Van Brussel et al., 2008).
This is a brief description of the physiotherapy intervention at each stage of development, the assessments and outcome measures used, practical treatment techniques and ongoing management. Some of the physiotherapy intervention at each stage of development must be carried on to ensure proper support during their growth and development. While we care for a baby, positioning, handling, dressing, bathing, developmental facilitation, rolling, prone lying, reclined sitting, and gradual sitting are important. For toddlers, moving on the floor, sitting at 90/90, sitting at a table and reaching for objects, sit to stand practice, wheelchair, Panthera micro, walking, malte walker, transfers, and post-op occupation therapy are the important training interventions to be planned. In older children helping them through the transition from nursery and schools, training them to use a wheelchair/walker, providing insoles, teaching exercise for muscle strength, endurance, and pacing activity are required. This age group also requires maintenance of appropriate posture, correction of leg length discrepancy and prevention of fractures.
2.11. Fetal Mesenchymal Stem Cell Therapy for Osteogenesis Imperfecta
Dr Cecilia Götherström, Department of Clinical Science, Intervention & Technology Karolinska Institutet, Sweden
Transplantation of fetal mesenchymal stem cells (MSC) early in life or before birth will ameliorate severe Osteogenesis Imperfecta (OI). MSC are chosen as the therapy as they are stromal precursor cells, bone-forming, has paracrine effects, immune evasive, having low risk, stable genotype, and easy to isolate and expand. Animal and initial case studies show a potential effect in the treatment of OI.
Fetal MSC are derived from the first-trimester liver because in fetal tissues, these cells are more prevalent, have a more primitive genetic signature, and a higher differentiative potential compared to MSC derived from adult tissues. Fetal MSC are easy to expand, not tumorigenic and are more osteogenic than adult MSC. Prenatal transplantation is performed with the aim to prevent the irreversible damage taking place during fetal life and to intervene before more pathology occurs. Furthermore, it is possible to administer a larger cell dose to the immunologically naïve fetus, and the transplanted and engrafted cells can take part in the large stem cell migration during fetal development. Lastly, the effect is possible better due to the delivery route that bypasses the fetal lungs. Postnatally infused cells are filtered away in the lungs, and a large part of the dose is therefore lost.
Mouse studies on MSC transplantation rescued the OI-fetuses from perinatal lethality, resulted in a reduction of circa 70% in the fracture frequency and increased bone strength, thickness, and length. Donor cell engraftment was around 2% after postnatal transplantation and 5% after prenatal transplantation. The donor MSC contributed to the osteoprogenitor population, and a 2% donor cell engraftment resulted in 20% production of human collagen.
Clinical studies by Professor Horwitz et al. in a group of six children with OI transplanting adult HLA-matched MSC showed a reduced fracture frequency of 80% and an increase in lengthwise growth (mean 1.25 vs. 7.5 cm) six months after transplantation compared to six months before transplantation. It also showed an increase in total body bone mineral content (45–77%) and new dense bone formation in five of six patients, showing a 1–2% engraftment during the same period.
The first patient was treated with fetal MSC in utero in 2002. The fetus was diagnosed with OI type III based on the clinical picture with multiple fractures, angulated bones, limbs < 5 SD, and the skull was compressible with the ultrasound transducer due to poor mineralization. In gestation week 32, a dose of 5 × 106/kg male HLA mismatched MSC isolated and expanded from one fetal liver from gestation week ten was administered into the umbilical vein. The procedure was uneventful, and the baby was delivered by elective Caesarean section in the 35th week of gestation after spontaneous rupture of the membranes. The baby girl had APGAR 9/10/10, weighed 1.669 kg and was 40 cm long. There were no complications during the neonatal period. On the OI focused examination, the baby presented with typical signs of severe OI; a soft calvarium and slightly asymmetric rib cage, generalized osteopenia, deformities indicating healed fractures, and an actual fracture of the right femoral diaphysis. The child was initiated with pamidronate from the fourth month of age due to vertebral compression fractures. In the first two years of life, the child experienced three fractures. The genetic analysis revealed that she had a mutation in COL1A2 (c.3008G>A; p.Gly1003Asp; Gly913Asp), which later has been reported to cause severe OI (type II/III). Her growth rate deteriorated from −5.0 to −6.5 SD between 6 and 8 years of life. At eight years of life, a booster dose of the same donor fetal MSC was administered intravenously. The re-transplantation was uneventful with no adverse events. Her lengthwise growth improved to −6.0 SD between 8 and 10 years of age, and she could walk 1000 m and again attend dance classes and school gymnastics. Immunologically she had no lymphocyte proliferation towards the donor MSC, no anti-HLA I or II antibodies, anti-IgG or IgM antibodies, or anti-Fetal Calf Serum antibodies before or after any of the doses. Low engraftment of male cells expressing osteogenic markers was demonstrated in bone (0.1–16.4%) at nine months of age. Three additional children have been treated with a similar set-up, and no adverse events have been documented, and there is a potential clinical effect (reduction of fractures and an increase in growth).
2.12. The Long and Winding Road to Cell Therapy for Children with Osteogenesis Imperfecta
Dr Edwin M. Horwitz, Professor of Pediatrics, Co-Director, Center for Pediatric Cellular Therapies, Member, Immunology and Molecular Pathogenesis Program, Emory University
Bone marrow transplant in children with osteogenesis imperfecta showed improved microarchitecture of the bone with donor-derived osteoblast cells, resulting in increased growth velocity compared to children without transplant. Particularly there is an improvement in linear growth. Animal studies showed an increase in collagen I, which is majorly present in bones after the MSC/BM+MSC transfusion. The mesenchymal cell therapy showed an engraftment percentage of <1% compared to bone marrow transplant, which is approximately 2%. The donor non-adherent BMC is 14% compared to MSC’s, which is only 2%. In Mesenchymal stem cell therapy, the osteoblastic activity lasts only for a week, and after that, there is a gradual decrease in osteoblast and osteocytes. Cytokine-stimulated osteoporosis showed increased osteocytes in metaphysic in GH medium compared to G-CSF, PTH, SCF, or saline. To analyze whether MSC alone is enough for the chondrocyte proliferation, three mice were injected with MSC, MSC with conditioned medium and fresh control medium. Animals were subjected to chondrocyte proliferation assay. The relative chondrocyte proliferation results are the same for MSC alone and MSC in conditioned medium, which is very low in the control medium. The growth plate PCNA expression shows increased chondrocyte cells in MSC than PBS (phosphate-buffered solution). Furthermore, there is much increment in the growth velocity, vertebral length, and body weight. One dose of MSC shows a short-lived effect; hence repeated doses of MSC’s every four months is planned. The results show a tremendous increase in linear growth and bone mineral content after MSC therapy.
2.13. BOOST TO BRITTLE BONES—A Collaborative Project between CMC Vellore and Karolinska University
Dr Vrisha Madhuri, Pediatric Orthopedics Department, Christian Medical College and Hospital, Vellore India
Osteogenesis imperfecta is a genetic disorder for which there is no effective treatment at present. Fetal-derived mesenchymal cells transfusion has been promising in a tiny cohort of patients so far and shown improvement in growth and fracture frequency in children with brittle bone disease. These cells isolated from fetal liver tissues and expanded have shown a greater colony-forming ability, higher proliferative capacity. They have high osteogenic capability with less immunogenic effects (Fetal MSC do not express HLA Class II on the cell surface, and the surface expression requires seven days of treatment with IFNγ (Götherström et al., 2004). Whereas, intracellular levels of HLA Class II can be detected in untreated adult MSC, and just one-day treatment with IFNγ induced the surface expression of HLA Class II)
The BOOST2B study is focused on children under the age of four years. The child is initiated with the treatment at an early age to prevent detrimental effects on respiration, improved healing of fractures at the younger age, reduced risk of additional fractures and bowing of long bones, improved growth pattern, and the rapid growth of the baby/child providing an opportunity for engraftment, expansion, and subsequent migration and distribution of the donor cells to different anatomical compartments. The cells are transfused via an intra-osseous route, allowing the cells to be delivered directly into the bone, avoiding cell entrapment by pulmonary filtration and reported to have fewer complications than infusion via central lines. It can be performed much faster than central or peripheral lines when vascular collapse is present. The intraosseous route avoids loss of MSC in pulmonary filtration. In a pig model of avascular necrosis of the femoral head, intraosseous injection of BM MSC has shown tropism of the engrafted MSC to the bone tissue and no MSC were found in the filter organs or other body tissues (Lebouvier et al., 2015). Murine BM stromal cells injected into the femoral cavity in an OI mouse model resulted in donor cell differentiation into osteoblasts and contributed to the bone formation (Li et al., 2010). Intraosseous BM MSC delivery in a bone defect model in beagle dog resulted in donor-specific chimerism with higher engraftment than that without BM MSC injection. Polymerized Chain Reaction (PCR) and Fluorescence in situ hybridization (FISH) results showed the presence of donor BM MSC in the femoral cavity of the contralateral limb (Liu et al., 2014).
Intra-osseous delivery provided a larger number of donor cells to the bone injury site than intravenous injection of cells. The infused MSC were detected in epiphysis or diaphysis of the distal femurs or proximal tibiae. Since long bone fractures are a significant problem in OI and require much treatment, it would be appropriate to inject MSC via the intraosseous route in the weight-bearing long bones. This strategy will ensure a high amount of cell engraftment in the more susceptible areas.
The study’s investigational product is the BOOST cells (allogeneic expanded human first-trimester liver-derived MSC), which are a cell suspension. The difference of BOOST2B from BOOSTB4 is mainly the age at the initiation of treatment between the child’s first to fourth years in BOOST2B, whereas it is during the prenatal and infancy period in BOOSTB4. Outcome assessment of the quality of life of children undergoing the study is using PedsQL in BOOST2B rather than ITQL.
