Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements

 ,
,  and
and 
Abstract
1. Introduction
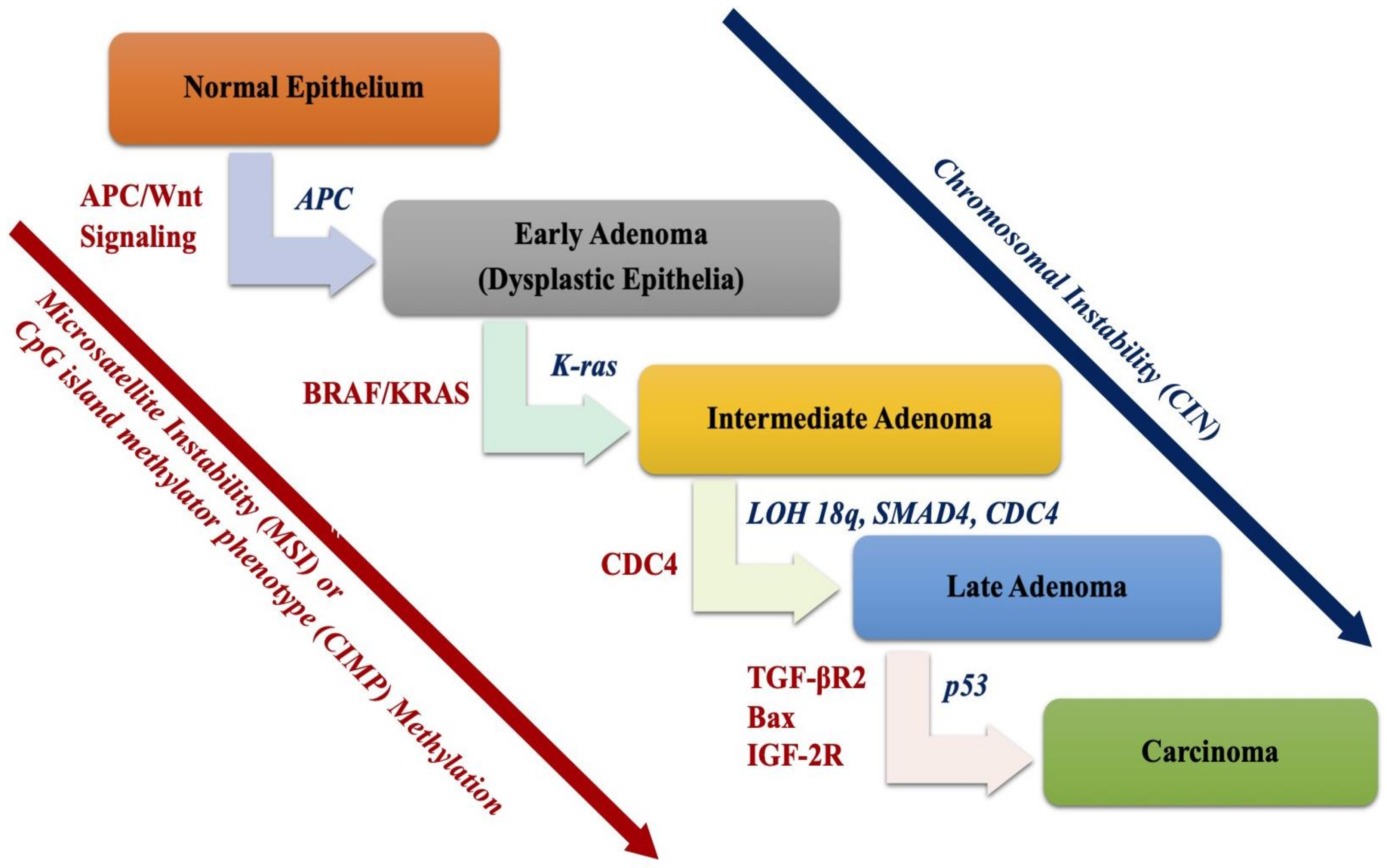
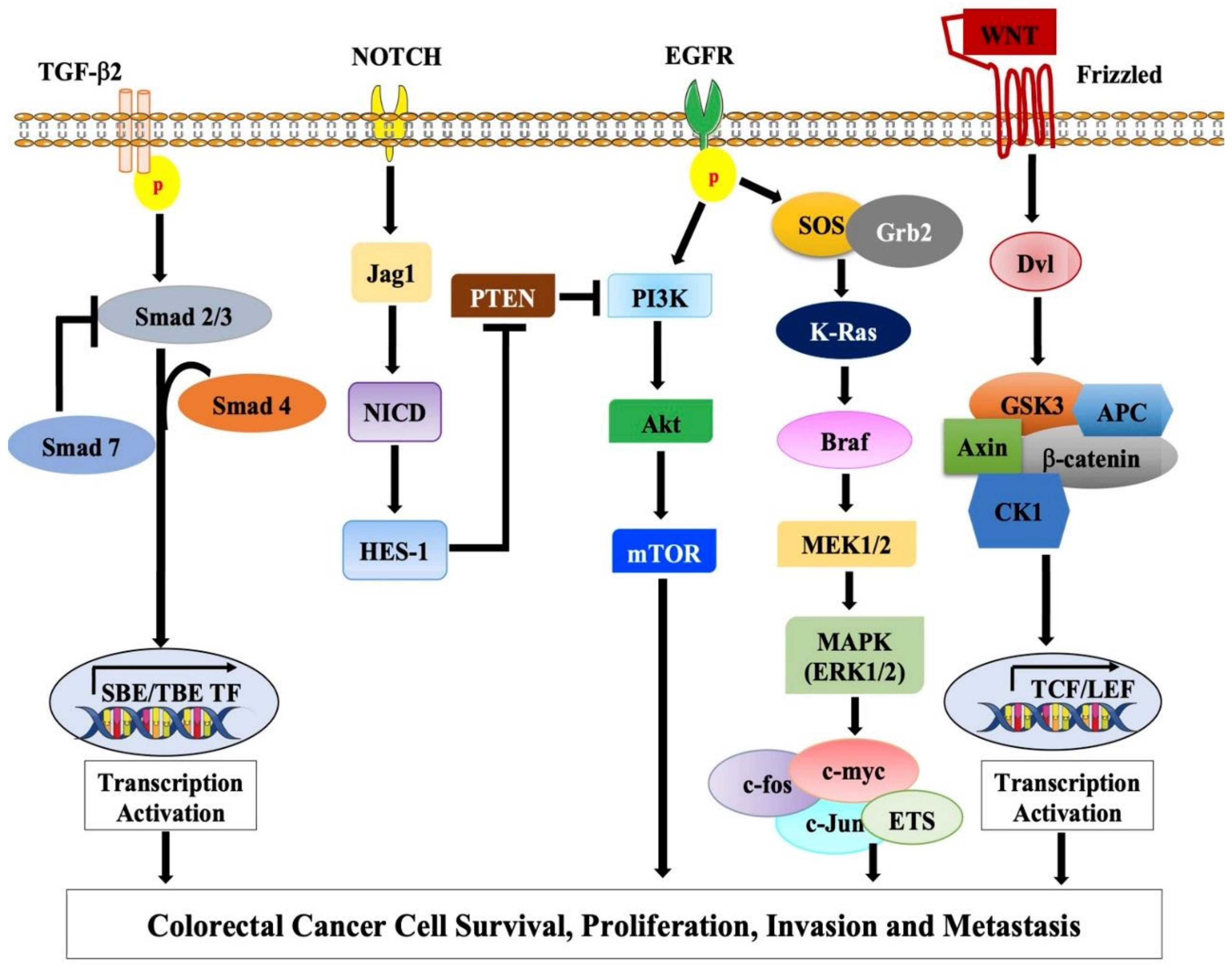
2. Molecular Basis of Colorectal Cancer
2.1. Chromosomal Instability (CIN) Pathway
2.1.1. Adenomatous Polyposis Coli (APC) Gene and Wnt Signaling Pathway
2.1.2. TP53 Pathway
2.1.3. The 18q Loss of Heterozygosity (LOH)
2.2. The Microsatellite Instability Pathway
2.3. CpG Island Methylator Phenotype (CIMP) Pathway
EGFR-KRAS-BRAF Pathway
3. Therapeutic Strategies
3.1. Targeting CIN Pathway
3.2. Targeting MSI Pathway
3.3. Targeting CIMP Pathway
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-FU | Fluorouracil |
| ACVR2 | Activin receptor type 2 |
| AFAP | Attenuated familial adenomatous polyposis |
| ALP | Alpelisib |
| AMPK | Adenosine monophosphate-activated protein kinase |
| APC | Adenomatous polyposis coli |
| BRAF | Serine/threonine-protein kinase B-Raf |
| Bub | Budding uninhibited by benzimidazoles |
| CDC20 | Cell division cycle 20 |
| CDKN | Cyclin-dependent kinases |
| CIMP | CpG island methylator phenotype |
| CIN | Chromosomal instability |
| CMS | Consensus molecular subtypes |
| COX | Cyclooxygenase |
| CRC | Colorectal cancer |
| CTNNB1 | Catenin beta-1 |
| DCC | Deleted in colorectal cancer |
| dMMR | Defective mismatch repair |
| EDM | Exonuclease domain |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal related kinase |
| FAP | Familial adenomatous polyposis |
| FDA | Food and Drug Administration |
| FOLFOX | Folinic acid, fluorouracil, and oxaliplatin |
| GSH | Glutathione |
| GSK3-β | Glycogen synthase kinase 3-β |
| HNPCC | Hereditary non-polyposis colorectal cancer |
| IBD | Inflammatory bowel disease; Ig: Immunoglobin |
| IGFBP7 | Insulin-like growth factor-binding protein 7 |
| JAK | Janus kinase |
| KRAS | Kirsten ras |
| LOH | Loss of heterozygosity |
| LV | Leucovorin |
| MAP | MUTYH-associated polyposis |
| MAP/MEK | Mitogen-activated protein kinase |
| MLH/MSH | MutL homolog |
| MSI | Microsatellite instability |
| MSS | Microsatellite stable |
| NF-κB | Nuclear factor kappa |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| OS | Overall survival |
| PD-1/PD-L1 | Programmed death ligand-1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PI3KCA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha |
| POLE/POLD1 | Polymerase epsilon or delta 1 |
| RAC1 | Rac family small GTPase 1 |
| ROS | Reactive oxygen species |
| SCNAs | Somatic copy number alterations |
| SMAD | Small mothers against decapentaplegic |
| STAT | Signal transducer and activator of transcription |
| TCGA | The Cancer Genome Atlas |
| TGF-β | Transforming growth factor-β |
| TIGAR | T53-induced glycolysis regulatory phosphatase |
| TNM | Tumor-nodes-metastasis |
| TP53 | Tumor protein 53 |
| TSG | Tumor suppressor gene |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| Wnt | Wingless |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- O’Connell, J.B.; Maggard, M.A.; Ko, C.Y. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J. Natl. Cancer Inst. 2004, 96, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9–14. [Google Scholar] [PubMed]
- Le Marchand, L.; Wilkens, L.R.; Hankin, J.H.; Kolonel, L.N.; Lyu, L.-C. A case-control study of diet and colorectal cancer in a multiethnic population in Hawaii (United States): Lipids and foods of animal origin. Cancer Causes Control 1997, 8, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Wirtzfeld, D.A.; Petrelli, N.J.; Rodriguez-Bigas, M.A. Hamartomatous polyposis syndromes: Molecular genetics, neoplastic risk, and surveillance recommendations. Ann. Surg. Oncol. 2001, 8, 319–327. [Google Scholar] [CrossRef]
- Xie, J.; Itzkowitz, S.H. Cancer in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 378–389. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Grady, W.M. Epigenetic events in the colorectum and in colon cancer. Biochem. Soc. Trans. 2005, 33, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Markowitz, S.D. Genetic and Epigenetic Alterations in Colon Cancer. Ann. Rev. Genom. Hum. Genet. 2002, 3, 101. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.K.; Chang, S.C.; Yang, Y.C.; Li, A.F. Loss of heterozygosity and DNA aneuploidy in colorectal adenocarcinoma. Ann. Surg. Oncol. 2003, 10, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Leary, R.J.; Lin, J.C.; Cummins, J.; Boca, S.; Wood, L.D.; Parsons, D.W.; Jones, S.; Sjöblom, T.; Park, B.H.; Parsons, R.; et al. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 16224–16229. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Tsang, A.H.; Cheng, K.H.; Wong, A.S.; Ng, S.S.; Ma, B.B.; Chan, C.M.; Tsui, N.B.; Chan, L.W.; Yung, B.Y.; Wong, S.C. Current and future molecular diagnostics in colorectal cancer and colorectal adenoma. World J. Gastroenterol. 2014, 20, 3847–3857. [Google Scholar] [CrossRef]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; van Tuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Thiagalingam, S.; Lengauer, C.; Leach, F.S.; Schutte, M.; Hahn, S.A.; Overhauser, J.; Willson, J.K.; Markowitz, S.; Hamilton, S.R.; Kern, S.E.; et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat. Genet. 1996, 13, 343–346. [Google Scholar] [CrossRef]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Diep, C.B.; Kleivi, K.; Ribeiro, F.R.; Teixeira, M.R.; Lindgjaerde, O.C.; Lothe, R.A. The order of genetic events associated with colorectal cancer progression inferred from meta-analysis of copy number changes. Genes Chromosom. Cancer 2006, 45, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Jasmine, F.; Rahaman, R.; Dodsworth, C.; Roy, S.; Paul, R.; Raza, M.; Paul-Brutus, R.; Kamal, M.; Ahsan, H.; Kibriya, M.G. A genome-wide study of cytogenetic changes in colorectal cancer using SNP microarrays: Opportunities for future personalized treatment. PLoS ONE 2012, 7, e31968. [Google Scholar] [CrossRef] [PubMed]
