
Cellular and Molecular Players in the Interplay between Adipose Tissue and Breast Cancer



Abstract
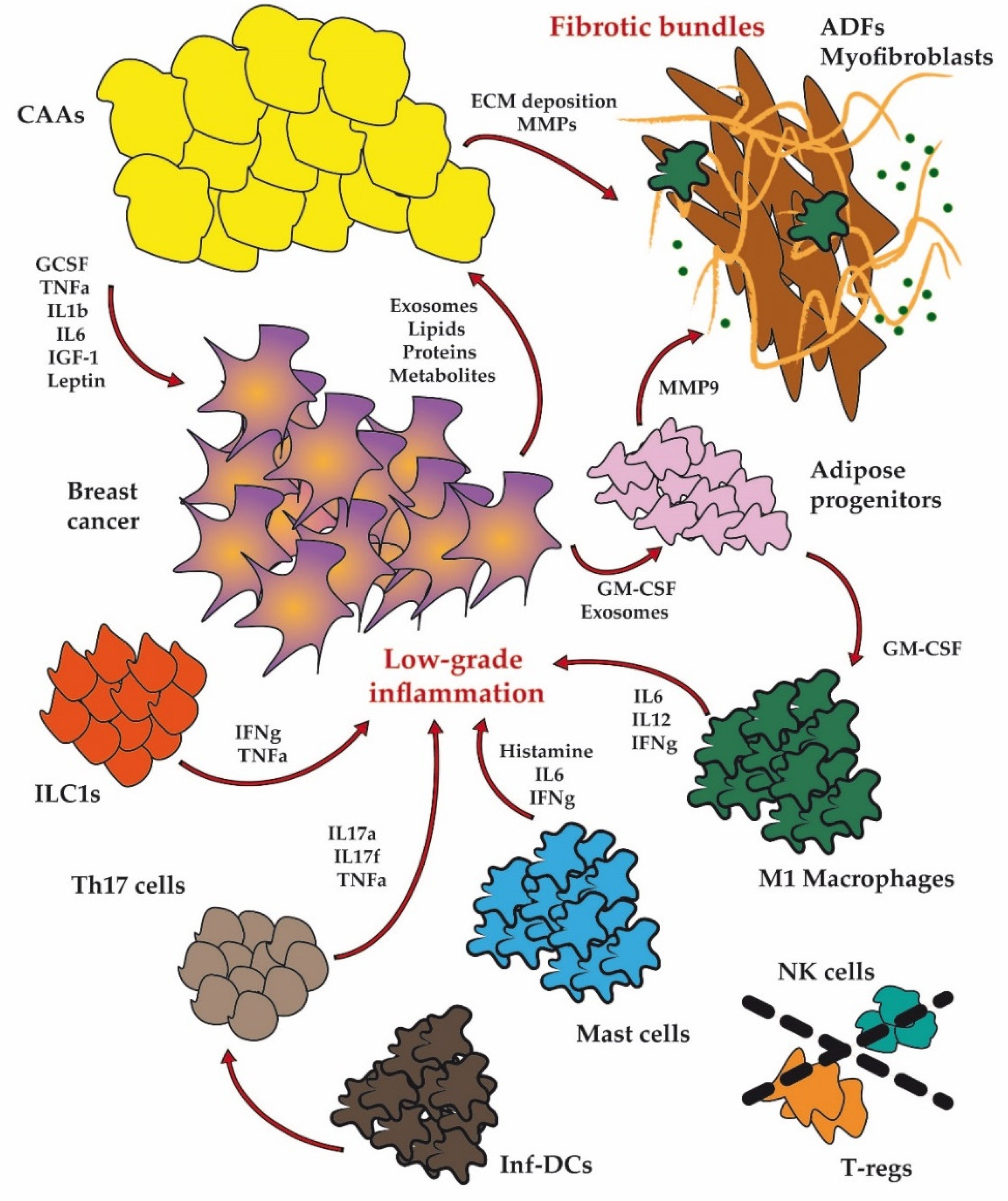
1. Introduction
2. Dysfunctional Adipose Cellular Players in BC
2.1. Adipocytes
2.2. Fibroblasts
2.3. Immune Cells
2.3.1. Macrophages
2.3.2. Dendritic Cells
2.3.3. Natural Killer Cells
2.3.4. Mast Cells
2.3.5. T-Regulatory Cells
2.3.6. Innate Lymphoid Cells
2.4. Adipose Progenitor Cells
3. Molecular Alterations in Obesity and BC
3.1. Inflammation
3.2. Metabolism
3.3. Granulocyte–Macrophage Colony Stimulating Factor (GM-CSF)
3.4. MiRNAs
4. Preclinical Models to Study Obesity
4.1. Genetically Modified Obese Models
4.2. Diet-Induced Obese (DIO) Models
5. Obesity and BC in Preclinical Models and Therapeutic Approaches
5.1. Investigation of Obesity in Murine Models of BC
5.2. Therapeutic Interventions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270. [Google Scholar] [CrossRef] [PubMed]
- Olshansky, S.J.; Passaro, D.J.; Hershow, R.C.; Layden, J.; Carnes, B.A.; Brody, J.; Hayflick, L.; Butler, R.N.; Allison, D.B.; Ludwig, D.S. A Potential Decline in Life Expectancy in the United States in the 21st Century. New Engl. J. Med. 2005, 352, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, E.A.; Khavjou, O.A.; Thompson, H.; Trogdon, J.G.; Pan, L.; Sherry, B.; Dietz, W. Obesity and Severe Obesity Forecasts through 2030. Am. J. Prev. Med. 2012, 42, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular Mechanisms of Cancer Development in Obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [PubMed]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The Obesity Paradox in Cancer: A Review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef]
- Endogenous Hormones Breast Cancer Collaborative Group. Body Mass Index, Serum Sex Hormones, and Breast Cancer Risk in Postmenopausal Women. J. Natl. Cancer Inst. 2003, 95, 1218–1226. [Google Scholar] [CrossRef]
- De Waard, F.; Halewijn, E.B.; Huizinga, J. The Bimodal Age Distribution of Patients with Mammary Carcinoma. Evidence for the Existence of 2 Types of Human Breast Cancer. Cancer 1964, 17, 141–151. [Google Scholar] [CrossRef]
- Bhaskaran, K.; dos-Santos-Silva, I.; Leon, D.A.; Douglas, I.J.; Smeeth, L. Association of BMI with Overall and Cause-Specific Mortality: A Population-Based Cohort Study of 3.6 Million Adults in the UK. Lancet Diabetes Endocrinol. 2018, 6, 944–953. [Google Scholar] [CrossRef]
- Parekh, N.; Chandran, U.; Bandera, E.V. Obesity in Cancer Survival. Annu. Rev. Nutr. 2012, 32, 311–342. [Google Scholar]
- Lashinger, L.; Rossi, E.; Hursting, S. Obesity and Resistance to Cancer Chemotherapy: Interacting Roles of Inflammation and Metabolic Dysregulation. Clin. Pharmacol. Ther. 2014, 96, 458–463. [Google Scholar]
- Sethi, J.K.; Vidal-Puig, A.J. Thematic Review Series: Adipocyte Biology. Adipose Tissue Function and Plasticity Orchestrate Nutritional Adaptation. J. Lipid Res. 2007, 48, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Benabdelkamel, H.; Masood, A.; Almidani, G.M.; Alsadhan, A.A.; Bassas, A.F.; Duncan, M.W.; Alfadda, A.A. Mature Adipocyte Proteome Reveals Differentially Altered Protein Abundances between Lean, Overweight and Morbidly Obese Human Subjects. Mol. Cell. Endocrinol. 2015, 401, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Masood, A.; Al-Naami, M.Y.; Chaurand, P.; Benabdelkamel, H. A Proteomics Based Approach Reveals Differential Regulation of Visceral Adipose Tissue Proteins between Metabolically Healthy and Unhealthy Obese Patients. Mol. Cells 2017, 40, 685. [Google Scholar] [PubMed]
- Quail, D.F.; Dannenberg, A.J. The Obese Adipose Tissue Microenvironment in Cancer Development and Progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Pyrina, I.; Chung, K.-J.; Michailidou, Z.; Koutsilieris, M.; Chavakis, T.; Chatzigeorgiou, A. Fate of Adipose Progenitor Cells in Obesity-Related Chronic Inflammation. Front. Cell Dev. Biol. 2020, 8, 644. [Google Scholar] [CrossRef]
- Liu, L.; Wu, Y.; Zhang, C.; Zhou, C.; Li, Y.; Zeng, Y.; Zhang, C.; Li, R.; Luo, D.; Wang, L. Cancer-Associated Adipocytes-Derived G-CSF Promotes Breast Cancer Malignancy via Stat3 Signaling. J. Mol. Cell Biol. 2020, 12, 723–737. [Google Scholar] [CrossRef]
- Iyengar, P.; Combs, T.P.; Shah, S.J.; Gouon-Evans, V.; Pollard, J.W.; Albanese, C.; Flanagan, L.; Tenniswood, M.P.; Guha, C.; Lisanti, M.P. Adipocyte-Secreted Factors Synergistically Promote Mammary Tumorigenesis through Induction of Anti-Apoptotic Transcriptional Programs and Proto-Oncogene Stabilization. Oncogene 2003, 22, 6408–6423. [Google Scholar]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol Lipase Regulates a Fatty Acid Network That Promotes Cancer Pathogenesis. Cell 2010, 140, 49–61. [Google Scholar]
- Crewe, C.; An, Y.A.; Scherer, P.E. The Ominous Triad of Adipose Tissue Dysfunction: Inflammation, Fibrosis, and Impaired Angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef]