The study’s primary objective is to assess the child’s safety and tolerability after intravenous and intraosseous infusion of four doses of allogeneic expanded human fetal liver-derived MSC every four months in individuals with OI type III or severe type IV. The expected treatment period is up to 16 months of follow-up after the first infusion. Secondary objectives are to assess the effect of intravenous and intraosseous infusions of four doses of allogeneic expanded human fetal liver-derived MSC every four months in individuals with OI type III or severe type IV on fracture frequency, time (days) to first fracture after MSC infusion, bone mineral density (BMD), growth, clinical status of OI, and biochemical bone turnover.
2.14. Role of DEXA in Osteogenesis Imperfecta Assessment
Dr Thomas V. Paul, Department of Endocrinology, CMC Vellore
Dexa scan is used for determining the bone mineral density (BMD) and bone mineral content (BMC) in children with osteogenesis imperfecta as T scores and Z scores are not applicable for very young children. T scores are used for adults predominantly. However, bone mineral density is the preferred term for pediatric DXA reports when BMC or areal BMD Z-score are less than or equal to −2.0 SD. Dexa scan is majorly used for Comprehensive skeletal health assessment in patients with increased fracture risk, treatment follows up and during the initial diagnosis. The main limitation being DXA should not be performed if the child’s appropriate positioning cannot be assured; the hip site cannot be used for growing children. In children with short stature, spine and TBLH BMC and areal BMD results should be adjusted. Adjustment is using either BMAD (bone mineral apparent density) or the height Z-score. Dexa is the appropriate method for clinical densitometry in infants and young children. Whole-body measurements are feasible and can provide reproducible measures of BMC and aBMD (areal BMD) for children > 3 y. Whole-body BMC measurements for children < 3 years there is limited clinical utility due to poor feasibility and lack of normative data. The preferred skeletal sites are the lumbar spine and total body less height (for children less than five years). Forearm and femur measurement are technically feasible, but normative data is insufficient for those sites. The minimal interval for taking a DEXA scan for follow up is 6–12 months. The four major components of the DEXA scan are BMD/BMC, VFA (vertebral fracture assessment), and Muscle mass. Trabecular bone score.
The accuracy and precision of the DEXA scan results depend on the Different DXA manufacturers (Lunar, or Hologic), Different reference population, Technical expertise (e.g., positioning), Patient-related factors (e.g., implants). Precision and precision error are calculated as the root mean square standard deviation in absolute terms (g/cm2). The least significant change, or LSC, is the least amount of BMD change that can be considered significant. The ISCD recommends calculating this for a 95% confidence level, which is done by multiplying the precision error by 2.77. The vertebral fracture assessment has very little radiation, and good reliability with morphogenic data is used for the extent and to determine the number of vertebral fractures. Studies show a decrease in fracture and surgical rate post bisphosphonate treatment, as the BMD measurements go higher in follow up scans. The other two components are muscle mass and trabecular bone score (TBS), which help assess OI children in treatment follow-up. TBS acts as a tool to differentiate antiresorptive impact on cortical and trabecular bone in children with osteogenesis imperfecta.
3. Day 2 Lectures: Regulatory and Ethical Aspects of Cell Therapy Clinical Trials
3.1. The Need for Dialogue—Between Regulators, Investigators, and Industry
Prof. Alok Srivastava, Head, Center for Stem Cell Research, CMC Vellore
With the increasing number of clinical trials, there should be a way to channelize and harmonize regulatory concerns in stem cell and gene therapy. Taking the example of establishing gene therapy for Hemophilia in India, initially, there were many concerns expressed regarding defining and setting standards for safety and efficacy. A Gene Therapy Working Group of the Factor VIII/IX and RBD (Rare bleeding disorders) Subcommittee was formed to address such issues. In India, a clinical trial proposal submitted by the academic investigator is reviewed by the Institutional Review Board, the Sponsor, Indian Council for Medical Research /Department of Health Research, and Drug Controller General of India-Cellular biology based therapeutic drug evaluation committee (CBBTDEC), while corporate sponsors can directly submit the proposals to DCGI for further processing. Based on the type of cell therapy, further recommendations are provided by the respective committees, and in some cases, National Apex Committee for Stem Cell Research and Therapy (NAC-SCRT) clearances are required for more than minimal manipulation of stem cells. There are already preexisting and emerging pathways of relevance to Advanced Therapy Medical Products (ATMPs) covering regulatory, reimbursement, access and new stakeholder dialogue platforms in EU, US, and the UK as examples of national jurisdiction. In India, there are many challenges involved in ATMP regulation (Figure 1). The regulators, along with the investigators and industry, are attempting to make this a seamless process.
3.2. Current Challenges in the Regulation of Production and Distribution of Innovative Cell Therapy Products in Europe
Dr. Gudmund Hedenskog, QP, Vecura, Karolinska Institutet, Sweden
In the EU, Eudralex is the body of European Union legislation where the rules governing medicinal products in the European Union are framed (Figure 2). An Active Pharmaceutical Substance (API) in a Phase I study is not required to be produced under Good Manufacturing Practice (GMP); however, in phase II, this is mandatory. Similarly, the validation of a method for analysis is not required (expect for sterility/endotoxin) for a phase I study while for phase II it is required. The European Medicines Agency (EMA) is a centralized agency of the European Union (EU) responsible for the scientific evaluation, supervision and safety monitoring of medicines in the EU.
When conducting a trial, the role of Quality Person is important as the product shall be released only after being authorized by the QP. It is important to perform risk assessment and risk control prior to the start.
3.3. Cell Therapy—Regulatory Changes in India’s Landscape: Emerging Concepts
Dr Velu Nair, Lt Gen, Hemato-Oncology & Bone Marrow Transplant, Comprehensive Blood & Cancer Center (CBCC), India
Cell therapy is defined as the administration of living whole cells for the patient for the treatment of a disease. The origin of the cells can be from the same individual (autologous source) or from another individual (allogeneic source). Cells can be derived from stem cells, such as bone marrow or induced pluripotent stem cells (iPSCs), reprogrammed from skin fibroblasts or adipocytes. Stem cells (HSCs) are applied in the context of bone marrow transplantation directly. In order to prevent the misuse of stem cells, regulators have framed guidelines. To monitor cell therapy, ICMR, NAC-SCRT, along with Central Drugs Standard Control Organisation (CDSCO) have formed screening committees (Figure 3).
For an investigational new drug discovered in India, pilot studies are mandatory; for drugs that are approved outside India, phase III studies need to be carried out to generate evidence of safety and efficacy in Indian population. Commercialization will be permitted only if results are found satisfactory. The draft guidelines for Stem Cell Research and Regulation was framed in 2002 and established in 2007; NAC-SCRT 2013 was revised in 2017. In India, research related to human germline gene therapy and reproductive cloning is restricted.
A clinical trial using human stem cells should be conducted in accordance with Schedule Y of Drugs and Cosmetic Act and Good Clinical Practice Guidelines of CDSCO and ICMR. The trial shall be registered in the CTRI and approved by the IC-SCR and institutional review board. The cell or cell-based products used in the trial must be processed in CDSCO certified Good Laboratory Practice and GMP facility. A minimum of two years of post-trial follow up is necessary to assess the safety of the drug. All participants should be consented and explained the procedures and the possible adverse events that are likely to occur. The trial data must be maintained for a period of at least 15 years.
3.4. Regulation of Cell-Based Therapies: Commercial Perspective (India)
Dr. Pawan Gupta, Stempeutics, Bangalore
According to the American Society for Blood and Marrow Transplantation guidelines, the approved indications for hematopoietic transplantation have been framed for adult and pediatric malignant, non-malignant, and solid tumors. The ICMR-DBT guidelines to conduct stem cell research in India have evolved since 2007. In 2017, the need for stem cell use as Clinical Trial (CT), levels of manipulations, CMC, release criteria, manufacturing facility—GMP and GLP certified, ICSCR registration mandatory was defined. To carry out a trial, CDSCO shall be issuing four categories of licenses/approval right from obtaining facility licenses (form 29), the conduct of CT, manufacture (form 46/import 45) and lastly, product for sale (form 28/form 10) (Figure 4). According to the guidelines, minimally manipulated cells can be used for stem cell therapy without any approval. Furthermore, it is not clear if a single IC-SCR or Multiple IC-SCR approval is required for undertaking multi-centric, DCGI approved clinical trials. The product has to undergo rigorous preclinical testing before commercialization and that incurs an additional cost. Some issues need to be addressed for rapid commercialization of Regenerative Medicine products and early patient access for major unmet medical need.
3.5. Regulation of ATMPs in Europe
Vera Franzen, Regulatory Affairs Consultant at Vera Franzén Consulting AB
The European legislation involves the European Commission, the European Parliament, and the European Council. According to Regulation (EC) No 726/2004, a centralized procedure is mandatory for ATMPs. Marketing authorization is granted in all EU countries at the same time. The EMA is responsible for coordinating the existing scientific resources put at its disposal by the Member States for evaluating, supervision, and pharmacovigilance of medicinal products. The Directive 2001/83/EC as amended is the community code used to medicinal products for human use. Regulation (EC) No 1394/2007 on ATMPs was published on 13 November 2007, and started to apply on 30 December 2008. The three categories of ATMPs are (i) gene therapy medicinal product, (ii) a somatic cell therapy medicinal product, (iii) a tissue engineered product. The Common Technical Document triangle is given as Figure 5.