- Baudis, M. Genomic imbalances in 5918 malignant epithelial tumors: An explorative meta-analysis of chromosomal CGH data. BMC Cancer 2007, 7, 226. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Béroud, C.; Soussi, T. APC gene: Database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1996, 24, 121–124. [Google Scholar] [CrossRef]
- Aoki, K.; Taketo, M.M. Adenomatous polyposis coli (APC): A multi-functional tumor suppressor gene. J. Cell Sci. 2007, 120, 3327–3335. [Google Scholar] [CrossRef]
- Kaplan, K.B.; Burds, A.A.; Swedlow, J.R.; Bekir, S.S.; Sorger, P.K.; Näthke, I.S. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat. Cell Biol. 2001, 3, 429–432. [Google Scholar] [CrossRef]
- Näthke, I.S.; Adams, C.L.; Polakis, P.; Sellin, J.H.; Nelson, W.J. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J. Cell Biol. 1996, 134, 165–179. [Google Scholar] [CrossRef]
- Browne, S.J.; MacFarlane, M.; Cohen, G.M.; Paraskeva, C. The adenomatous polyposis coli protein and retinoblastoma protein are cleaved early in apoptosis and are potential substrates for caspases. Cell Death Differ. 1998, 5, 206–213. [Google Scholar] [CrossRef]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Baeg, G.H.; Matsumine, A.; Kuroda, T.; Bhattacharjee, R.N.; Miyashiro, I.; Toyoshima, K.; Akiyama, T. The tumour suppressor gene product APC blocks cell cycle progression from G0/G1 to S phase. EMBO J. 1995, 14, 5618–5625. [Google Scholar] [CrossRef] [PubMed]
- Grossi, V.; Fasano, C.; Celestini, V.; Lepore Signorile, M.; Sanese, P.; Simone, C. Chasing the FOXO3: Insights into Its New Mitochondrial Lair in Colorectal Cancer Landscape. Cancers 2019, 11, 414. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, E.; Hiraga, N.; Imamura, M.; Uchida, T.; Kan, H.; Tsuge, M.; Abe-Chayama, H.; Hayes, C.N.; Makokha, G.N.; Serikawa, M.; et al. Interferon alpha treatment stimulates interferon gamma expression in type I NKT cells and enhances their antiviral effect against hepatitis C virus. PLoS ONE 2017, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Franovic, A.; Holterman, C.E.; Payette, J.; Lee, S. Human cancers converge at the HIF-2α oncogenic axis. Proc. Natl. Acad. Sci. USA 2009, 106, 21306–21311. [Google Scholar] [CrossRef] [PubMed]
- Chittenden, T.W.; Howe, E.A.; Culhane, A.C.; Sultana, R.; Taylor, J.M.; Holmes, C.; Quackenbush, J. Functional classification analysis of somatically mutated genes in human breast and colorectal cancers. Genomics 2008, 91, 508–511. [Google Scholar] [CrossRef][Green Version]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Bonk, T.; Humeny, A.; Sutter, C.; Gebert, J.; von Knebel Doeberitz, M.; Becker, C.-M. Molecular diagnosis of familial adenomatous polyposis (FAP): Genotyping of adenomatous polyposis coli (APC) alleles by MALDI-TOF mass spectrometry. Clin. Biochem. 2002, 35, 87. [Google Scholar] [CrossRef]
- Powell, S.M.; Petersen, G.M.; Krush, A.J.; Booker, S.; Jen, J.; Giardiello, F.M.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Molecular Diagnosis of Familial Adenomatous Polyposis. N. Engl. J. Med. 1993, 329, 1982–1987. [Google Scholar] [CrossRef]
- Ishiguro, K.; Yoshida, T.; Yagishita, H.; Numata, Y.; Okayasu, T. Epithelial and stromal genetic instability contributes to genesis of colorectal adenomas. Gut 2006, 55, 695–702. [Google Scholar] [CrossRef]
- Donger, Z.; Liu, Y.; Liangtao, Z.; Weiting, G.; Dan, L.; Yong, Z.; Xueda, H.; Zhibo, G.; Jinghong, X.; Yanqin, H.; et al. Exome Capture Sequencing of Adenoma Reveals Genetic Alterations in Multiple Cellular Pathways at the Early Stage of Colorectal Tumorigenesis. PLoS ONE 2013, 8, 1–8. [Google Scholar] [CrossRef]
- Cheng, T.H.T.; Gorman, M.; Martin, L.; Barclay, E.; Casey, G.; Saunders, B.; Thomas, H.; Clark, S.; Tomlinson, I. Common colorectal cancer risk alleles contribute to the multiple colorectal adenoma phenotype, but do not influence colonic polyposis in FAP. Eur. J. Hum. Genet. 2015, 23, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Vaqué, J.P.; Martínez, N.; Varela, I.; Fernández, F.; Mayorga, M.; Derdak, S.; Beltrán, S.; Moreno, T.; Almaraz, C.; De las Heras, G.; et al. Colorectal Adenomas Contain Multiple Somatic Mutations That Do Not Coincide with Synchronous Adenocarcinoma Specimens. PLoS ONE 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar] [PubMed]