- Marcelin, G.; Ferreira, A.; Liu, Y.; Atlan, M.; Aron-Wisnewsky, J.; Pelloux, V.; Botbol, Y.; Ambrosini, M.; Fradet, M.; Rouault, C. A PDGFRα-Mediated Switch toward CD9high Adipocyte Progenitors Controls Obesity-Induced Adipose Tissue Fibrosis. Cell Metab. 2017, 25, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Divoux, A.; Tordjman, J.; Lacasa, D.; Veyrie, N.; Hugol, D.; Aissat, A.; Basdevant, A.; Guerre-Millo, M.; Poitou, C.; Zucker, J.-D. Fibrosis in Human Adipose Tissue: Composition, Distribution, and Link with Lipid Metabolism and Fat Mass Loss. Diabetes 2010, 59, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, G.; Silveira, A.L.M.; Martins, L.B.; Ferreira, A.V.; Clément, K. Deciphering the Cellular Interplays Underlying Obesity-Induced Adipose Tissue Fibrosis. J. Clin. Investig. 2019, 129. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.R.; Bhardwaj, P.; Choi, S.; Gonzalez, J.; Eguiluz, R.C.A.; Wang, K.; Mohanan, S.; Morris, P.G.; Du, B.; Zhou, X.K. Obesity-Dependent Changes in Interstitial ECM Mechanics Promote Breast Tumorigenesis. Sci. Transl. Med. 2015, 7, 301ra130. [Google Scholar] [CrossRef]
- Spencer, M.; Yao-Borengasser, A.; Unal, R.; Rasouli, N.; Gurley, C.M.; Zhu, B.; Peterson, C.A.; Kern, P.A. Adipose Tissue Macrophages in Insulin-Resistant Subjects Are Associated with Collagen VI and Fibrosis and Demonstrate Alternative Activation. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E1016–E1027. [Google Scholar] [CrossRef]
- Sun, K.; Tordjman, J.; Clément, K.; Scherer, P.E. Fibrosis and Adipose Tissue Dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S. Adipocyte-Derived Fibroblasts Promote Tumor Progression and Contribute to the Desmoplastic Reaction in Breast Cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Mehner, C.; Hockla, A.; Miller, E.; Ran, S.; Radisky, D.C.; Radisky, E.S. Tumor Cell-Produced Matrix Metalloproteinase 9 (MMP-9) Drives Malignant Progression and Metastasis of Basal-like Triple Negative Breast Cancer. Oncotarget 2014, 5, 2736. [Google Scholar] [CrossRef]
- Reggiani, F.; Labanca, V.; Mancuso, P.; Rabascio, C.; Talarico, G.; Orecchioni, S.; Manconi, A.; Bertolini, F. Adipose Progenitor Cell Secretion of GM-CSF and MMP9 Promotes a Stromal and Immunological Microenvironment That Supports Breast Cancer Progression. Cancer Res. 2017, 77, 5169–5182. [Google Scholar] [CrossRef]
- Motrescu, E.R.; Rio, M.-C. Cancer Cells, Adipocytes and Matrix Metalloproteinase 11: A Vicious Tumor Progression Cycle. Biol. Chem. 2008, 389, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F. Normalization of Obesity-Associated Insulin Resistance through Immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Pandya, S.K.; Varshney, S.; Shankar, K.; Rajan, S.; Srivastava, A.; Gupta, A.; Gupta, S.; Vishwakarma, A.L.; Misra, A. Temporal Immmunometabolic Profiling of Adipose Tissue in HFD-Induced Obesity: Manifestations of Mast Cells in Fibrosis and Senescence. Int. J. Obes. 2019, 43, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar]
- Lumeng, C.N.; DeYoung, S.M.; Bodzin, J.L.; Saltiel, A.R. Increased Inflammatory Properties of Adipose Tissue Macrophages Recruited during Diet-Induced Obesity. Diabetes 2007, 56, 16–23. [Google Scholar] [CrossRef]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic Switching of Adipose Tissue Macrophages with Obesity Is Generated by Spatiotemporal Differences in Macrophage Subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef]
- Boutens, L.; Stienstra, R. Adipose Tissue Macrophages: Going off Track during Obesity. Diabetologia 2016, 59, 879–894. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Valentić, S.; Šestan, M.; Turk Wensveen, T.; Polić, B. The “Big Bang” in Obese Fat: Events Initiating Obesity-induced Adipose Tissue Inflammation. Eur. J. Immunol. 2015, 45, 2446–2456. [Google Scholar]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S. Metabolic Dysfunction Drives a Mechanistically Distinct Proinflammatory Phenotype in Adipose Tissue Macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Xu, X.; Grijalva, A.; Skowronski, A.; van Eijk, M.; Serlie, M.J.; Ferrante Jr, A.W. Obesity Activates a Program of Lysosomal-Dependent Lipid Metabolism in Adipose Tissue Macrophages Independently of Classic Activation. Cell Metab. 2013, 18, 816–830. [Google Scholar] [CrossRef]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J. Identification of Adipose Tissue Dendritic Cells Correlated with Obesity-Associated Insulin-Resistance and Inducing Th17 Responses in Mice and Patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, T.D.; Schmidt-Christensen, A.; Nilsson, J.; Fransén-Pettersson, N.; Hansen, L.; Holmberg, D. Deficiency in Plasmacytoid Dendritic Cells and Type I Interferon Signalling Prevents Diet-Induced Obesity and Insulin Resistance in Mice. Diabetologia 2017, 60, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Zlotnikov-Klionsky, Y.; Nathansohn-Levi, B.; Shezen, E.; Rosen, C.; Kagan, S.; Bar-On, L.; Jung, S.; Shifrut, E.; Reich-Zeliger, S.; Friedman, N. Perforin-Positive Dendritic Cells Exhibit an Immuno-Regulatory Role in Metabolic Syndrome and Autoimmunity. Immunity 2015, 43, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Bähr, I.; Spielmann, J.; Quandt, D.; Kielstein, H. Obesity-Associated Alterations of Natural Killer Cells and Immunosurveillance of Cancer. Front. Immunol. 2020, 11, 245. [Google Scholar] [CrossRef]
- Spielmann, J.; Hanke, J.; Knauf, D.; Ben-Eliyahu, S.; Jacobs, R.; Stangl, G.; Bähr, I.; Kielstein, H. Significantly Enhanced Lung Metastasis and Reduced Organ NK Cell Functions in Diet-Induced Obese Rats. BMC Obes. 2017, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, J.; Mattheis, L.; Jung, J.-S.; Rauße, H.; Glaß, M.; Bähr, I.; Quandt, D.; Oswald, J.; Kielstein, H. Effects of Obesity on NK Cells in a Mouse Model of Postmenopausal Breast Cancer. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Lamas, B.; Nachat-Kappes, R.; Goncalves-Mendes, N.; Mishellany, F.; Rossary, A.; Vasson, M.; Farges, M. Dietary Fat without Body Weight Gain Increases in Vivo MCF-7 Human Breast Cancer Cell Growth and Decreases Natural Killer Cell Cytotoxicity. Mol. Carcinog. 2015, 54, 58–71. [Google Scholar] [CrossRef]