3.6. Manufacture and Quality Assurance of Stem Cell Products
Dr. Lilian Walther-Jallow, Department of Clinical Science, Intervention & Technology Karolinska Institutet, Sweden
Good Manufacturing Practice (GMP) ensures that products are consistently produced and controlled according to quality standards designed to minimize the risks involved in any pharmaceutical production that cannot be eliminated by testing the final product. It covers all aspects of production from the starting materials, premises, and equipment to staff training and personal hygiene. Detailed, written procedures are essential for each process that could affect the quality of the finished product (Figure 6). There must be systems to provide documented proof that correct procedures are consistently followed at each step in the manufacturing process—every time a product is made each batch of the produced drug must be approved by a Qualified Person (QP). Inside a GMP laboratory there should not be any quick movements or excessive talking. It is recommended to change gloves frequently and also not to touch any exposed sterile surface. The airflow in the room has to be monitored and frequent cleaning of the room is mandatory. The personnel working inside the GMP facility should have undergone a basic entry/exit course/validation to ensure the following of aseptic techniques. Any product used inside GMP should be triple packed, and where applicable, validated before entering the clean room. ATMPs are medicine for a human use based on genes, tissue or cells. They can be classified as gene therapy medicine, somatic cell therapy medicines, or tissue-engineered medicines. In the case of manufacture of an ATMP, the quality testing at several stages of the production process has to be validated. This includes obtaining informed consent from the patients and ensuring that the sample has gone through the initial criteria for processing. The quality control during manufacturing of drug product is process monitoring test (routine culture check), in-process controls (criteria for termination of culture) and release criteria of the manufactured product. Routine testing of cells could for example, be based on appearance, sterility, mycoplasma, viability, recovery, population doubling, cell immunophenotyping, a functional assay displaying some of the desired properties of the product, chromosomal abnormality, and where appropriate screening for infectious agents. A clean product should be free of any virus, bacteria, mycoplasma, and endotoxins; express the correct surface markers, have the right morphology and be viable. Every step in the process has to be documented right from the manufacturing process until the analysis. The working protocols for production, analysis, and submethods, such as antibody titration and preparation of DNA, have to be documented along with stability program and reports. Validation of the manufacturing process with a validated protocol has to be approved by the QP. This needs to be done on approximately three approved batches in a row. If using an in-house analysis method, such as cell count, differentiation, flow cytometry, this has to be validated to ensure intermediate precision, accuracy, and, linearity. The variation between operators and between instruments also needs to be investigated. Long term stability during cryopreservation needs to be tested at 1–2 weeks, 3, 6, 9, and 12 months and then bi-annually. Furthermore, the stability of prepared product after washing and formulation has to be checked at, e.g., 0 h, 1, 2, and 4 h after thawing. This is to ensure that viable cells are being injected at the time of transplantation. Some of the other characterization required for a cell product would be to study tumorigenicity in mice, check for pluripotent markers, telomerase activity, soft agar assay, and Hayflick limit testing.
3.7. Reconstitution of Cell Therapy Products
Annika Goos, Department of Clinical Science, Intervention & Technology, Karolinska Institutet, Sweden
For European standard, GMP production of Advanced Therapy Medical Products, ATMP, is regulated in Eudralex—volume 4—GMP guidelines part IV—GMP requirements for Advanced Therapy Medical Products, ATMP (May 2018). Chapter 16 talks about reconstitution of product after batch release.
Reconstitution of an ATMP can be performed at the administration site (e.g., hospital pharmacies) outside a GMP environment. The term “reconstitution” covers activities required after batch release and prior to administering the ATMP to the patient, which cannot be considered a manufacturing step. All substantial manipulations (e.g., cultivation) must be performed in a GMP environment. Examples of reconstitution activities are:
Thawing, washing, buffer exchange, centrifugations steps necessary to remove preservation solutions (e.g., Dimethylsulphoxide), and removal of process related impurities (residual amount of preservation solution, and dead cells) including filtering.
(Re)suspension, dissolution or dilution with solvent/buffer, dispersion; Splitting the product and use in separate doses, adaptation of dose (e.g., cell count); Loading into delivery systems/surgical devices, transfer to an infusion bag/syringe; The compliance of the administration site with the defined reconstitution process falls outside of the responsibility of the manufacturer and is also outside of the scope of GMP.
The above steps can only be considered a part of the reconstitution process if it is appropriately justified that it cannot be performed as part of the manufacturing process before batch release without negative impact on the product.
The manufacturer is obliged to provide the administration site with a detailed instruction for the reconstitution process including equipment to be used and requirements at the administration site. For authorized ATMP the reconstitution process needs to be validated from batch release to the administration to the patient. The validation must demonstrate the reconstitution process is sufficiently robust and consistent without negative impact on the quality/safety/efficacy of the ATMP.
3.8. Putting the Cell into Cell Therapy
Dr Edwin M Horowitz, Emory University, USA
In the classical framework of clinical research, the idea has to undergo preclinical research. A phase I is then performed to look for any adverse events by performing dose escalation studies and mainly from the view-point of safety, while a phase II study focuses on the benefit with a set cell dose. A phase III trial is conducted as a randomized trial to test the efficacy against a standard of care of placebo and a phase IV trial mainly focuses on the long-term monitoring usually a post-marketing study. The major points to consider are the type of cell used and whether the cells are minimally manipulated or not. In the USA, the FDA recommends a specific application for the conduct of a clinical trial (Figure 7). From standard operating protocol, validations, release criteria to stability are required.
3.9. Regulatory Requirements to Conduct a New Cell-Scaffold Product Clinical Trial in India: Challenges and Implementation.
Dr Annie John, Sree Chitra Tirunal Institute of Medical Sciences, Kerala
A tissue engineering construct must be developed according to GLP guidelines, qualify ISO and National Accreditation Board for Testing and Calibration Laboratories (NABL) standards. The material has to be biocompatible after undergoing toxicological tests in animals. The process of medical device evaluation has to pass through material qualification, design verification, preclinical studies, and product release studies. The figure below shows the necessary validation that needs to be performed during preclinical studies (Figure 8).
3.10. Fetal Mesenchymal Stem Cell Therapy for Osteogenesis Imperfecta
Dr Cecilia Götherström, Department of Clinical Science, Intervention & Technology Karolinska Institutet, Sweden
The Boost Brittle Bones Before Birth (BOOSTB4) study is based on the hypothesis that transplantation of fetal mesenchymal stem cells (MSC) early in life or before birth will ameliorate severe osteogenesis imperfecta. Several in vitro studies have been carried out, which show that fetal MSC is better for bone regeneration than adult MSC. Animal studies give proof of principle that transplanted MSC contribute to the osteoprogenitor population with the reduced manifestation of the disease phenotype. A clinical study by Prof. Horwitz show that adult bone marrow-derived MSC reduce the fracture rate and improve patients’ growth for up to six months. Based on this existing data, the first patient was treated in utero, which showed no adverse events and improved growth. Three additional children have been treated with potential clinical effect. Now a Phase I/II study, the multicentric trial (BOOSTB4) is being carried out in Europe along with a sister study in Vellore, India (Boost to Brittle Bones, BOOST2B) to evaluate the safety and efficacy of prenatal or postnatal intravenous administration of multiple doses of fetal MSC.
3.11. Conduct of Cell/Cell Scaffold Clinical Trials: Roadblocks and Challenges
Dr. Vrisha Madhuri, Prof. Pediatric Orthopedics, Adjunct Scientist, CSCR, CMC Vellore
Any new investigational drug must undergo rigorous in vitro characterization and preclinical testing before moving on to human subjects. Examples of how the scenario has changed over the last ten years in terms of Indian regulatory bodies are presented here.
Autologous transplantation of chondrocytes for human articular cartilage defects has been carried out in Singapore. We adapted the culture techniques for articular cartilage to physeal chondrocytes harvested from iliac crest and designed our own technique for the transplantation of cells. After extensive in vitro cell characterization, we moved on to preclinical testing (2008–2010) in a goat model which has a similar size and growth as a young child. With enough preliminary data showing the safety and efficacy of the technique, we tested the same in five children as a pilot study. Currently point, there were no regulatory restrictions pertaining to cell therapy other than hematopoietic stem cells. After the trial was approved by the Institutional Review Board in 2010, the study was carried out in 2012 with institutional Data Safety Monitoring Board, no clearance from DCGI was required. The trial was applied for in 2010 early but retrospectively registered by the CTRI in 2012. The type of cells used was from an autologous source which was minimally manipulated according to the ICMR guidelines. We made our own GMP protocol and prepared our own release criteria for the transplant of cells. As of 2017, we had a follow-up data of five years from these five children.
Based on the findings, we went ahead to conduct a phase I clinical trial in 15 children. We obtained a sponsor in the department of health research (DHR) after IRB clearance in 2017. At this juncture, a lot of changes have taken place in the regulatory body. The CDSCO has come up with new regulations where all cell therapy trials have to obtain a No-objection certificateOC from the regulatory body. The time taken from the application submission to obtaining NOC is challenging. After a review process of the technical committee, a joint inspection for manufacturing facility was conducted by the south zone and the north zone CDSCO, after which, the NOC for the conduct of the clinical trial was granted. However, there was a requirement for obtaining manufacturing license clearance by the South Zone. This along with manufacture to license the product is necessary to initiate the trial.
The major issues with this are that after the sponsor clearance the CDCSO has taken now nearly two years for granting the license. Will the rules of the funding agency allow the extension of the trial by two years and if we do a clinical trial, we need to compress the trial into a shorter time frame adding a lot of pressure on recruitment. Navigating the regulatory landscape especially in the early days of changes, is a challenge and we are still struggling to get through. A lot of pressure is thus on the investigator as the main motive would be to finish the trial in the limited time frame instead of performing an interim analysis of the study data and then proceed to completion.
Another example of navigating the changing regulatory landscape is the use of cell-scaffold products for bone tissue engineering. After generating preclinical data in a large animal, we applied for funding in 2009. This was granted in 2010, but the application was kept on hold till 2013 by the human studies committee of ICMR. Since that was when regulatory authorities were framing the rules, our trial was further kept on hold until 2014. After one year we obtained NOC from CDSCO while human studies of ICMR committee took more than two years. The patient transplants were carried out in one year and data was submitted to DSMB appointed by the DBT for the interim analysis. After which, we recruited the remaining five patients. The follow-up duration for these patients was all beyond the duration of the actual trial period. A further DSMB was held one year after the last recruitment. This highlights the need for a proper channel of regulatory bodies who keep periodic checks on all the academic trials.
More recently, we applied for another clinical trial for fetal liver derived MSC transplantation in children. This trial is the first of its kind in India. We obtained IRB, IC-SCRT clearance and registered the trial in CTRI after securing funds from the sponsor. A Health Ministry’s Screening Committee (HMSC) clearance was required for the import of cells and a clinical trial clearance from the CDCSO. This has another type of challenge as we are not the manufacturer of the cell product. The cells are being prepared according to GMP guidelines in Sweden while we are importing the cryopreserved cells for transplantation upon reconstitution. We obtained NOC for importing cells for research purpose from CDSCO. However, our clinical trial is still awaiting clearance as the CBBTDEC meeting has not taken place for eight months due to restructuring.