- Sieber, O.M.; Lipton, L.; Crabtree, M.; Heinimann, K.; Fidalgo, P.; Phillips, R.K.S.; Bisgaard, M.-L.; Orntoft, T.F.; Aaltonen, L.A.; Hodgson, S.V.; et al. Multiple Colorectal Adenomas, Classic Adenomatous Polyposis, and Germ-Line Mutations in MYH. N. Engl. J. Med. 2003, 348, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef]
- Rowan, A.J.; Lamlum, H.; Ilyas, M.; Wheeler, J.; Straub, J.; Papadopoulou, A.; Bicknell, D.; Bodmer, W.F.; Tomlinson, I.P. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. USA 2000, 97, 3352–3357. [Google Scholar] [CrossRef]
- Cottrell, S.; Bicknell, D.; Kaklamanis, L.; Bodmer, W.F. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet 1992, 340, 626–630. [Google Scholar] [CrossRef]
- Christie, M.; Jorissen, R.N.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Day, F.; Tsui, C.; Lipton, L.; Desai, J.; Jones, I.T.; et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/β-catenin signalling thresholds for tumourigenesis. Oncogene 2013, 32, 4675–4682. [Google Scholar] [CrossRef]
- Miller, J.R.; Moon, R.T. Signal transduction through beta-catenin and specification of cell fate during embryogenesis. Genes Dev. 1996, 10, 2527–2539. [Google Scholar] [CrossRef]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Albert, I.; Porfiri, E.; Fiol, C.; Munemitsu, S.; Polakis, P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 1996, 272, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Pronobis, M.I.; Rusan, N.M.; Peifer, M. A novel GSK3-regulated APC:Axin interaction regulates Wnt signaling by driving a catalytic cycle of efficient βcatenin destruction. Elife 2015, 4, e08022. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Sekiya, T.; Adachi, S.; Kohu, K.; Yamada, T.; Higuchi, O.; Furukawa, Y.; Nakamura, Y.; Nakamura, T.; Tashiro, K.; Kuhara, S.; et al. Identification of BMP and Activin Membrane-bound Inhibitor (BAMBI), an Inhibitor of Transforming Growth Factor-β Signaling, as a Target of the β-Catenin Pathway in Colorectal Tumor Cells. J. Biol. Chem. 2004, 279, 6840–6846. [Google Scholar] [CrossRef]
- He, L.; Lu, N.; Dai, Q.; Zhao, Y.; Zhao, L.; Wang, H.; Li, Z.; You, Q.; Guo, Q. Wogonin induced G1 cell cycle arrest by regulating Wnt/β-catenin signaling pathway and inactivating CDK8 in human colorectal cancer carcinoma cells. Toxicology 2013, 312, 36–47. [Google Scholar] [CrossRef]
- Bolanos-Garcia, V.M.; Blundell, T.L. BUB1 and BUBR1: Multifaceted kinases of the cell cycle. Trends Biochem. Sci. 2011, 36, 141–150. [Google Scholar] [CrossRef]
- Tasaki, T.; Sohr, R.; Zanxian, X.; Heliweg, R.; Hörtnagi, H.; Varshavsky, A.; Yong Tae, K. Biochemical and Genetic Studies of UBR3, a Ubiquitin Ligase with a Function in Olfactory and Other Sensory Systems. J. Biol. Chem. 2007, 282, 18510–18520. [Google Scholar] [CrossRef]
- Nilsson, J.; Yekezare, M.; Minshull, J.; Pines, J. The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat. Cell Biol. 2008, 10, 1411–1420. [Google Scholar] [CrossRef]
- Shin, H.-J.; Baek, K.-H.; Jeon, A.-H.; Park, M.-T.; Lee, S.-J.; Kang, C.-M.; Lee, H.-S.; Yoo, S.-H.; Chung, D.-H.; Sung, Y.-C.; et al. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell 2003, 4, 483. [Google Scholar] [CrossRef]
- Takayama, O.; Yamamoto, H.; Damdinsuren, B.; Sugita, Y.; Ngan, C.Y.; Xu, X.; Tsujino, T.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; et al. Expression of PPARδ in multistage carcinogenesis of the colorectum: Implications of malignant cancer morphology. Br. J. Cancer 2006, 95, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; O’Dwyer, S.T.; Haboubi, N.J.; Potten, C.S. Early cellular events in colorectal carcinogenesis. Colorectal Dis. 2002, 4, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Luebeck, E.G.; Moolgavkar, S.H. Multistage carcinogenesis and the incidence of colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 15095. [Google Scholar] [CrossRef] [PubMed]
- An, N.; Zhao, C.; Yu, Z.; Yang, X. Identification of prognostic genes in colorectal cancer through transcription profiling of multi-stage carcinogenesis. Oncol. Lett. 2019, 17, 432–441. [Google Scholar] [CrossRef]
- Tetsu, O.; McCormick, F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398, 422–426. [Google Scholar] [CrossRef]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Soohyun, P.; Jie, C.; Wangsheng, Y.; Ling, W.; Carmon, K.S.; Qingyun, J.L. Differential activities and mechanisms of the four R-spondins in potentiating Wnt/β-catenin signaling. J. Biol. Chem. 2018, 293, 9759–9769. [Google Scholar] [CrossRef]
- Heinen, C.D.; Goss, K.H.; Cornelius, J.R.; Babcock, G.F.; Knudsen, E.S.; Kowalik, T.; Groden, J. The APC tumor suppressor controls entry into S-phase through its ability to regulate the cyclin D/RB pathway. Gastroenterology 2002, 123, 751–763. [Google Scholar] [CrossRef]
- Arber, N.; Hibshoosh, H.; Moss, S.F.; Sutter, T.; Zhang, Y.; Begg, M.; Wang, S.; Weinstein, I.B.; Holt, P.R. Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology 1996, 110, 669–674. [Google Scholar] [CrossRef]
- Van Es, J.H.; van Gijn, M.E.; Riccio, O.; van den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. Notch/γ-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef]