- Lautenbach, A.; Wrann, C.D.; Jacobs, R.; Müller, G.; Brabant, G.; Nave, H. Altered Phenotype of NK Cells from Obese Rats Can Be Normalized by Transfer into Lean Animals. Obesity 2009, 17, 1848–1855. [Google Scholar] [CrossRef]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R. Metabolic Reprogramming of Natural Killer Cells in Obesity Limits Antitumor Responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef]
- Wang, J.; Shi, G. Mast Cell Stabilization: Novel Medication for Obesity and Diabetes. Diabetes/Metab. Res. Rev. 2011, 27, 919–924. [Google Scholar] [CrossRef]
- Divoux, A.; Moutel, S.; Poitou, C.; Lacasa, D.; Veyrie, N.; Aissat, A.; Arock, M.; Guerre-Millo, M.; Clément, K. Mast Cells in Human Adipose Tissue: Link with Morbid Obesity, Inflammatory Status, and Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, E1677–E1685. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Divoux, A.; Sun, J.; Zhang, J.; Clément, K.; Glickman, J.N.; Sukhova, G.K.; Wolters, P.J.; Du, J.; Gorgun, C.Z. Genetic Deficiency and Pharmacological Stabilization of Mast Cells Reduce Diet-Induced Obesity and Diabetes in Mice. Nat. Med. 2009, 15, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, D.A.; Muralidhar, S.; Feyerabend, T.B.; Herzig, S.; Rodewald, H.-R. Hematopoietic Kit Deficiency, Rather than Lack of Mast Cells, Protects Mice from Obesity and Insulin Resistance. Cell Metab. 2015, 21, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Chmelař, J.; Chatzigeorgiou, A.; Chung, K.-J.; Prucnal, M.; Voehringer, D.; Roers, A.; Chavakis, T. No Role for Mast Cells in Obesity-Related Metabolic Dysregulation. Front. Immunol. 2016, 7, 524. [Google Scholar] [CrossRef] [PubMed]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ Is a Major Driver of the Accumulation and Phenotype of Adipose Tissue T Reg Cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S. Lean, but Not Obese, Fat Is Enriched for a Unique Population of Regulatory T Cells That Affect Metabolic Parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Cipolletta, D.; Cohen, P.; Spiegelman, B.M.; Benoist, C.; Mathis, D. Appearance and Disappearance of the MRNA Signature Characteristic of Treg Cells in Visceral Adipose Tissue: Age, Diet, and PPARγ Effects. Proc. Natl. Acad. Sci. USA 2015, 112, 482–487. [Google Scholar] [CrossRef]
- O’Sullivan, T.E.; Rapp, M.; Fan, X.; Weizman, O.-E.; Bhardwaj, P.; Adams, N.M.; Walzer, T.; Dannenberg, A.J.; Sun, J.C. Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity 2016, 45, 428–441. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Kim, B.S.; Saenz, S.A.; Stine, R.R.; Monticelli, L.A.; Sonnenberg, G.F.; Thome, J.J.; Farber, D.L.; Lutfy, K.; Seale, P. Group 2 Innate Lymphoid Cells Promote Beiging of White Adipose Tissue and Limit Obesity. Nature 2015, 519, 242–246. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.J.; Chang, Y.-J.; Pichavant, M.; Shore, S.A.; Fitzgerald, K.A.; Iwakura, Y.; Israel, E.; Bolger, K.; Faul, J. Interleukin-17–Producing Innate Lymphoid Cells and the NLRP3 Inflammasome Facilitate Obesity-Associated Airway Hyperreactivity. Nat. Med. 2014, 20, 54–61. [Google Scholar] [CrossRef]
- Everaere, L.; Ait-Yahia, S.; Molendi-Coste, O.; Vorng, H.; Quemener, S.; LeVu, P.; Fleury, S.; Bouchaert, E.; Fan, Y.; Duez, C. Innate Lymphoid Cells Contribute to Allergic Airway Disease Exacerbation by Obesity. J. Allergy Clin. Immunol. 2016, 138, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Martin-Padura, I.; Gregato, G.; Marighetti, P.; Mancuso, P.; Calleri, A.; Corsini, C.; Pruneri, G.; Manzotti, M.; Lohsiriwat, V.; Rietjens, M. The White Adipose Tissue Used in Lipotransfer Procedures Is a Rich Reservoir of CD34+ Progenitors Able to Promote Cancer Progression. Cancer Res. 2012, 72, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, S.; Gregato, G.; Martin-Padura, I.; Reggiani, F.; Braidotti, P.; Mancuso, P.; Calleri, A.; Quarna, J.; Marighetti, P.; Aldeni, C. Complementary Populations of Human Adipose CD34+ Progenitor Cells Promote Growth, Angiogenesis, and Metastasis of Breast Cancer. Cancer Res. 2013, 73, 5880–5891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Daquinag, A.C.; Amaya-Manzanares, F.; Sirin, O.; Tseng, C.; Kolonin, M.G. Stromal Progenitor Cells from Endogenous Adipose Tissue Contribute to Pericytes and Adipocytes That Populate the Tumor Microenvironment. Cancer Res. 2012, 72, 5198–5208. [Google Scholar] [CrossRef]
- Razmkhah, M.; Jaberipour, M.; Erfani, N.; Habibagahi, M.; Talei, A.; Ghaderi, A. Adipose Derived Stem Cells (ASCs) Isolated from Breast Cancer Tissue Express IL-4, IL-10 and TGF-Β1 and Upregulate Expression of Regulatory Molecules on T Cells: Do They Protect Breast Cancer Cells from the Immune Response? Cell. Immunol. 2011, 266, 116–122. [Google Scholar] [CrossRef]
- Engela, A.U.; Hoogduijn, M.J.; Boer, K.; Litjens, N.H.; Betjes, M.G.; Weimar, W.; Baan, C.C. Human Adipose-tissue Derived Mesenchymal Stem Cells Induce Functional De-novo Regulatory T Cells with Methylated FOXP3 Gene DNA. Clin. Exp. Immunol. 2013, 173, 343–354. [Google Scholar]