In summary, we have learned from the past that we need to apply for funding and prepare documents as per Schedule Y for CDSCO; prepare the release criteria, register in CTRI and identify patients in parallel. Although this process may sound simple, each step is time bound. We also learned that the academic trials do not have the resources to manage the GMP, which may need to be delegated. There should be a dedicated team of regulatory bodies who work for stem cell-related clinical trial in an academic setting.
3.12. Clinical Concerns (GCP) and Regulatory Strategies for Implementation: European Standards
Lilian Walther- Jallow, Department of Clinical Science, Intervention & Technology Karolinska Institutet, Sweden
The concept of GCP was developed by the international conference on Harmonization (ICH) based on the Declaration of Helsinki. GCP is defined as an international ethical and quality’ standard for the design, conduct, performance, monitoring, auditing, recording, analyses and reporting of clinical trials that provides assurance that the data and reported results are credible and accurate, and that the rights, safety, and well-being of study participants are protected. This has become a legal obligation in Europe since 2004. The EU clinical research framework follows several professional codes that are being imposed on human research, clinical trials, data protection, and bio-banking (Figure 9). At the outset, the ICH GCP has 13 core principle components which ensure that the clinical trial is conducted in a safe and sound manner.
A Good Clinical Practice is primarily dependent on the sponsor who directly links the regulatory bodies and the clinical investigator; this, together with institutional ethics committee approval, works in parallel to initiate the clinical trial on the subjects. In the context of regulatory affairs, the investigator is the chief person responsible for the conduct of the clinical trial, while a sponsor takes up the responsibility of managing or providing financial support to the clinical trial. A study subject is a person who is recruited after informed consent/assent to take part in the clinical trial after obtaining the necessary clearance from the institutional review board. The important documents that are to be prepared before the start of the study are the Case Report Form and site master file. This ensures that the data collection is performed in a similar fashion across the subjects by dedicated personnel. According to the GCP standards, the process of monitoring or overseeing the progress of the clinical trial is important. This is mainly to ensure that the well-being of the human subjects is protected. A monitor of a clinical trial should not be involved in the execution of the trial. This meticulous auditing of related activities will help during the inspection by the regulatory authorities for a review of the study or facility. If any serious adverse events result in death or are life-threatening, the trial should be suspended immediately with notice to all relevant bodies explaining the reason. Close monitoring of all the participants should be performed in that case. GCP guidelines underscore the importance of having a quality assurance and control which consists of detailed protocol or SOP for all procedures to avoid variation in data collection. This is usually performed by having an electronic Case Report Form which is prepared for each participant. This trial-related record can anytime be verified by the Monitor, Audits, and the Inspectors. The most common audit findings include improper informed consent, lack of data safety monitoring board, deviations from the original protocol, the missing source of original documentation. To ensure that the clinical trial is conducted according to GCP standards, there are agreements between the various coordinating parties.
Author Contributions
V.M., S.R., R.R., A.S., and L.L. compiled the report based on the lectures. All authors have read and agreed to the published version of the manuscript.
Funding
This meeting was funded by the Indo-Swedish project, Department of Biotechnology, Government of India BT/IN/Sweden/09/VM/2017-18.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Figure 1.
Clinical trial with stem cell in India.
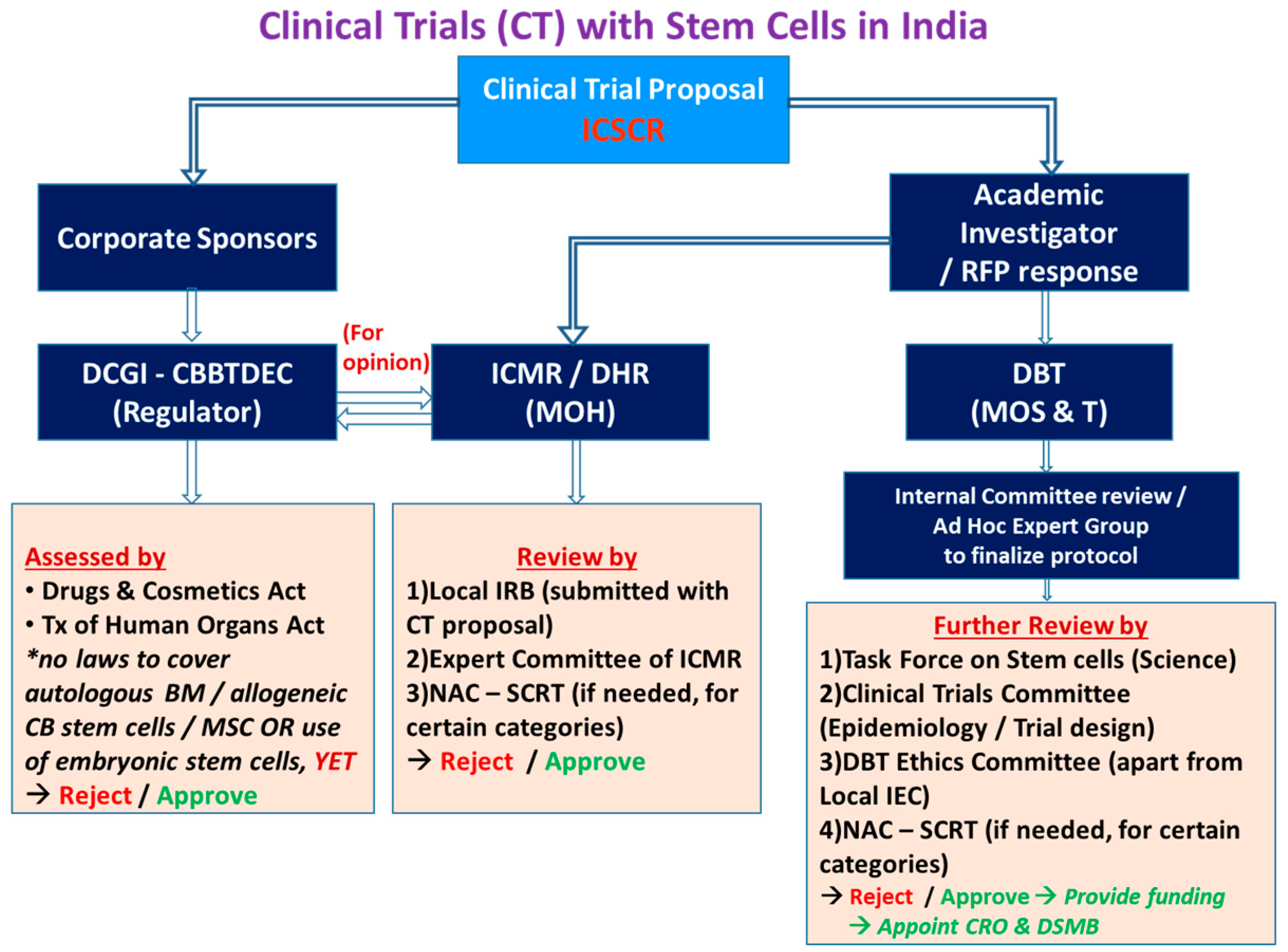
Figure 2.
European Union Legislation.
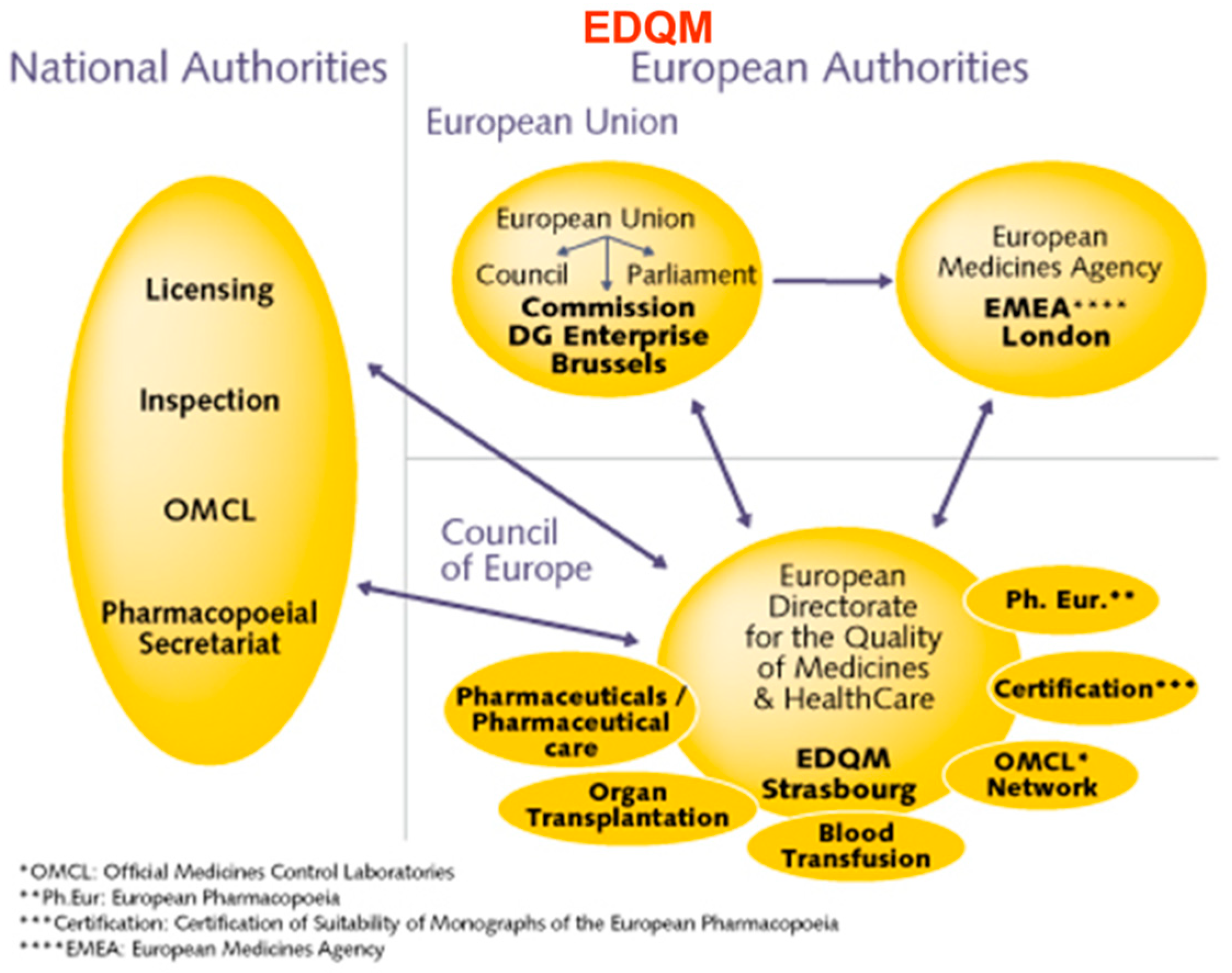
Figure 3.