- Hai, Z.; Pritchard, D.M.; Xiangdong, Y.; Bennett, E.; Gang, L.; Chunming, L.; Ai, W. KLF4 gene expression is inhibited by the notch signaling pathway that controls goblet cell differentiation in mouse gastrointestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G490–G498. [Google Scholar] [CrossRef][Green Version]
- Kwon, C.; Cheng, P.; King, I.N.; Andersen, P.; Shenje, L.; Nigam, V.; Srivastava, D. Notch post-translationally regulates ?-catenin protein in stem and progenitor cells. Nat. Cell Biol. 2011, 13, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Bass, A.J.; Young, K.S.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates β-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Fryer, C.J.; White, J.B.; Jones, K.A. Mastermind Recruits CycC:CDK8 to Phosphorylate the Notch ICD and Coordinate Activation with Turnover. Mol. Cell 2004, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Smith, G.; Carey, F.A.; Beattie, J.; Wilkie, M.J.V.; Lightfoot, T.J.; Coxhead, J.; Garner, R.C.; Steele, R.J.C.; Wolf, C.R. Mutations in APC, Kirsten-ras, and p53—Alternative genetic pathways to colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 9433. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar]
- Liu, Y.; Bodmer, W.F. Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 976–981. [Google Scholar] [CrossRef]
- Oikawa, T.; Okuda, M.; Ma, Z.; Goorha, R.; Tsujimoto, H.; Inokuma, H.; Fukasawa, K. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol. Cell. Biol. 2005, 25, 4046–4061. [Google Scholar] [CrossRef]
- Colussi, D.; Brandi, G.; Bazzoli, F.; Ricciardiello, L. Molecular Pathways Involved in Colorectal Cancer: Implications for Disease Behavior and Prevention. Int. J. Mol. Sci. 2013, 14, 16365–16385. [Google Scholar] [CrossRef]
- Zhang, J.; Su, G.; Lin, Y.; Meng, W.; Lai, J.K.L.; Qiao, L.; Li, X.; Xie, X. Targeting cyclin-dependent kinases in gastrointestinal cancer therapy. Discov. Med. 2019, 27, 27–36. [Google Scholar] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Pasz-Walczak, G.; Kordek, R.; Faflik, M. P21 (WAF1) Expression in Colorectal Cancer: Correlation with P53 and Cyclin D1 Expression, Clinicopathological Parameters and Prognosis. Pathol. Res. Pract. 2001, 197, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Abukhdeir, A.M.; Park, B.H. P21 and p27: Roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Kawasaki, T.; Chan, A.T.; Ohnishi, M.; Suemoto, Y.; Kirkner, G.J.; Fuchs, C.S.; Ogino, S. Cyclin D1 is frequently overexpressed in microsatellite unstable colorectal cancer, independent of CpG island methylator phenotype. Histopathology 2008, 53, 588–598. [Google Scholar] [CrossRef]
- Ogino, S.; Brahmandam, M.; Kawasaki, T.; Kirkner, G.J.; Loda, M.; Fuchs, C.S. Combined Analysis of COX-2 and p53 Expressions Reveals Synergistic Inverse Correlations with Microsatellite Instability and CpG Island Methylator Phenotype in Colorectal Cancer. Neoplasia 2006, 8, 458–464. [Google Scholar] [CrossRef][Green Version]
- Venkatesan, P.; Bhutia, S.K.; Singh, A.K.; Das, S.K.; Dash, R.; Chaudhury, K.; Sarkar, D.; Fisher, P.B.; Mandal, M. AEE788 potentiates celecoxib-induced growth inhibition and apoptosis in human colon cancer cells. Life Sci. 2012, 91, 789–799. [Google Scholar] [CrossRef]
- Park, G.-B.; Jin, D.-H.; Kim, D. Sequential treatment with celecoxib and bortezomib enhances the ER stress-mediated autophagy-associated cell death of colon cancer cells. Oncol. Lett. 2018, 16, 4526–4536. [Google Scholar] [CrossRef]
- Schwarzenbach, H. Loss of Heterozygosity. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 271–273. [Google Scholar] [CrossRef]
- Sheffer, M.; Bacolod, M.D.; Zuk, O.; Giardina, S.F.; Pincas, H.; Barany, F.; Paty, P.B.; Gerald, W.L.; Notterman, D.A.; Domany, E. Association of survival and disease progression with chromosomal instability: A genomic exploration of colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 7131–7136. [Google Scholar] [CrossRef]
- Jen, J.; Kim, H.; Piantadosi, S.; Liu, Z.F.; Levitt, R.C.; Sistonen, P.; Kinzler, K.W.; Vogelstein, B.; Hamilton, S.R. Allelic loss of chromosome 18q and prognosis in colorectal cancer. N. Engl. J. Med. 1994, 331, 213–221. [Google Scholar] [CrossRef]
- Fearon, E.R.; Cho, K.R.; Nigro, J.M.; Kern, S.E.; Simons, J.W.; Ruppert, J.M.; Hamilton, S.R.; Preisinger, A.C.; Thomas, G.; Kinzler, K.W.; et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 1990, 247, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Fearon, E.R. Role of the dependence receptor DCC in colorectal cancer pathogenesis. J. Clin. Oncol. 2004, 22, 3420–3428. [Google Scholar] [CrossRef] [PubMed]
- Mazelin, L.; Bernet, A.; Bonod-Bidaud, C.; Pays, L.; Arnaud, S.; Gespach, C.; Bredesen, D.E.; Scoazec, J.Y.; Mehlen, P. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature 2004, 431, 80–84. [Google Scholar] [CrossRef]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (SMAD4) and Apc genes. Cell 1998, 92, 645. [Google Scholar] [CrossRef]
- Ritterhouse, L.L.; Wu, E.Y.; Kim, W.G.; Dillon, D.A.; Hirsch, M.S.; Sholl, L.M.; Agoston, A.T.; Setia, N.; Lauwers, G.Y.; Park, D.Y.; et al. Loss of SMAD4 protein expression in gastrointestinal and extra-gastrointestinal carcinomas. Histopathology 2019, 75, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hata, A.; Lo, R.S.; Massagué, J.; Pavletich, N.P. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 1997, 388, 87–93. [Google Scholar] [CrossRef]