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from Breast Cancer Cells Can Convert Adipose Tissue-Derived Mesenchymal Stem Cells into Myofibroblast-like Cells. Int. J. Oncol. 2011, 40, 130–138. [Google Scholar]
- Raajendiran, A.; Ooi, G.; Bayliss, J.; O’Brien, P.E.; Schittenhelm, R.B.; Clark, A.K.; Taylor, R.A.; Rodeheffer, M.S.; Burton, P.R.; Watt, M.J. Identification of Metabolically Distinct Adipocyte Progenitor Cells in Human Adipose Tissues. Cell Rep. 2019, 27, 1528–1540. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Morris, P.G.; Howe, L.R.; Giri, D.D.; Morrow, M.; Wang, H.; Pollak, M.; Jones, L.W. Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin. Cancer Res. 2016, 22, 2283–2289. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Morris, P.G.; Hudis, C.A.; Giri, D.; Morrow, M.; Falcone, D.J.; Zhou, X.K.; Du, B.; Brogi, E.; Crawford, C.B.; Kopelovich, L. Inflammation and Increased Aromatase Expression Occur in the Breast Tissue of Obese Women with Breast Cancer. Cancer Prev. Res. 2011, 4, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Conforti-Andreoni, C.; Ricciardi-Castagnoli, P.; Mortellaro, A. The Inflammasomes in Health and Disease: From Genetics to Molecular Mechanisms of Autoinflammation and Beyond. Cell. Mol. Immunol. 2011, 8, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Flier, J.S. Obesity and Insulin Resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Martyn, J.A.J.; Kaneki, M.; Yasuhara, S.; Warner, D.S.; Warner, M.A. Obesity-Induced Insulin Resistance and Hyperglycemia: Etiologic Factors and Molecular Mechanisms. J. Am. Soc. Anesthesiol. 2008, 109, 137–148. [Google Scholar]
- D’Souza, K.; Kane, D.A.; Touaibia, M.; Kershaw, E.E.; Pulinilkunnil, T.; Kienesberger, P.C. Autotaxin Is Regulated by Glucose and Insulin in Adipocytes. Endocrinology 2017, 158, 791–803. [Google Scholar] [CrossRef]
- Brindley, D.N.; Tang, X.; Meng, G.; Benesch, M.G.K. Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 5938. [Google Scholar] [CrossRef]
- Brandon, J.A.; Kraemer, M.; Vandra, J.; Halder, S.; Ubele, M.; Morris, A.J.; Smyth, S.S. Adipose-Derived Autotaxin Regulates Inflammation and Steatosis Associated with Diet-Induced Obesity. PLoS ONE 2019, 14, e0208099. [Google Scholar] [CrossRef]
- Fitzgibbons, T.P.; Kogan, S.; Aouadi, M.; Hendricks, G.M.; Straubhaar, J.; Czech, M.P. Similarity of Mouse Perivascular and Brown Adipose Tissues and Their Resistance to Diet-Induced Inflammation. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1425–H1437. [Google Scholar] [CrossRef]
- Tews, D.; Pula, T.; Funcke, J.-B.; Jastroch, M.; Keuper, M.; Debatin, K.-M.; Wabitsch, M.; Fischer-Posovszky, P. Elevated UCP1 Levels Are Sufficient to Improve Glucose Uptake in Human White Adipocytes. Redox Biol. 2019, 26, 101286. [Google Scholar] [CrossRef]
- Whittle, A.J.; López, M.; Vidal-Puig, A. Using Brown Adipose Tissue to Treat Obesity–the Central Issue. Trends Mol. Med. 2011, 17, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-H.; Lundh, M.; Fu, A.; Kriszt, R.; Huang, T.L.; Lynes, M.D.; Leiria, L.O.; Shamsi, F.; Darcy, J.; Greenwood, B.P. CRISPR-Engineered Human Brown-like Adipocytes Prevent Diet-Induced Obesity and Ameliorate Metabolic Syndrome in Mice. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R. Brown Remodeling of White Adipose Tissue by SirT1-Dependent Deacetylation of Pparγ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.; Wang, X.; Wu, Y.; Xu, C.; Xia, Z.; Dai, J.; Shao, M.; Zhao, F.; He, S.; Yang, L. The Metabolic ER Stress Sensor IRE1α Suppresses Alternative Activation of Macrophages and Impairs Energy Expenditure in Obesity. Nat. Immunol. 2017, 18, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, J.; Osborne, O.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; Quehenberger, O. Increased Adipocyte O2 Consumption Triggers HIF-1α, Causing Inflammation and Insulin Resistance in Obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef]
- Engin, A. Adipose tissue hypoxia in obesity and its impact on preadipocytes and macrophages: Hypoxia hypothesis. In Obesity and Lipotoxicity; Springer International Publishing: New York, NY, USA, 2017; Volume 960, pp. 305–326. [Google Scholar]
- Bournat, J.C.; Brown, C.W. Mitochondrial Dysfunction in Obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446. [Google Scholar] [CrossRef]
- Sparks, L.M.; Xie, H.; Koza, R.A.; Mynatt, R.; Hulver, M.W.; Bray, G.A.; Smith, S.R. A High-Fat Diet Coordinately Downregulates Genes Required for Mitochondrial Oxidative Phosphorylation in Skeletal Muscle. Diabetes 2005, 54, 1926–1933. [Google Scholar]
- Ban, J.-J.; Ruthenborg, R.J.; Cho, K.W.; Kim, J. Regulation of Obesity and Insulin Resistance by Hypoxia-Inducible Factors. Hypoxia 2014, 2, 171. [Google Scholar]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased Oxidative Stress in Obesity and Its Impact on Metabolic Syndrome. J. Clin. Investig. 2017, 114, 1752–1761. [Google Scholar] [CrossRef]
- Quail, D.F.; Olson, O.C.; Bhardwaj, P.; Walsh, L.A.; Akkari, L.; Quick, M.L.; Chen, I.-C.; Wendel, N.; Ben-Chetrit, N.; Walker, J. Obesity Alters the Lung Myeloid Cell Landscape to Enhance Breast Cancer Metastasis through IL5 and GM-CSF. Nat. Cell Biol. 2017, 19, 974–987. [Google Scholar]
- Reggiani, F.; Bertolini, F. GM-CSF Promotes a Supportive Adipose and Lung Microenvironment in Metastatic Breast Cancer. Oncoscience 2017, 4, 126. [Google Scholar] [CrossRef] [PubMed]