Process of regulatory clearance in India.
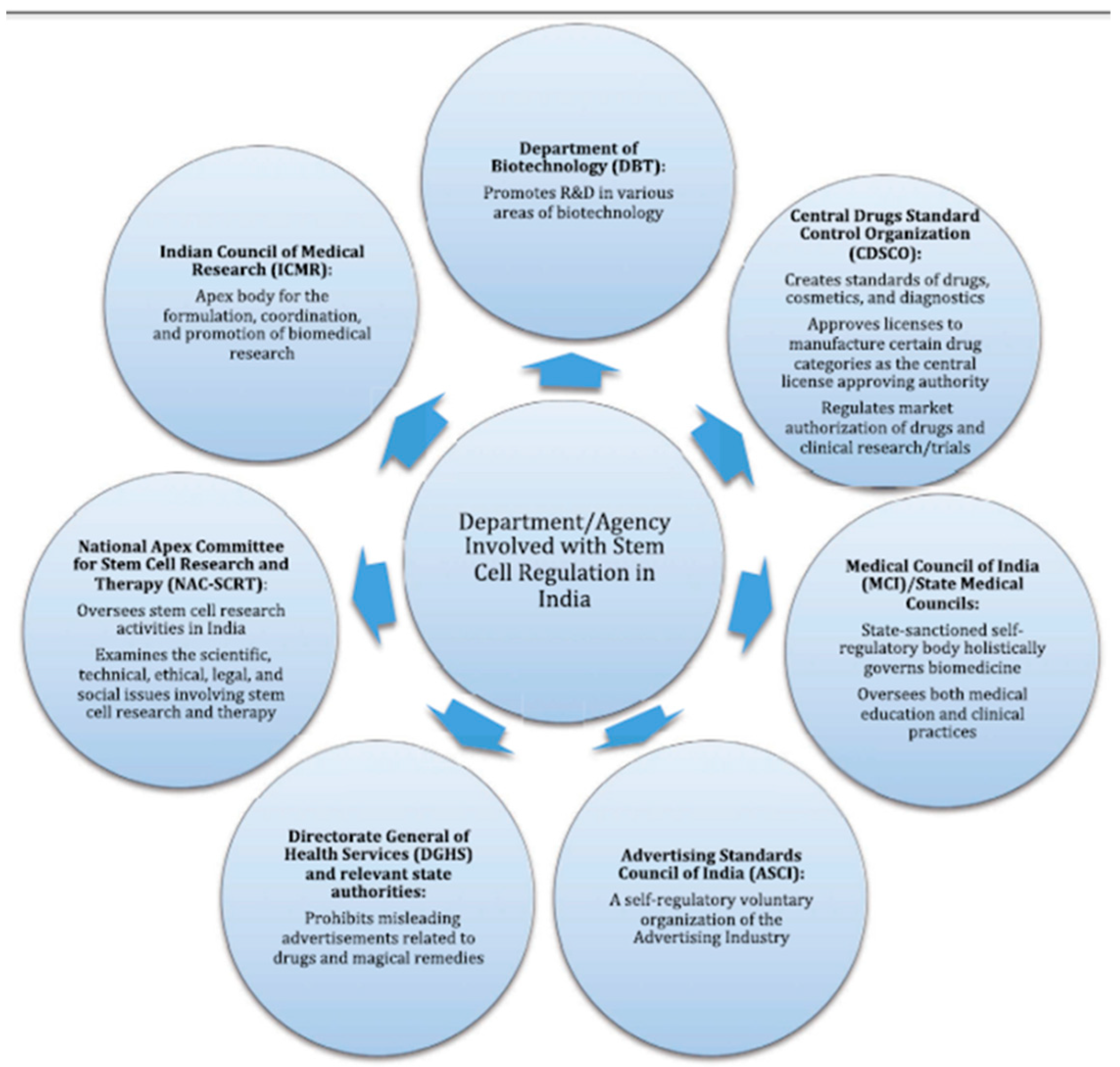
Figure 4.
Process workflow clinical trial licenses and approvals.
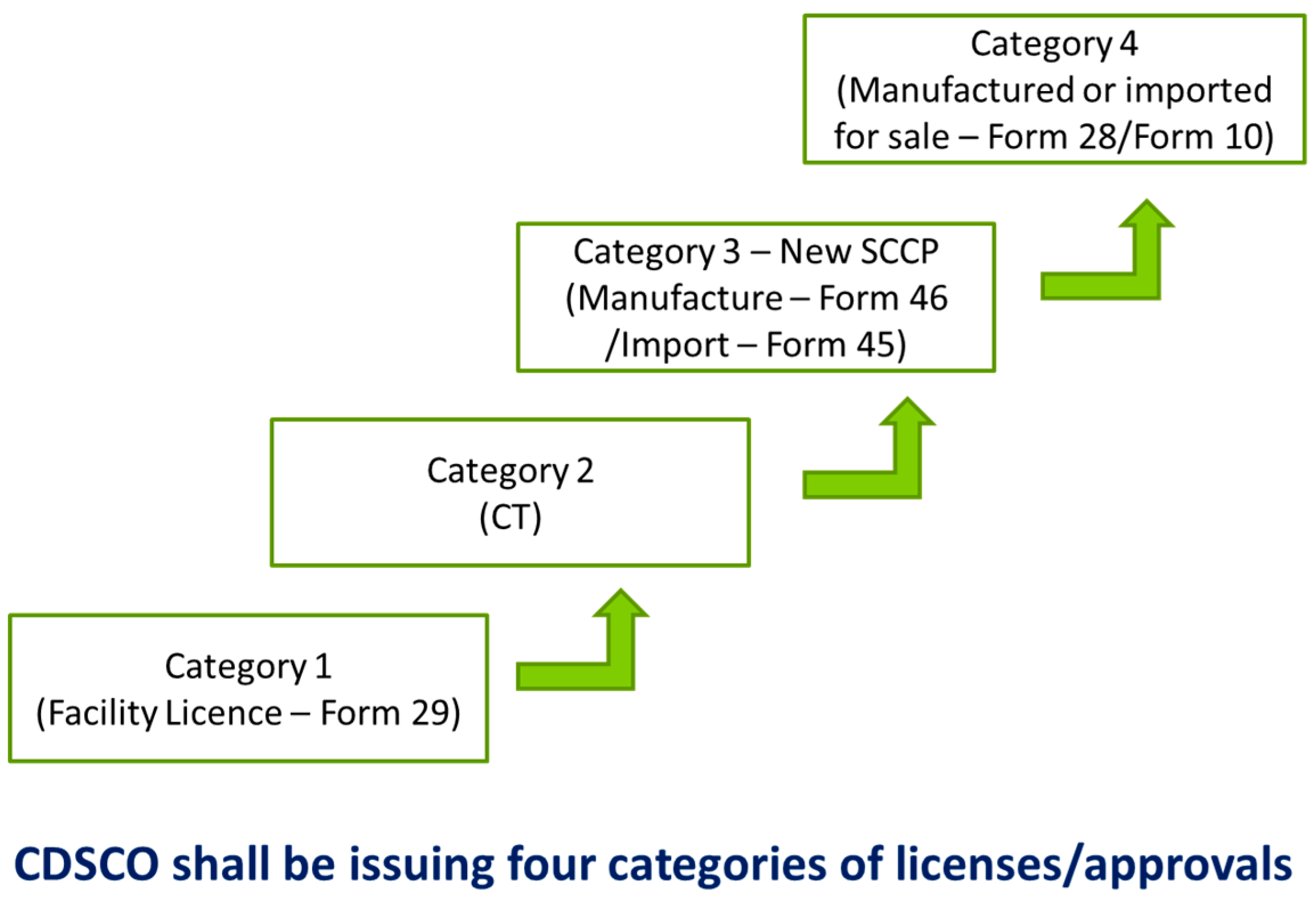
Figure 5.
European regulation—Advanced Therapy Medical Products (ATMP).

Figure 6.
Validation of drug product.