- Eppert, K.; Scherer, S.W. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally. Cell 1996, 86, 544. [Google Scholar] [CrossRef]
- Grady, D. How to Halve the Death Rate from Colon Cancer. New York Times 2007, 156, H5. [Google Scholar]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Geiersbach, K.B.; Samowitz, W.S. Microsatellite instability and colorectal cancer. Arch. Pathol. Lab. Med. 2011, 135, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nosho, K.; Irahara, N.; Shima, K.; Baba, Y.; Kirkner, G.J.; Meyerhardt, J.A.; Fuchs, C.S. Prognostic significance and molecular associations of 18q loss of heterozygosity: A cohort study of microsatellite stable colorectal cancers. J. Clin. Oncol. 2009, 27, 4591–4598. [Google Scholar] [CrossRef] [PubMed]
- Perucho, M. Cancer of the microsatellite mutator phenotype. Biol. Chem. 1996, 377, 675–684. [Google Scholar]
- Mori, Y.; Yin, J.; Rashid, A.; Leggett, B.A.; Young, J.; Simms, L.; Kuehl, P.M.; Langenberg, P.; Meltzer, S.J.; Stine, O.C. Instabilotyping: Comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res. 2001, 61, 6046–6049. [Google Scholar]
- Parsons, R.; Myeroff, L.L.; Liu, B.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1995, 55, 5548–5550. [Google Scholar]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Velho, S.; Moutinho, C.; Cirnes, L.; Albuquerque, C.; Hamelin, R.; Schmitt, F.; Carneiro, F.; Oliveira, C.; Seruca, R. BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: Primary or secondary genetic events in colorectal carcinogenesis? BMC Cancer 2008, 8, 1–6. [Google Scholar] [CrossRef]
- Kim, J.H.; Rhee, Y.Y.; Kim, K.J.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Annexin A10 expression correlates with serrated pathway features in colorectal carcinoma with microsatellite instability. APMIS 2014, 122, 1187–1195. [Google Scholar] [CrossRef]
- Meyer, L.A.; Broaddus, R.R.; Lu, K.H. Endometrial cancer and Lynch syndrome: Clinical and pathologic considerations. Cancer Control 2009, 16, 14–22. [Google Scholar] [CrossRef]
- Kaiser, J.C.; Meckbach, R.; Jacob, P. Genomic Instability and Radiation Risk in Molecular Pathways to Colon Cancer. PLoS ONE 2014, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huaizeng, C.; Dafeng, Y.; Xing, X.; Weiguo, L.; Changkun, Z.; Xiaodong, C. Mismatch repair gene promoter methylation and expression in hydatidiform moles. Arch. Gynecol. Obstet. 2005, 272, 35–39. [Google Scholar] [CrossRef]
- Manes, M.; Garcia-Gomes, M.d.S.A.; Sandini, T.M.; Zaccarelli-Magalhães, J.; Florio, J.C.; Alexandre-Ribeiro, S.R.; Wadt, D.; Bernardi, M.M.; Massironi, S.M.G.; Mori, C.M.C. Behavioral and neurochemical characterization of the mlh mutant mice lacking otoconia. Behav. Brain Res. 2019, 359, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Takehara, Y.; Nagasaka, T.; Nyuya, A.; Haruma, T.; Haraga, J.; Mori, Y.; Nakamura, K.; Fujiwara, T.; Boland, C.R.; Goel, A. Accuracy of four mononucleotide-repeat markers for the identification of DNA mismatch-repair deficiency in solid tumors. J. Transl. Med. 2018, 16, 1-N.PAG. [Google Scholar] [CrossRef]
- O’Brien, M.J.; Yang, S.; Mack, C.; Xu, H.; Huang, C.S.; Mulcahy, E.; Amorosino, M.; Farraye, F.A. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am. J. Surg. Pathol. 2006, 30, 1491–1501. [Google Scholar] [CrossRef]
- Domingo, E.; Niessen, R.C.; Oliveira, C.; Alhopuro, P.; Moutinho, C.; Espín, E.; Armengol, M.; Sijmons, R.H.; Kleibeuker, J.H.; Seruca, R.; et al. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene 2005, 24, 3995–3998. [Google Scholar] [CrossRef]
- Frouws, M.A.; Reimers, M.S.; Swets, M.; Bastiaannet, E.; Prinse, B.; van Eijk, R.; Lemmens, V.E.P.P.; van Herk-Sukel, M.P.P.; van Wezel, T.; Kuppen, P.J.K.; et al. The Influence of BRAF and KRAS Mutation Status on the Association between Aspirin Use and Survival after Colon Cancer Diagnosis. PLoS ONE 2017, 12, 1–12. [Google Scholar] [CrossRef]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef]
- Wang, Y.; Loree, J.M.; Yu, C.; Tschautscher, M.; Briggler, A.M.; Overman, M.J.; Broaddus, R.; Meric-Bernstam, F.; Jones, J.C.; Balcom, J.; et al. Distinct impacts of KRAS, NRAS and BRAF mutations on survival of patients with metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 3513. [Google Scholar] [CrossRef]
- Roth, A.D.; Tejpar, S.; Delorenzi, M.; Yan, P.; Fiocca, R.; Klingbiel, D.; Dietrich, D.; Biesmans, B.; Bodoky, G.; Barone, C.; et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: Results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J. Clin. Oncol. 2010, 28, 466–474. [Google Scholar] [CrossRef]
- Manthravadi, S.; Sun, W.; Saeed, A. Prognostic impact of BRAF V600E mutation in patients with non-metastatic colorectal cancer with microsatellite instability: A systematic review and meta-analysis. J. Clin. Oncol. 2018, 36, 3597. [Google Scholar] [CrossRef]