- Plubell, D.L.; Fenton, A.M.; Wilmarth, P.A.; Bergstrom, P.; Zhao, Y.; Minnier, J.; Heinecke, J.W.; Yang, X.; Pamir, N. GM-CSF Driven Myeloid Cells in Adipose Tissue Link Weight Gain and Insulin Resistance via Formation of 2-Aminoadipate. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Catellani, C.; Sartori, C.; Lazzeroni, P.; Amarri, S.; Street, M.E. Obesity, Insulin Resistance, and Colorectal Cancer: Could MiRNA Dysregulation Play a Role? Int. J. Mol. Sci. 2019, 20, 2922. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Kulyté, A. MicroRNA Regulatory Networks in Human Adipose Tissue and Obesity. Nat. Rev. Endocrinol. 2015, 11, 276. [Google Scholar] [CrossRef]
- Heneghan, H.; Miller, N.; McAnena, O.; O’brien, T.; Kerin, M. Differential MiRNA Expression in Omental Adipose Tissue and in the Circulation of Obese Patients Identifies Novel Metabolic Biomarkers. J. Clin. Endocrinol. Metab. 2011, 96, E846–E850. [Google Scholar] [CrossRef]
- Herrero-Aguayo, V.; Jiménez-Vacas, J.M.; Sáez-Martínez, P.; Gómez-Gómez, E.; López-Cánovas, J.L.; Garrido-Sánchez, L.; Herrera-Martínez, A.D.; García-Bermejo, L.; Macías-González, M.; López-Miranda, J. Influence of Obesity in the MiRNome: MiR-4454, a Key Regulator of Insulin Response via Splicing Modulation in Prostate. J. Clin. Endocrinol. Metab. 2021, 106, e469–e484. [Google Scholar] [CrossRef]
- Meerson, A.; Traurig, M.; Ossowski, V.; Fleming, J.; Mullins, M.; Baier, L. Human Adipose MicroRNA-221 Is Upregulated in Obesity and Affects Fat Metabolism Downstream of Leptin and TNF-α. Diabetologia 2013, 56, 1971–1979. [Google Scholar] [CrossRef]
- Hwang, M.S.; Yu, N.; Stinson, S.Y.; Yue, P.; Newman, R.J.; Allan, B.B.; Dornan, D. MiR-221/222 Targets Adiponectin Receptor 1 to Promote the Epithelial-to-Mesenchymal Transition in Breast Cancer. PLoS ONE 2013, 8, e66502. [Google Scholar] [CrossRef]
- Capobianco, V.; Nardelli, C.; Ferrigno, M.; Iaffaldano, L.; Pilone, V.; Forestieri, P.; Zambrano, N.; Sacchetti, L. MiRNA and Protein Expression Profiles of Visceral Adipose Tissue Reveal MiR-141/YWHAG and MiR-520e/RAB11A as Two Potential MiRNA/Protein Target Pairs Associated with Severe Obesity. J. Proteome Res. 2012, 11, 3358–3369. [Google Scholar] [CrossRef]
- Song, Y.; Wu, L.; Li, M.; Xiong, X.; Fang, Z.; Zhou, J.; Yan, G.; Chen, X.; Yang, J.; Li, Y. Down-Regulation of MicroRNA-592 in Obesity Contributes to Hyperglycemia and Insulin Resistance. EBioMedicine 2019, 42, 494–503. [Google Scholar] [CrossRef]
- Ge, Q.; Brichard, S.; Yi, X.; Li, Q. MicroRNAs as a New Mechanism Regulating Adipose Tissue Inflammation in Obesity and as a Novel Therapeutic Strategy in the Metabolic Syndrome. J. Immunol. Res. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Gholami, M.; Larijani, B.; Zahedi, Z.; Mahmoudian, F.; Bahrami, S.; Omran, S.P.; Saadatian, Z.; Hasani-Ranjbar, S.; Taslimi, R.; Bastami, M. Inflammation Related MiRNAs as an Important Player between Obesity and Cancers. J. Diabetes Metab. Disord. 2019, 18, 675–692. [Google Scholar] [CrossRef] [PubMed]
- Hamam, R.; Hamam, D.; Alsaleh, K.A.; Kassem, M.; Zaher, W.; Alfayez, M.; Aldahmash, A.; Alajez, N.M. Circulating MicroRNAs in Breast Cancer: Novel Diagnostic and Prognostic Biomarkers. Cell Death Dis. 2017, 8, e3045. [Google Scholar] [CrossRef]
- Kasiappan, R.; Rajarajan, D. Role of MicroRNA Regulation in Obesity-Associated Breast Cancer: Nutritional Perspectives. Adv. Nutr. 2017, 8, 868–888. [Google Scholar] [CrossRef] [PubMed]
- Lorente-Cebrián, S.; González-Muniesa, P.; Milagro, F.I.; Martínez, J.A. MicroRNAs and Other Non-Coding RNAs in Adipose Tissue and Obesity: Emerging Roles as Biomarkers and Therapeutic Targets. Clin. Sci. 2019, 133, 23–40. [Google Scholar] [CrossRef]
- Barrett, P.; Mercer, J.G.; Morgan, P.J. Preclinical Models for Obesity Research. Dis. Models Mech. 2016, 9, 1245–1255. [Google Scholar] [CrossRef]
- Wang, B.; Charukeshi Chandrasekera, P.; J Pippin, J. Leptin-and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef]
- Lang, P.; Hasselwander, S.; Li, H.; Xia, N. Effects of Different Diets Used in Diet-Induced Obesity Models on Insulin Resistance and Vascular Dysfunction in C57BL/6 Mice. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Ingalls, A. Dickie MM, and Snell GD. ObeseA New Mutat. House Mouse. J. Hered. 1950, 41, 317–318. [Google Scholar]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Maffei, M.; Stoffel, M.; Barone, M.; Moon, B.; Dammerman, M.; Ravussin, E.; Bogardus, C.; Ludwig, D.S.; Flier, J.S.; Talley, M. Absence of Mutations in the Human OB Gene in Obese/Diabetic Subjects. Diabetes 1996, 45, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Osei, S.Y. Leptin Signaling. Physiol. Behav. 2004, 81, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G. Development of Hypothalamic Circuits That Control Food Intake and Energy Balance. In Appetite and Food Intake: Central Control; CRC Press: Boca Raton, FL, USA, 2017; pp. 135–154. [Google Scholar]
- Bates, S.H.; Kulkarni, R.N.; Seifert, M.; Myers Jr, M.G. Roles for Leptin Receptor/STAT3-Dependent and-Independent Signals in the Regulation of Glucose Homeostasis. Cell Metab. 2005, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Saadat, N.; IglayReger, H.B.; Myers Jr, M.G.; Bodary, P.; Gupta, S.V. Differences in Metabolomic Profiles of Male Db/Db and s/s, Leptin Receptor Mutant Mice. Physiol. Genom. 2012, 44, 374–381. [Google Scholar] [CrossRef][Green Version]