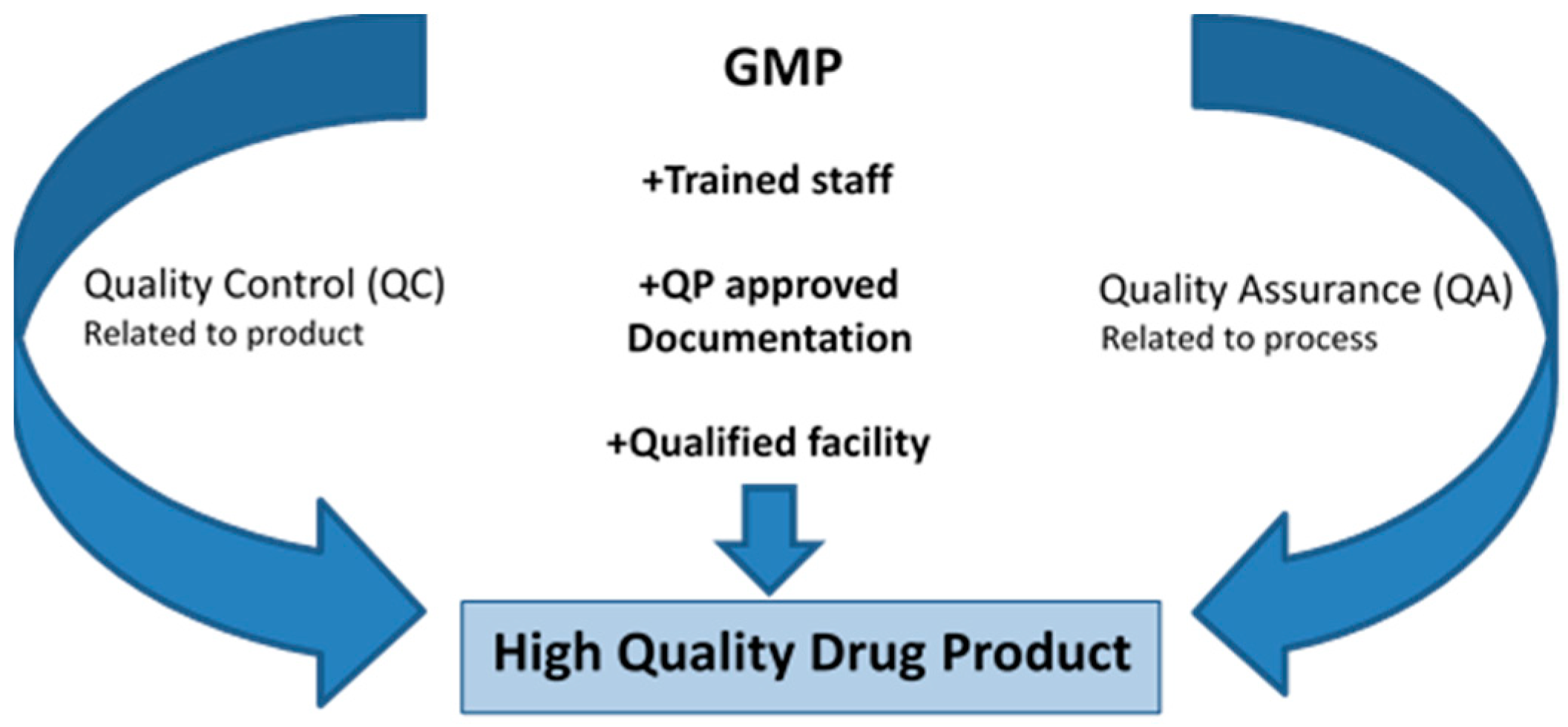
Figure 7.
From Concept to the clinics: Workflow.
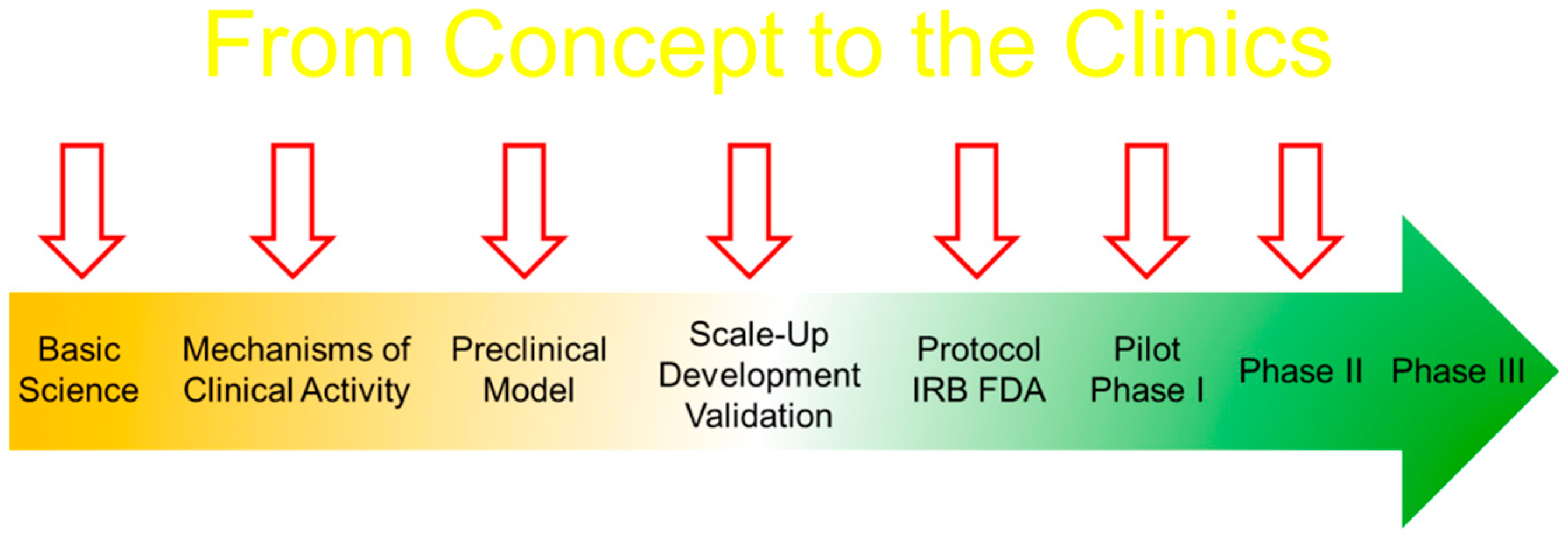
Figure 8.
Preclinical evaluation of drugs and devices.
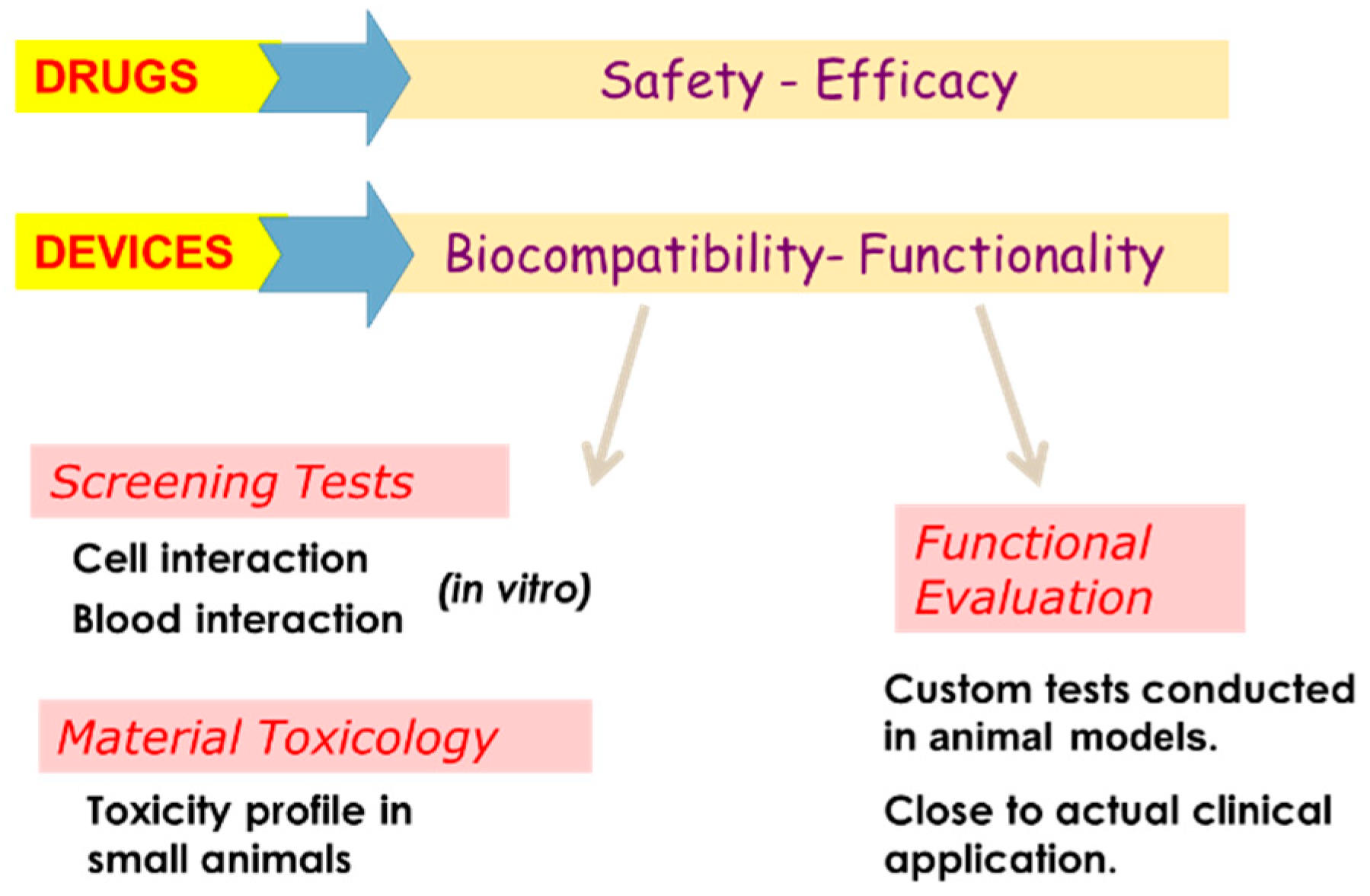
Figure 9.
European Union clinical research framework.