- Tosi, F.; Magni, E.; Amatu, A.; Mauri, G.; Bencardino, K.; Truini, M.; Veronese, S.; De Carlis, L.; Ferrari, G.; Nichelatti, M.; et al. Effect of KRAS and BRAF Mutations on Survival of Metastatic Colorectal Cancer After Liver Resection: A Systematic Review and Meta-Analysis. Clin. Colorectal Cancer 2017, 16, e153–e163. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Di Bartolomeo, M.; Amatu, A.; Antoniotti, C.; Moretto, R.; Berenato, R.; Perrone, F.; Tamborini, E.; Aprile, G.; Lonardi, S.; et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann. Oncol. 2015, 26, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Tajima, Y.; Nagahashi, M.; Ichikawa, H.; Oyanagi, H.; Okuda, S.; Takabe, K.; Wakai, T. Clinical Significance of BRAF Non-V600E Mutations in Colorectal Cancer: A Retrospective Study of Two Institutions. J. Surg. Res. 2018, 232, 72–81. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, D.; Jin, X.; Song, H.; Lou, G. Genome-wide characterization of aberrant DNA methylation patterns and the potential clinical implications in patients with endometrial cancer. Pathol. Res. Pract. 2019, 215, 137–143. [Google Scholar] [CrossRef]
- González-Ramírez, I.; García-Cuellar, C.; Sánchez-Pérez, Y.; Granados-García, M. DNA methylation in oral squamous cell carcinoma: Molecular mechanisms and clinical implications. Oral Dis. 2011, 17, 771–778. [Google Scholar] [CrossRef]
- Magzoub, M.M.; Prunello, M.; Brennan, K.; Gevaert, O. The impact of DNA methylation on the cancer proteome. PLoS Comput. Biol. 2019, 15, 1–19. [Google Scholar] [CrossRef]
- Bastian, P.J.; Ellinger, J.; Heukamp, L.C.; Kahl, P.; Müller, S.C.; von Rücker, A. Prognostic Value of CpG Island Hypermethylation at PTGS2, RAR-beta, EDNRB, and Other Gene Loci in Patients Undergoing Radical Prostatectomy. Eur. Urol. 2007, 51, 665–674. [Google Scholar] [CrossRef]
- Hesson, L.B.; Wilson, R.; Morton, D.; Adams, C.; Walker, M.; Maher, E.R.; Latif, F. CpG island promoter hypermethylation of a novel Ras-effector gene RASSF2A is an early event in colon carcinogenesis and correlates inversely with K-ras mutations. Oncogene 2005, 24, 3987–3994. [Google Scholar] [CrossRef]
- Cohen, Y.; Merhavi-Shoham, E.; Avraham, R.B.; Frenkel, S.; Pe’er, J.; Goldenberg-Cohen, N. Hypermethylation of CpG island loci of multiple tumor suppressor genes in retinoblastoma. Exp. Eye Res. 2008, 86, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Puccini, A.; Berger, M.D.; Naseem, M.; Tokunaga, R.; Battaglin, F.; Cao, S.; Hanna, D.L.; McSkane, M.; Soni, S.; Zhang, W.; et al. Colorectal cancer: Epigenetic alterations and their clinical implications. BBA Rev. Cancer 2017, 1868, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Suemoto, Y.; Meyerhardt, J.A.; Fuchs, C.S. Molecular correlates with MGMT promoter methylation and silencing support CpG island methylator phenotype-low (CIMP-low) in colorectal cancer. Gut 2007, 56, 1564–1571. [Google Scholar] [CrossRef]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef]
- Ogino, S.; Cantor, M.; Kawasaki, I.; Brahmandam, M.; Kirkner, G.J.; Weisenberger, D.J.; Campan, M.; Laird, P.W.; Loda, M.; Fuchs, C.S. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut 2006, 55, 1000–1006. [Google Scholar] [CrossRef]
- Suzuki, H.; Yamamoto, E.; Maruyama, R.; Niinuma, T.; Kai, M. Biological significance of the CpG island methylator phenotype. Biochem. Biophys. Res. Commun. 2014, 455, 35–42. [Google Scholar] [CrossRef]
- Suzuki, H.; Igarashi, S.; Nojima, M.; Maruyama, R.; Yamamoto, E.; Kai, M.; Akashi, H.; Watanabe, Y.; Yamamoto, H.; Sasaki, Y.; et al. IGFBP7 is a p53-responsive gene specifically silenced in colorectal cancer with CpG island methylator phenotype. Carcinogenesis 2010, 31, 342–349. [Google Scholar] [CrossRef]
- Hanon, B.M.; Al-Mohaimen Mohammad, N.A.; Mahmood, A.S. CpG Island Methylator Phenotype (CIMP) Correlation with Clinical and Morphological Feature of Colorectal Cancer in Iraq patients. Pan Arab J. Oncol. 2015, 8, 6–13. [Google Scholar]
- Shen, L.; Kondo, Y.; Yi, G.; Jiexin, Z.; Li, Z.; Ahmed, S.; Jingmin, S.; Xinli, C.; Waterland, R.A.; Issa, J.-P.J. Genome-Wide Profiling of DNA Methylation Reveals a Class of Normally Methylated CpG Island Promoters. PLoS Genet. 2007, 3, e181–e2036. [Google Scholar] [CrossRef]
- Suzuki, M.; Sato, S.; Arai, Y.; Shinohara, T.; Tanaka, S.; Greally, J.M.; Hattori, N.; Shiota, K. A new class of tissue-specifically methylated regions involving entire CpG islands in the mouse. Genes Cells 2007, 12, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Rigal, M.; Kevei, Z.; Pélissier, T.; Mathieu, O. DNA methylation in an intron of the IBM1 histone demethylase gene stabilizes chromatin modification patterns. EMBO J. 2012, 31, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Zee, B.M.; Dibona, A.B.; Alekseyenko, A.A.; French, C.A.; Kuroda, M.I. The Oncoprotein BRD4-NUT Generates Aberrant Histone Modification Patterns. PLoS ONE 2016, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fuminori, S.; Yoshikuni, I.; Masamichi, H.; Yasuhiro, K.; Shuji, N. Epigenetic modulation associated with carcinogenesis and prognosis of human gastric cancer (Review). Oncol. Lett. 2017, 13, 3363–3368. [Google Scholar] [CrossRef][Green Version]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, N.; Pedersen, M.W.; Johns, T.G. Targeting the ERBB family in cancer: Couples therapy. Nat. Rev. Cancer 2013, 13, 663–673. [Google Scholar] [CrossRef]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacolog. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- Vecchione, L.; Jacobs, B.; Normanno, N.; Ciardiello, F.; Tejpar, S. EGFR-targeted therapy. Exp. Cell Res. 2011, 317, 2765–2771. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef]