- Phillips, M.S.; Liu, Q.; Hammond, H.A.; Dugan, V.; Hey, P.J.; Caskey, C.T.; Hess, J.F. Leptin Receptor Missense Mutation in the Fatty Zucker Rat. Nat. Genet. 1996, 13, 18–19. [Google Scholar] [CrossRef]
- Koletsky, S. Obese Spontaneously Hypertensive Rats—a Model for Study of Atherosclerosis. Exp. Mol. Pathol. 1973, 19, 53–60. [Google Scholar] [CrossRef]
- Challis, B.; Coll, A.P.; Yeo, G.S.; Pinnock, S.B.; Dickson, S.L.; Thresher, R.R.; Dixon, J.; Zahn, D.; Rochford, J.J.; White, A.; et al. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY (3–36). Proc. Natl. Acad. Sci. USA 2004, 101, 4695–4700. [Google Scholar]
- O’Rahilly, S. Human Genetics Illuminates the Paths to Metabolic Disease. Nature 2009, 462, 307–314. [Google Scholar]
- Mul, J.D.; Van Boxtel, R.; Bergen, D.J.; Brans, M.A.; Brakkee, J.H.; Toonen, P.W.; Garner, K.M.; Adan, R.A.; Cuppen, E. Melanocortin Receptor 4 Deficiency Affects Body Weight Regulation, Grooming Behavior, and Substrate Preference in the Rat. Obesity 2012, 20, 612–621. [Google Scholar] [CrossRef]
- Butler, A.A.; Kesteson, R.A.; Khong, K.; Cullen, M.J.; Pelleymounter, M.A.; Dekoning, J.; Baetscher, M.; Cone, R.D. A Unique Metalolic Sysdrone Causes Obesity in the Melanocortin-3 Receptor-Deficient Mouse. Endocrinology 2000, 141, 3518–3521. [Google Scholar] [CrossRef]
- Rowland, N.E.; Schaub, J.W.; Robertson, K.L.; Andreasen, A.; Haskell-Luevano, C. Effect of MTII on Food Intake and Brain C-Fos in Melanocortin-3, Melanocortin-4, and Double MC3 and MC4 Receptor Knockout Mice. Peptides 2010, 31, 2314–2317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hawkes, C. Uneven Dietary Development: Linking the Policies and Processes of Globalization with the Nutrition Transition, Obesity and Diet-Related Chronic Diseases. Glob. Health 2006, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial Dysfunction in NASH: Causes, Consequences and Possible Means to Prevent It. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Dubouchaud, H.; Couturier, K.; Cottet-Rousselle, C.; Taleux, N.; Athias, A.; Galinier, A.; Casteilla, L.; Leverve, X.M. Effects of a High-Fat Diet on Energy Metabolism and ROS Production in Rat Liver. J. Hepatol. 2011, 54, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, N.J.; Stock, M.J. A Role for Insulin in the Diet-Induced Thermogenesis of Cafeteria-Fed Rats. Metabolism 1981, 30, 673–678. [Google Scholar] [PubMed]
- Rothwell, N.J.; Saville, M.E.; Stock, M.J. Effects of Feeding a “Cafeteria” Diet on Energy Balance and Diet-Induced Thermogenesis in Four Strains of Rat. J. Nutr. 1982, 112, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, N.J.; Stock, M.J. The Cafeteria Diet as a Tool for Studies of Thermogenesis. J. Nutr. 1988, 118, 925–928. [Google Scholar] [PubMed]
- Martire, S.I.; Westbrook, R.F.; Morris, M.J. Effects of Long-Term Cycling between Palatable Cafeteria Diet and Regular Chow on Intake, Eating Patterns, and Response to Saccharin and Sucrose. Physiol. Behav. 2015, 139, 80–88. [Google Scholar] [CrossRef]
- de Zwaan, M. Binge Eating Disorder and Obesity. Int. J. Obes. 2001, 25, S51–S55. [Google Scholar] [CrossRef]
- Fairburn, C.G.; Cooper, Z.; Doll, H.A.; Norman, P.; O’Connor, M. The Natural Course of Bulimia Nervosa and Binge Eating Disorder in Young Women. Arch. Gen. Psychiatry 2000, 57, 659–665. [Google Scholar] [CrossRef]
- Yanovski, S.Z.; Nelson, J.E.; Dubbert, B.K.; Spitzer, R.L. Association of Binge Eating Disorder and Psychiatric Comorbidity in Obese Subjects. Am. J. Psychiatry 1993, 150, 1472–1479. [Google Scholar] [PubMed]
- Wilson, G.T.; Wilfley, D.E.; Agras, W.S.; Bryson, S.W. Psychological Treatments of Binge Eating Disorder. Arch. Gen. Psychiatry 2010, 67, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Forrest, L.N.; Zuromski, K.L.; Dodd, D.R.; Smith, A.R. Suicidality in Adolescents and Adults with Binge-eating Disorder: Results from the National Comorbidity Survey Replication and Adolescent Supplement. Int. J. Eat. Disord. 2017, 50, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Berner, L.A.; Avena, N.M.; Hoebel, B.G. Bingeing, Self-restriction, and Increased Body Weight in Rats with Limited Access to a Sweet-fat Diet. Obesity 2008, 16, 1998–2002. [Google Scholar] [PubMed]
- Valdivia, S.; Patrone, A.; Reynaldo, M.; Perello, M. Acute High Fat Diet Consumption Activates the Mesolimbic Circuit and Requires Orexin Signaling in a Mouse Model. PLoS ONE 2014, 9, e87478. [Google Scholar]
- Bake, T.; Murphy, M.; Morgan, D.; Mercer, J. Large, Binge-Type Meals of High Fat Diet Change Feeding Behaviour and Entrain Food Anticipatory Activity in Mice. Appetite 2014, 77, 62–73. [Google Scholar] [CrossRef]
- Bake, T.; Morgan, D.; Mercer, J. Feeding and Metabolic Consequences of Scheduled Consumption of Large, Binge-Type Meals of High Fat Diet in the Sprague–Dawley Rat. Physiol. Behav. 2014, 128, 70–79. [Google Scholar] [CrossRef]
- Waxler, S.; Leef, M. Augmentation of Mammary Tumors in Castrated Obese C3H Mice. Cancer Res. 1966, 26, 860–862. [Google Scholar]
- Waxler, S.H.; Tabar, P.; Melcher, L.R. Obesity and the Time of Appearance of Spontaneous Mammary Carcinoma in C3H Mice. Cancer Res. 1953, 13, 276–278. [Google Scholar]
- Cleary, M.P.; Grossmann, M.E.; Ray, A. Effect of Obesity on Breast Cancer Development. Vet. Pathol. 2010, 47, 202–213. [Google Scholar] [CrossRef]