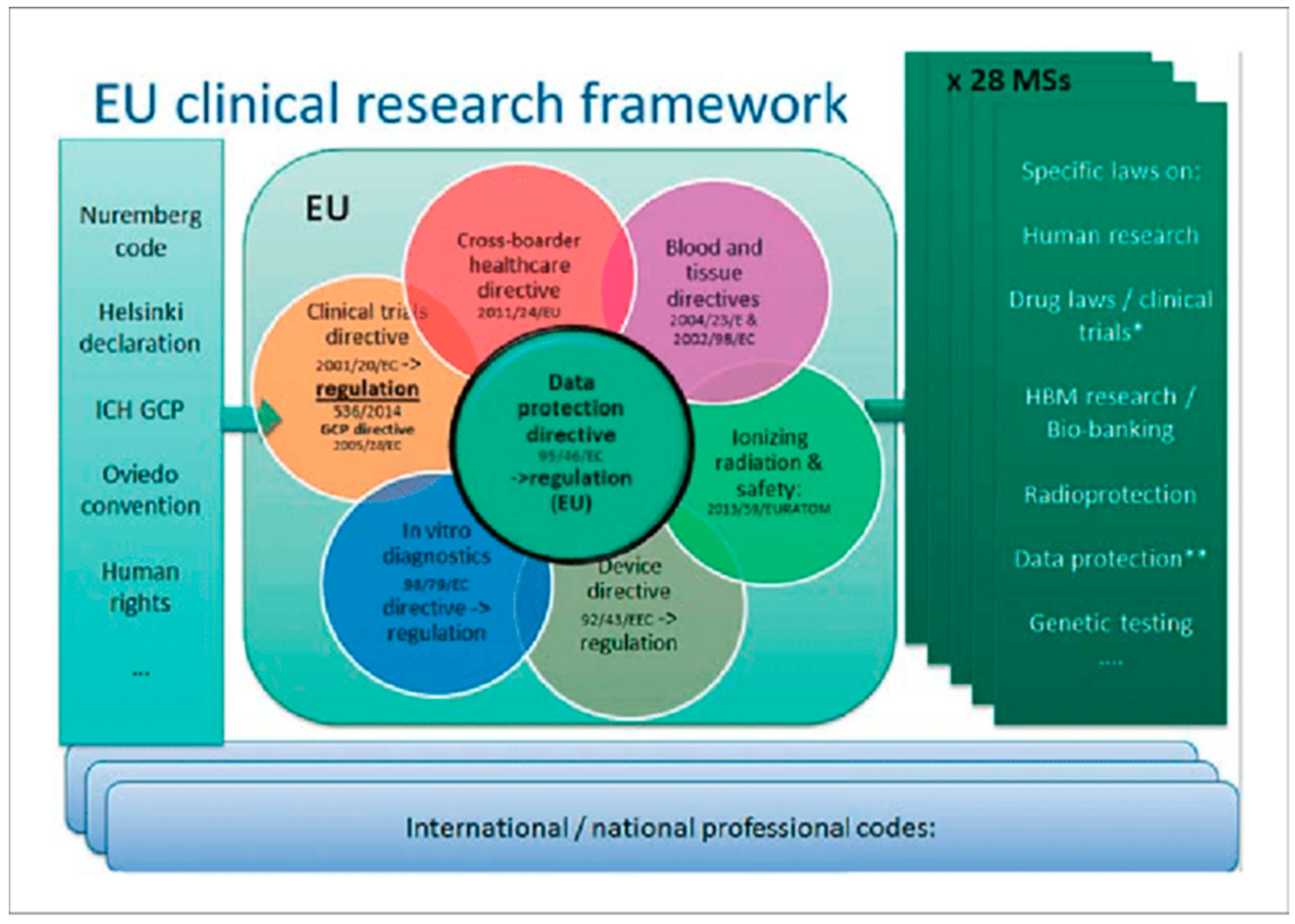

{kind=link}
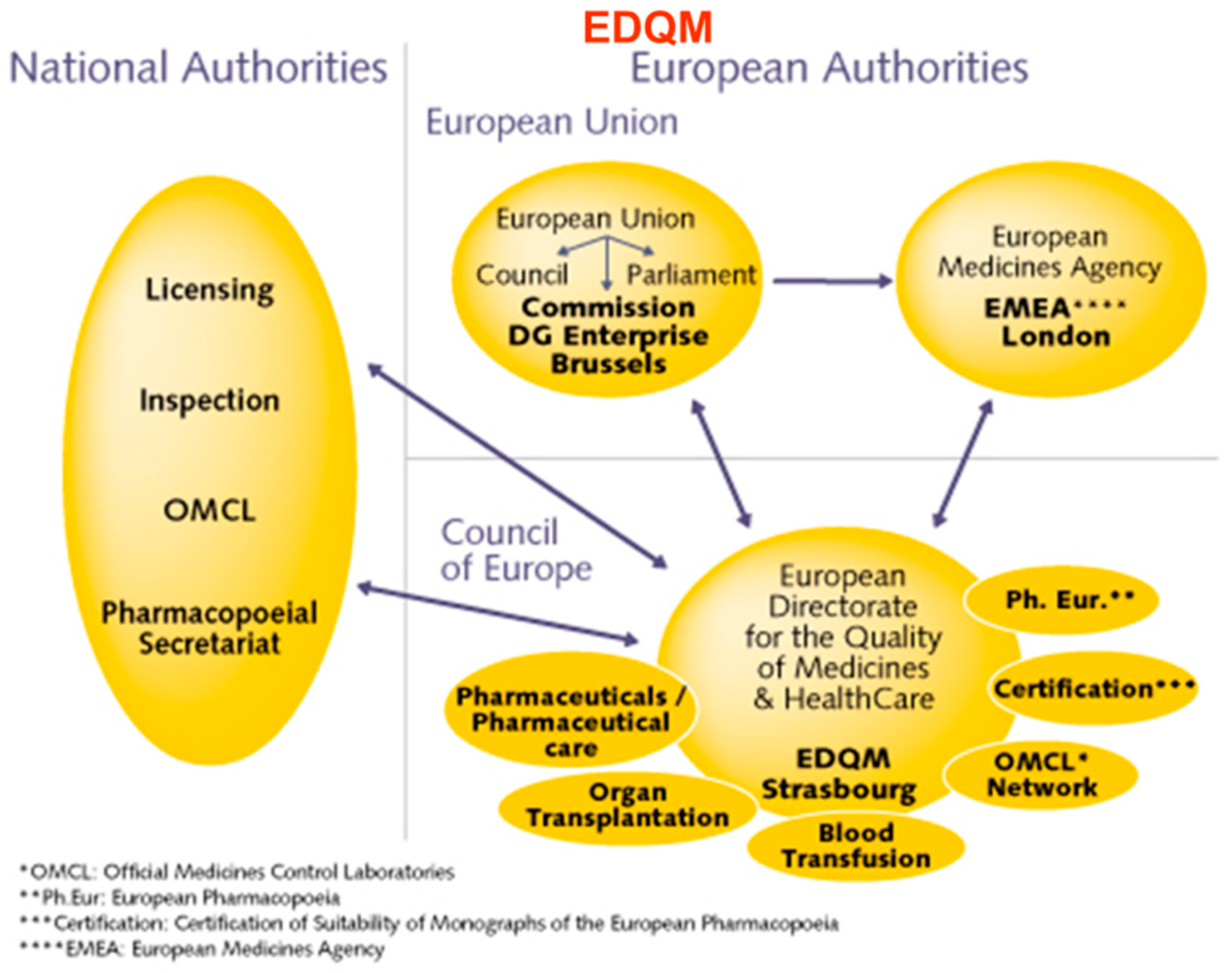{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The Osteogenesis Imperfecta (OI) Nomenclature 2015—International Nomenclature Committee for Constitutional Disorders of the Skeleton (Updated January 2019).
Table 1.
The Osteogenesis Imperfecta (OI) Nomenclature 2015—International Nomenclature Committee for Constitutional Disorders of the Skeleton (Updated January 2019).
| Syndrome Name | Equivalent Numerical Type | Subtypes |
|---|---|---|
| Classic Non-deforming OI (with Blue Sclerae) | 1 | 2 |
| Common variable OI (with normal sclerae) | 4 | 10 |
| OI with calcification in interosseous membranes | 5 | 1 |
| Progressively Deforming OI with normal sclerae | 3 | 18 |
| Perinatally Lethal OI | 2 | 5 |
Table 2.
Patterns of Inheritance in Osteogenesis Imperfecta.
| Modes of Inheritance | Number |
|---|---|
| Autosomal Dominant | 4(+3) |
| Autosomal Recessive | 16 |
| X-linked | 2 |
| Mosaicism (Somatic plus germinal cell) | - |
| Chromosomal microdeletion- Includes 7q, 17q, Tetrasomy 9p | A number reported |
| Polygenic | Unknown but likely |
Table 3.
Genomic variants (mutations) resulting in various types of OI.
| Type of Mutation | Genomic Sequence Variants |
|---|---|
| Nonsense | Premature stop codons |
| Missense | Single nucleotide substitutions |
| Splicing mutations | In-frame and frameshift deletions |
| Insertions/deletions | Frameshift or in-frame sequence variation |
Table 4.
Genes in which mutations result in at least one type of Osteogenesis Imperfecta 2019.
| Gene | Protein | Function |
|---|---|---|
| Genes encoding Type 1 collagens and post translational modifications | ||
| COL1A1 | Collagen 1 alpha 1 chain | Structural polypeptide |
| COL1A2 | Collagen 1 alpha 2 chain | Structural polypeptide |
| CRTAP | Cartilage associated protein | Prolyl 3—hydrolation |
| LEPRE1 | Leucine proline-enriched proteoglycan (leprecan) 1 | Prolyl 3’- hydrolation |
| PPIB | Peptidylprolyl isomerase B | Prolyl 3’- hydrolation |
| P4HB | Prolyl 4-hydrolase, beta-subunit Procollagen lysyl hydrolase 2 | Prolyl 4’- hydrolation Lysylhydrolation |
| PLOD2 | ||
| SERPINH1 | Serpin peptidase inhibitor, clade H, member 1 | Collagen specific ER chaperone (Colligin/HSP) |
| SERPINF1 | Serpin peptidase inhibitor, clade F, member 1 | PEDF involved in osteoblast/osteoclast homeostasis |
| FKBP10 | FK506 binding protein 10 | Type I procollagen chaperone C terminal cleavage, Type I procollagens |
| BMP1 | Bone morphogenetic protein 1 | Calcium flux in ER disturbs collagen folding |
| TMEM38B | Transmembrane protein 38B/TRIC | |
| Genes encoding transcriptional regulator | ||
| CREB3L1 | OASIS | Unfolded Protein Response |
| MBTPS2 | Membrane-bound transcription factor peptidase, Site-2 | Regulated intra-membrane proteolysis |
| FAM46A | Family with sequence similarity, Member A | Nucleotidyltransferase fold protein |
| Genes encoding signaling processes | ||
| LRP5 | LDL-receptor related protein 5 | WNT-β-catenin signalling pathway |
| WNT-1 | Wingless-type MMTV integration site family, member 1 | WNT-β-catenin signalling pathway |
| Genes encoding processes of Osteogenesis | ||
| PLS3 | Plastin 3 | Bone Cell migration |
| IFITM5 | Interferon-Induced-Transmembrane protein 5 | Modulator of bone formation Osteoblast specific transcription factor |
| SP7/OSX | SP7 transcription factor (Osterix) | |
| Genes encoding matrix proteins or modifications | ||
| XYLT2 | Xylosyltransferase 2 | Tetrasaccharide linkage to proteoglycan core proteins |
| SEC24D | SEC24-related gene family, member D | Component of COP II machinery and transport of proteins through ER |
| SPARC | Secreted protein, Acidic, Cysteine-Rich | Osteonectin |
Table 5.