- Hsu, J.L.; Hung, M.C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacolog. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wee, P.; Jiang, J.; Chen, X.; Wang, Z. Differential regulation of transcription factors by location-specific EGF receptor signaling via a spatio-temporal interplay of ERK activation. PLoS ONE 2012, 7, e41354. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Gaslightwala, I.; Birnbaum, M.J.; Rustgi, A.K.; Nakagawa, H. Akt/protein kinase B isoforms are differentially regulated by epidermal growth factor stimulation. J. Biol. Chem. 2000, 275, 30934–30942. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Verlaan-de Vries, M.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef]
- Neumann, J.; Zeindl-Eberhart, E.; Kirchner, T.; Jung, A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol. Res. Pract. 2009, 205, 858–862. [Google Scholar] [CrossRef]
- Kosmidou, V.; Oikonomou, E.; Vlassi, M.; Avlonitis, S.; Katseli, A.; Tsipras, I.; Mourtzoukou, D.; Kontogeorgos, G.; Zografos, G.; Pintzas, A. Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum. Mutat. 2014, 35, 329–340. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef]
- Forrester, K.; Almoguera, C.; Han, K.; Grizzle, W.E.; Perucho, M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature 1987, 327, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Morikawa, T.; Liao, X.; Lochhead, P.; Kuchiba, A.; Yamauchi, M.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; Haigis, K.M.; et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin. Cancer Res. 2012, 18, 4753–4763. [Google Scholar] [CrossRef] [PubMed]
- Conlin, A.; Smith, G.; Carey, F.A.; Wolf, C.R.; Steele, R.J.C. The prognostic significance of K-ras, p53, and APC mutations in colorectal carcinoma. Gut 2005, 54, 1283–1286. [Google Scholar] [CrossRef]
- Phipps, A.I.; Buchanan, D.D.; Makar, K.W.; Win, A.K.; Baron, J.A.; Lindor, N.M.; Potter, J.D.; Newcomb, P.A. KRAS-mutation status in relation to colorectal cancer survival: The joint impact of correlated tumour markers. Br. J. Cancer 2013, 108, 1757–1764. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Iacopetta, B.; Li, W.Q.; Grieu, F.; Ruszkiewicz, A.; Kawakami, K. BRAF mutation and gene methylation frequencies of colorectal tumours with microsatellite instability increase markedly with patient age. Gut 2006, 55, 1213–1214. [Google Scholar] [CrossRef]
- Caputo, F.; Santini, C.; Bardasi, C.; Cerma, K.; Casadei-Gardini, A.; Spallanzani, A.; Andrikou, K.; Cascinu, S.; Gelsomino, F. BRAF-Mutated Colorectal Cancer: Clinical and Molecular Insights. Int. J. Mol. Sci. 2019, 20, 5369. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Lopez, A.; Harada, K.; Vasilakopoulou, M.; Shanbhag, N.; Ajani, J.A. Targeting Angiogenesis in Colorectal Carcinoma. Drugs 2019, 79, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Guba, M.; Seeliger, H.; Kleespies, A.; Jauch, K.W.; Bruns, C. Vascular endothelial growth factor in colorectal cancer. Int. J. Colorectal Dis. 2004, 19, 510–517. [Google Scholar] [CrossRef]
- Amelio, I.; Melino, G. The p53 family and the hypoxia-inducible factors (HIFs): Determinants of cancer progression. Trends Biochem. Sci. 2015, 40, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Hossain, U.; Mandal, A.; Sil, P.C. The Wnt signaling pathway: A potential therapeutic target against cancer. Ann. N. Y. Acad. Sci. 2019, 1443, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Dihlmann, S.; Siermann, A.; von Knebel Doeberitz, M. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate β-catenin/TCF-4 signaling. Oncogene 2001, 20, 645–653. [Google Scholar] [CrossRef]
- Tuynman, J.B.; Vermeulen, L.; Boon, E.M.; Kemper, K.; Zwinderman, A.H.; Peppelenbosch, M.P.; Richel, D.J. Cyclooxygenase-2 inhibition inhibits c-Met kinase activity and Wnt activity in colon cancer. Cancer Res. 2008, 68, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Hamilton, S.R.; Krush, A.J.; Piantadosi, S.; Hylind, L.M.; Celano, P.; Booker, S.V.; Robinson, C.R.; Offerhaus, G.J. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N. Engl. J. Med. 1993, 328, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, G.; Lynch, P.M.; Phillips, R.K.; Wallace, M.H.; Hawk, E.; Gordon, G.B.; Wakabayashi, N.; Saunders, B.; Shen, Y.; Fujimura, T.; et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N. Engl. J. Med. 2000, 342, 1946–1952. [Google Scholar] [CrossRef]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef]