- Núñez, N.P.; Perkins, S.N.; Smith, N.C.; Berrigan, D.; Berendes, D.M.; Varticovski, L.; Barrett, J.C.; Hursting, S.D. Obesity Accelerates Mouse Mammary Tumor Growth in the Absence of Ovarian Hormones. Nutr. Cancer 2008, 60, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Ecker, B.L.; Lee, J.Y.; Sterner, C.J.; Solomon, A.C.; Pant, D.K.; Shen, F.; Peraza, J.; Vaught, L.; Mahendra, S.; Belka, G.K. Impact of Obesity on Breast Cancer Recurrence and Minimal Residual Disease. Breast Cancer Res. 2019, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Giles, E.D.; Wellberg, E.A. Preclinical Models to Study Obesity and Breast Cancer in Females: Considerations, Caveats, and Tools. J. Mammary Gland Biol. Neoplasia 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Eliassen, A.H.; Colditz, G.A.; Rosner, B.; Willett, W.C.; Hankinson, S.E. Adult Weight Change and Risk of Postmenopausal Breast Cancer. JAMA 2006, 296, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bousquenaud, M.; Fico, F.; Solinas, G.; Rüegg, C.; Santamaria-Martínez, A. Obesity Promotes the Expansion of Metastasis-Initiating Cells in Breast Cancer. Breast Cancer Res. 2018, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cranford, T.L.; Velázquez, K.T.; Enos, R.T.; Sougiannis, A.T.; Bader, J.E.; Carson, M.S.; Bellone, R.R.; Chatzistamou, I.; Nagarkatti, M.; Murphy, E.A. Effects of High Fat Diet-Induced Obesity on Mammary Tumorigenesis in the PyMT/MMTV Murine Model. Cancer Biol. Ther. 2019, 20, 487–496. [Google Scholar]
- Cleary, M.P.; Grande, J.P.; Maihle, N.J. Effect of High Fat Diet on Body Weight and Mammary Tumor Latency in MMTV-TGF-α Mice. Int. J. Obes. 2004, 28, 956–962. [Google Scholar] [CrossRef]
- Dogan, S.; Hu, X.; Zhang, Y.; Maihle, N.J.; Grande, J.P.; Cleary, M.P. Effects of High-Fat Diet and/or Body Weight on Mammary Tumor Leptin and Apoptosis Signaling Pathways in MMTV-TGF-α Mice. Breast Cancer Res. 2007, 9, R91. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Howe, L.R.; Bhardwaj, P.; Du, B.; Gravaghi, C.; Yantiss, R.K.; Zhou, X.K.; Blaho, V.A.; Hla, T.; Yang, P. Obesity Is Associated with Inflammation and Elevated Aromatase Expression in the Mouse Mammary Gland. Cancer Prev. Res. 2011, 4, 329–346. [Google Scholar] [CrossRef]
- Cleary, M.P.; Phillips, F.C.; Getzin, S.C.; Jacobson, T.L.; Jacobson, M.K.; Christensen, T.A.; Juneja, S.C.; Grande, J.P.; Maihle, N.J. Genetically Obese MMTV-TGF-α/Lep Ob Lep Ob Female Mice Do Not Develop Mammary Tumors. Breast Cancer Res. Treat. 2003, 77, 205–215. [Google Scholar] [CrossRef]
- Zheng, Q.; Dunlap, S.M.; Zhu, J.; Downs-Kelly, E.; Rich, J.; Hursting, S.D.; Berger, N.A.; Reizes, O. Leptin Deficiency Suppresses MMTV-Wnt-1 Mammary Tumor Growth in Obese Mice and Abrogates Tumor Initiating Cell Survival. Endocr.-Relat. Cancer 2011, 18, 491–503. [Google Scholar] [CrossRef]
- Rossi, E.L.; De Angel, R.E.; Bowers, L.W.; Khatib, S.A.; Smith, L.A.; Van Buren, E.; Bhardwaj, P.; Giri, D.; Estecio, M.R.; Troester, M.A. Obesity-Associated Alterations in Inflammation, Epigenetics, and Mammary Tumor Growth Persist in Formerly Obese Mice. Cancer Prev. Res. 2016, 9, 339–348. [Google Scholar]
- Swami, S.; Krishnan, A.V.; Williams, J.; Aggarwal, A.; Albertelli, M.A.; Horst, R.L.; Feldman, B.J.; Feldman, D. Vitamin D Mitigates the Adverse Effects of Obesity on Breast Cancer in Mice. Endocr.-Relat. Cancer 2016, 23, 251. [Google Scholar] [CrossRef] [PubMed]
- Incio, J.; Ligibel, J.A.; McManus, D.T.; Suboj, P.; Jung, K.; Kawaguchi, K.; Pinter, M.; Babykutty, S.; Chin, S.M.; Vardam, T.D. Obesity Promotes Resistance to Anti-VEGF Therapy in Breast Cancer by up-Regulating IL-6 and Potentially FGF-2. Sci. Transl. Med. 2018, 10, eaag0945. [Google Scholar] [CrossRef]
- Kolb, R.; Kluz, P.; Tan, Z.W.; Borcherding, N.; Bormann, N.; Vishwakarma, A.; Balcziak, L.; Zhu, P.; Davies, B.S.; Gourronc, F. Obesity-Associated Inflammation Promotes Angiogenesis and Breast Cancer via Angiopoietin-like 4. Oncogene 2019, 38, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Lee, Y.S.; Mayoral, R.; Oh, D.Y.; Siu, J.T.; Webster, N.J.; Sears, D.D.; Olefsky, J.M.; Ellies, L.G. Omega-3 fatty acids reduce obesity-induced tumor progression independent of GPR120 in a mouse model of postmenopausal breast cancer. Oncogene 2015, 34, 3504–3513. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.L.; Khatib, S.A.; Doerstling, S.S.; Bowers, L.W.; Pruski, M.; Ford, N.A.; Glickman, R.D.; Niu, M.; Yang, P.; Cui, Z. Resveratrol Inhibits Obesity-associated Adipose Tissue Dysfunction and Tumor Growth in a Mouse Model of Postmenopausal Claudin-low Breast Cancer. Mol. Carcinog. 2018, 57, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R. Paradoxical Effects of Obesity on T Cell Function during Tumor Progression and PD-1 Checkpoint Blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Du, B.; Zhou, X.K.; Sue, E.; Harbus, M.D.; Falcone, D.J.; Giri, D.; Hudis, C.A.; Kopelovich, L.; Subbaramaiah, K. Caloric Restriction Reverses Obesity-Induced Mammary Gland Inflammation in Mice. Cancer Prev. Res. 2013, 6, 282–289. [Google Scholar]
- Zhu, Z.; Haegele, A.; Thompson, H. Effect of Caloric Restriction on Pre-Malignant and Malignant Stages of Mammary Carcinogenesis. Carcinogenesis 1997, 18, 1007–1012. [Google Scholar] [CrossRef][Green Version]