Classic Non-deforming OI with Blue Sclerae (OI Type I)–Autosomal dominant (AD), autosomal recessive (AR).
Table 5.
Classic Non-deforming OI with Blue Sclerae (OI Type I)–Autosomal dominant (AD), autosomal recessive (AR).
| Disorder—Gene | Mode of Inheritance | MIM of Condition | Gene Product |
|---|---|---|---|
| OI type 1—COLIA1 | AD | 16200 | Collagen alpha 1 (I) |
| 166240 | |||
| OI type 1—COLIA2 | AD | 166200 | Collagen alpha 2 (I) |
| 166240 |
Table 6.
Common variable OI with normal sclerae (OI Type 4).
| Disorder—Gene | Mode of Inheritance | MIM of Condition |
|---|---|---|
| OI type 4—COLIA1 | AD | 166220 |
| OI type 4—COLIA2 | AD | 166220 |
| OI type 4—WNT1 | AD | 615220 |
| OI type 4—CRTAP | AR | 610854 |
| OI type 4—PPIB | AR | 259440 |
| OI type 4—SP7 | AR | 606633 |
| OI type 4—PLOD2 | AR | 609220 |
| OI type 4—FKBP10 | AR | 610968 |
| OI type 4—PLS3 | XL | 300912 |
| OI type 4—MBTPS2 | XL | 605927 |
Table 7.
Common variable OI with normal sclerae (OI Type 4).
| Disorder—Gene | OMIM | Mode of Inheritance | MIM of Condition | Gene Product |
|---|---|---|---|---|
| OI type 2—COL1A1 | AD | 166210 | Collagen alpha 1(I) | |
| OI type 2—COL1A2 | AD, AR | 166210, 259400 | Collagen alpha 2(I) | |
| OI type 2—CRTAP | VII | AR | 610854 | Cartilage- associated protein |
| OI type 2—P3H1/LEPRE 1 | VIII | AR | 610915 | Prolyl-3 hydroxyl/LEPRE1 |
| OI type 2—PIPB | IX | AR | 259440 | Cyclophilin B |
Table 8.
Potentially Progressively Deforming OI with usually sclerae (OI Type 3).
| Disorder—Gene | OMIM | Mode of Inheritance | MIM of Condition | Orphanet |
|---|---|---|---|---|
| OI type 3—COLIA1 | III | AD, AR | 259420 | 216812 |
| OI type 3—COLIA2 | VI | AD, AR | 216812 | |
| OI type 3—SEPINF1 | VII | AR | 613982 | 216812 |
| OI type 3—CRTAP | VIII | AR | 610682 | 216812 |
| OI type 3—P3H1/LEPRE1 | IX | AR | 610915 | 216812 |
| OI type 3—PIPB | X | AR | 259440 | 216812 |
| OI type 3—SERPINH1 | XI | AR | 613848 | 216812 |
| OI type 3—FKBP10 | XII | AR | 610968 | 216812 |
| OI type 3—SP7/OSX | XIII | AR | 606633 | 216812 |
| OI type 3—TMEM38B | XIV | AR | 615066 | 216812 |
| OI type 3—BMP1 | XV | AR | 112264 | 216812 |
| OI type 3—WNT1 | XVI | AD, AR | 615220 | 216812 |
| OI type 3—CREB3L1 | XVII | AR | 616229 | 216812 |
| OI type 3—SPARC | XVIII | AR | 616507 | 216812 |
| OI type 3—FAM24A | AR | 617952 | 216812 | |
| OI type 3—PLOD2 * | AR | 609220 | 216812 | |
| OI type 3—MBTPS2 | XLR | 300294 | 216820 | |
| OI type 3—SEC24D ** | AR | 616294 | 216812 |
* Mutations in SEC24D result in OI with craniosynostosis like features, a Cole–Carpenter syndrome-like presentation. ** Mutations in PLOD2 result in Bruck syndrome type 2 (Osteogenesis Imperfecta with Congenital Joint Contractures.
Table 9.
Familial Osteoporosis and Special Syndromes with Phenotypic features overlapping OI.
| Disorder—Gene | Mode of Inheritance | Gene |
|---|---|---|
| Osteoporosis X-linked | XL | PLS3 |
| Osteoporosis X-linked | XL | MBTPS2 |
| Osteoporosis—Autosomal dominant | AD | WNT1 |
| Osteoporosis—autosomal dominant | AD | LRP5 |
| Osteoporosis—pseudoglioma | AR | LRP5 |
| Osteoporosis, cataracts, and retinal dysplasia (spondylo-ocular) | AR | XYLT2 |
| OI with congenital joint contractures type 1(Bruck syndrome) | AR | FKBP10 |
| OI with congenital joint contractures type 2(Bruck syndrome) | AR | PLOD2 |
| Osteoporosis with radiolucent lesions of the mandible—Gnathodiaphyseal Dysplasia | AD | ANO5 |
| Osteogenesis Imperfecta with craniosynostosis (Cole–Carpenter) | AD | P4HB |
| Osteogenesis Imperfecta with craniosynostosis (Cole–Carpenter Like) | AR | SEC24D |
| Singleton–Merten syndrome type 1 | AD | IFIH1 |
| Singleton–Merten syndrome type 2 | AD | DDX58 |
| Short Stature, Bone fragility, DD and Immunodeficiency | AR | NBAS * |
* Neuroblastoma Amplified Sequence (see SOPH syndrome).
Table 10.
Comparison Nosology OMIM vs. International Nosology Committee.
| OI Type | INCDS 2014 | Genes | Mutations |
|---|---|---|---|
| I | 1 | COL1A1, COL1A2 COL1A1, COL1A2, CRTAP, LEPRE1, PPIB COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, FKBP10, SERPINF1, SERPINH1, SP7/OSX, BMP1, TMEM38B, WNT1, CREB3L1, SPARC, PLOD2, P4HB, SEC24D COL1A1, COL1A2, WNT1, CRTAP, PPIB, SP7, PLOD2, FKBP10, PLS3, MBTPS2 IFITM5 SERPINF1 CRTAP LEPRE1 PPIB SERPINH1 FKBP10 SP7/OSX TMEM38B BMP1 WNT1 CREB3L1 SPARC FAM46A PLOD2 P4HB SEC24D PLS3 MBTPS2 LRP5 XYLT2 | Nonsense, frameshift, split-site Missense, multiexon deletion Various Various. Normally these are gly Insertion cryptic initiationSite:- c.-14C>T Various AR Various AR Various AR Various AR Various AR Various AR Various AR Various AR Various AR Various, both heterozygous and homozygous Various AR Various AR AR Various AR Heterozygous AR X-Linked hemizygous, and heterozygous X-Linked hemizygous AR (but AD gain of function) AR (usually Spondylo-ocular) |
| II | 2 | ||
| III | 3 | ||
| IV | 4 | ||
| V | 5 | ||
| VI | 6 | ||
| VII | 7 | ||
| VIII | 8 | ||
| IX | 9 | ||
| X | 10 | ||
| XI | 11 | ||
| XII | 12 | ||
| XIII | 13 | ||
| XIV | 14 | ||
| XV | 15 | ||
| XVI | 16 | ||
| XVII | 17 | ||
| XVIII | 18 |
Table 11.
Comparison Nosology OMIM vs. International Nosology Committee.
| Disorder | Mode of Inheritance | Gene | Gene Product |
|---|---|---|---|
| Progeria (Hutchinson-Gilford) | AD | LMNA | Lamin A/C |
| Mandibulo-Acral Dysplasia A | AR | LMNA | Lamin A/C |
| Mandibulo-Acral Dysplasia B | AR | ZMPSTE24 | Zinc metalloproteinase2 |
| Wrinkly skin syndrome | AR | ATP6VOA2 | H+ ATPase |
| Gerodermaosteodysplasticum | AR | GORAB | GORAB |
| Cutis Laxa with ProgeroidFeatures | AR | PYCR1 | Pyrroline-5-carboxylatereductase1 |
| Wiedemann–Rautenstrauch | AR | ||
| Werner Syndrome | AR | RECQL2 | WRN (DNA helicase) |
| Metageria (Acrogeria, Gottrontype) | AR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Madhuri, V.; Ramesh, S.; Raymond, R.; Selina, A.; Loganathan, L. Translational Research in Osteogenesis Imperfecta and Cell Therapy. Proceedings 2021, 72, 3. https://doi.org/10.3390/proceedings2021072003
AMA Style
Madhuri V, Ramesh S, Raymond R, Selina A, Loganathan L. Translational Research in Osteogenesis Imperfecta and Cell Therapy. Proceedings. 2021; 72(1):3. https://doi.org/10.3390/proceedings2021072003
Chicago/Turabian StyleMadhuri, Vrisha, Sowmya Ramesh, Renita Raymond, Agnes Selina, and Lakshmi Loganathan. 2021. "Translational Research in Osteogenesis Imperfecta and Cell Therapy" Proceedings 72, no. 1: 3. https://doi.org/10.3390/proceedings2021072003