- Padovani, M.; Lavigne, J.A.; Chandramouli, G.V.; Perkins, S.N.; Barrett, J.C.; Hursting, S.D.; Bennett, L.M.; Berrigan, D. Distinct Effects of Calorie Restriction and Exercise on Mammary Gland Gene Expression in C57BL/6 Mice. Cancer Prev. Res. 2009, 2, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Aalders, K.C.; Tryfonidis, K.; Senkus, E.; Cardoso, F. Anti-Angiogenic Treatment in Breast Cancer: Facts, Successes, Failures and Future Perspectives. Cancer Treat. Rev. 2017, 53, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, S.; Reggiani, F.; Talarico, G.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Noonan, D.M.; Dallaglio, K.; Albini, A. The Biguanides Metformin and Phenformin Inhibit Angiogenesis, Local and Metastatic Growth of Breast Cancer by Targeting Both Neoplastic and Microenvironment Cells. Int. J. Cancer 2015, 136, E534–E544. [Google Scholar] [CrossRef] [PubMed]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M. Paradoxic Effects of Metformin on Endothelial Cells and Angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Giles, E.D.; Jindal, S.; Wellberg, E.A.; Schedin, T.; Anderson, S.M.; Thor, A.D.; Edwards, D.P.; MacLean, P.S.; Schedin, P. Metformin Inhibits Stromal Aromatase Expression and Tumor Progression in a Rodent Model of Postmenopausal Breast Cancer. Breast Cancer Res. 2018, 20, 50. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Dale, D.C.; Crawford, J. Incidence and Predictors of Low Dose-Intensity in Adjuvant Breast Cancer Chemotherapy: A Nationwide Study of Community Practices. J. Clin. Oncol. 2003, 21, 4524–4531. [Google Scholar] [CrossRef]
- Sheng, X.; Parmentier, J.-H.; Tucci, J.; Pei, H.; Cortez-Toledo, O.; Dieli-Conwright, C.M.; Oberley, M.J.; Neely, M.; Orgel, E.; Louie, S.G. Adipocytes Sequester and Metabolize the Chemotherapeutic Daunorubicin. Mol. Cancer Res. 2017, 15, 1704–1713. [Google Scholar]
- Liu, Z.; Shi, A.; Song, D.; Han, B.; Zhang, Z.; Ma, L.; Liu, D.; Fan, Z. Resistin Confers Resistance to Doxorubicin-Induced Apoptosis in Human Breast Cancer Cells through Autophagy Induction. Am. J. Cancer Res. 2017, 7, 574. [Google Scholar]
- Lehuédé, C.; Li, X.; Dauvillier, S.; Vaysse, C.; Franchet, C.; Clement, E.; Esteve, D.; Longué, M.; Chaltiel, L.; Le Gonidec, S. Adipocytes Promote Breast Cancer Resistance to Chemotherapy, a Process Amplified by Obesity: Role of the Major Vault Protein (MVP). Breast Cancer Res. 2019, 21, 1–17. [Google Scholar] [CrossRef]
- Duong, M.N.; Cleret, A.; Matera, E.-L.; Chettab, K.; Mathé, D.; Valsesia-Wittmann, S.; Clémenceau, B.; Dumontet, C. Adipose Cells Promote Resistance of Breast Cancer Cells to Trastuzumab-Mediated Antibody-Dependent Cellular Cytotoxicity. Breast Cancer Res. 2015, 17, 1–14. [Google Scholar] [CrossRef]


{kind=link}
| Mouse Model | Immuno-Competent | BC Subtype | Obesity Model | Menopausal State | Reference |
|---|---|---|---|---|---|
| FVB/N | Yes | MMTV-ErbB2+ (Her2+) | DIO | pre | [30] |
| BALB/c | Yes | 4T1 (ER−, PR−, Her2−) | DIO | pre | [30] |
| BALB/c | Yes | 4T1 (ER−, PR−, Her2−) | DIO | post | [46] |
| BALB/c Nude | No | MCF-7 (ER+) | DIO | pre | [47] |
| RAG-1 | No | MDA-MB-231 (ER−, PR−, Her2−) | DIO | pre | [64] |
| C57BL/6 | Yes | MMTV-PyMT (99LN, 86R2 Her2+) | DIO | pre | [91] |
| C3H | Yes | Spontaneous | Chemical induced (thioglucose) | post | [140,141] |
| C57BL/6 | Yes | MMTV-Wnt1 (basal-like, ER−, PR−, Her2−) | DIO | post | [143] |
| FVB/N | Yes | MMTV-rtTA;TetO-HER2/neu (Her2+) | DIO | pre | [144] |
| C57BL/6 | Yes | E0771 (luminal B, ER+, Her2+, PR+); Py230 (luminal, ER+/−, PR+/−) | DIO | post | [147] |
| FVB/N | Yes | MMTV-PyMT (Her2+) | DIO | post | [147] |
| C57BL/6 | Yes | MMTV-PyMT (Her2+) | DIO | pre and post | [148] |
| C57BL/6 | Yes | MMTV-TGF-α (ER+) | DIO | post | [149,150] |
| C57BL/6 | Yes | MMTV-TGF-α (ER+) | Genetic (Lepob/ob) | pre | [152] |
| C57BL/6J | Yes | MMTV-Wnt1 (basal-like, ER−, PR−, Her2−) | Genetic (Lepob/ob; Lepdb/db) | pre | [153] |
| C57BL/6 | Yes | MMTV-Wnt1 (basal-like, ER−, PR−, Her2−) | DIO | pre | [154] |
| C57BL/6 | Yes | MMTV-Wnt1 (basal-like, ER−, PR−, Her2−) | DIO | post | [155] |
| C3H; C57BL/6 | Yes | E0771 (luminal B, ER+, Her2+, PR+) | DIO | pre | [156] |
| C57BL/6N | Yes | Py8119 (ER−, PR−, Her2−); E0771 (luminal B, ER+, Her2+, PR+) | DIO | pre | [157] |
| C57BL/6J | Yes | Py230 (luminal, ER+/−, PR+/−) | DIO and genetic (Lepob/ob) | post | [158] |
| C57BL/6 | Yes | MMTV-Wnt1 (basal-like, ER-, PR−, Her2−) | DIO | post | [159] |
| BALB/c | Yes | 4T1 (ER−, PR−, Her2−) | DIO | pre | [160] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reggiani, F.; Falvo, P.; Bertolini, F. Cellular and Molecular Players in the Interplay between Adipose Tissue and Breast Cancer. Int. J. Mol. Sci. 2021, 22, 1359. https://doi.org/10.3390/ijms22031359
Reggiani F, Falvo P, Bertolini F. Cellular and Molecular Players in the Interplay between Adipose Tissue and Breast Cancer. International Journal of Molecular Sciences. 2021; 22(3):1359. https://doi.org/10.3390/ijms22031359
Chicago/Turabian StyleReggiani, Francesca, Paolo Falvo, and Francesco Bertolini. 2021. "Cellular and Molecular Players in the Interplay between Adipose Tissue and Breast Cancer" International Journal of Molecular Sciences 22, no. 3: 1359. https://doi.org/10.3390/ijms22031359
APA StyleReggiani, F., Falvo, P., & Bertolini, F. (2021). Cellular and Molecular Players in the Interplay between Adipose Tissue and Breast Cancer. International Journal of Molecular Sciences, 22(3), 1359. https://doi.org/10.3390/ijms22031359