- Huang, S.-M.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Chen, S.; Guttridge, D.C.; You, Z.; Zhang, Z.; Fribley, A.; Mayo, M.W.; Kitajewski, J.; Wang, C.Y. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001, 152, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chua, M.S.; Grepper, S.; So, S.K. Soluble Frizzled-7 receptor inhibits Wnt signaling and sensitizes hepatocellular carcinoma cells towards doxorubicin. Mol. Cancer 2011, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Pode-Shakked, N.; Harari-Steinberg, O.; Haberman-Ziv, Y.; Rom-Gross, E.; Bahar, S.; Omer, D.; Metsuyanim, S.; Buzhor, E.; Jacob-Hirsch, J.; Goldstein, R.S.; et al. Resistance or sensitivity of Wilms’ tumor to anti-FZD7 antibody highlights the Wnt pathway as a possible therapeutic target. Oncogene 2011, 30, 1664–1680. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Fernandes, C.; Sugden, D.; Lovestone, S. Transcriptomic profiles of Wnt3a and insulin in primary cultured rat cortical neurones. J. Neurochem. 2011, 118, 512–520. [Google Scholar] [CrossRef]
- Jeremy, J.Y.; Thompson, C.S.; Mikhailidis, D.P.; Dandona, P. Experimental diabetes mellitus inhibits prostacyclin synthesis by the rat penis: Pathological implications. Diabetologia 1985, 28, 365–368. [Google Scholar] [CrossRef]
- Lavergne, E.; Hendaoui, I.; Coulouarn, C.; Ribault, C.; Leseur, J.; Eliat, P.A.; Mebarki, S.; Corlu, A.; Clément, B.; Musso, O. Blocking Wnt signaling by SFRP-like molecules inhibits in vivo cell proliferation and tumor growth in cells carrying active β-catenin. Oncogene 2011, 30, 423–433. [Google Scholar] [CrossRef]
- Boulay, J.L.; Mild, G.; Lowy, A.; Reuter, J.; Lagrange, M.; Terracciano, L.; Laffer, U.; Herrmann, R.; Rochlitz, C. SMAD4 is a predictive marker for 5-fluorouracil-based chemotherapy in patients with colorectal cancer. Br. J. Cancer 2002, 87, 630. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, L.; Zhou, J.; Zheng, J. Silencing Smad4 attenuates sensitivity of colorectal cancer cells to cetuximab by promoting epithelial-mesenchymal transition. Mol. Med. Res. 2019, 20, 3735–3745. [Google Scholar] [CrossRef]
- Oyanagi, H.; Shimada, Y.; Nagahashi, M.; Ichikawa, H.; Tajima, Y.; Abe, K.; Nakano, M.; Kameyama, H.; Takii, Y.; Kawasaki, T.; et al. SMAD4 alteration associates with invasive-front pathological markers and poor prognosis in colorectal cancer. Histopathology 2019, 74, 873–882. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Yeatman, T.; Weber, J.; Coppola, D.; Schell, M.J.; Han, G.; Almhanna, K.; Kim, R.; Valone, T.; Jump, H.; et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur. J. Cancer 2012, 48, 997–1003. [Google Scholar] [CrossRef]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Wallmark, J.M.; Lorente, D.; Elez, E.; Raimbourg, J.; Gomez-Roca, C.; Ejadi, S.; Piha-Paul, S.A.; Stein, M.N.; Abdul Razak, A.R.; et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS ONE 2017, 12, e0189848. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Atkinson, V.; Cebon, J.S.; Jameson, M.B.; Fitzharris, B.M.; McNeil, C.M.; Hill, A.G.; Ribas, A.; Atkins, M.B.; Thompson, J.A.; et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): An open-label, phase 1b trial. Lancet Oncol. 2017, 18, 1202–1210. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Lenz, H.J.J.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.; et al. Durable clinical benefit with nivolumab (NIVO) plus low-dose ipilimumab (IPI) as first-line therapy in microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC). Ann. Oncol. 2018, 29, viii714. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Zhou, E.; Huang, Q.; Wang, J.; Fang, C.; Yang, L.; Zhu, M.; Chen, J.; Chen, L.; Dong, M. Up-regulation of Tim-3 is associated with poor prognosis of patients with colon cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8018–8027. [Google Scholar]
- Yu, X.; Huang, X.; Chen, X.; Liu, J.; Wu, C.; Pu, Q.; Wang, Y.; Kang, X.; Zhou, L. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. mABs 2019, 11, 1139–1148. [Google Scholar] [CrossRef]
- Mendelsohn, J.; Prewett, M.; Rockwell, P.; Goldstein, N.I. CCR 20th anniversary commentary: A chimeric antibody, C225, inhibits EGFR activation and tumor growth. Clin. Cancer Res. 2015, 21, 227–229. [Google Scholar] [CrossRef][Green Version]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [PubMed]
- Yarom, N.; Jonker, D.J. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 2011, 11, 95–105. [Google Scholar]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Final results from PRIME: Randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 2014, 25, 1346–1355. [Google Scholar] [CrossRef]
- Modest, D.P.; Rivera, F.; Bachet, J.B.; de Braud, F.; Pietrantonio, F.; Koukakis, R.; Demonty, G.; Douillard, J.Y. Panitumumab-based maintenance after oxaliplatin discontinuation in metastatic colorectal cancer: A retrospective analysis of two randomised trials. Int. J. Cancer 2019, 145, 576–585. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Morano, F.; Corallo, S.; Lonardi, S.; Cremolini, C.; Rimassa, L.; Sartore-Bianchi, A.; Tampellini, M.; Bustreo, S.; Clavarezza, M.; et al. First-line FOLFOX plus panitumumab (Pan) followed by 5FU/LV plus Pan or single-agent Pan as maintenance therapy in patients with RAS wild-type metastatic colorectal cancer (mCRC): The VALENTINO study. J. Clin. Oncol. 2018, 36, 3505. [Google Scholar] [CrossRef]
- Price, T.J.; Peeters, M.; Kim, T.W.; Li, J.; Cascinu, S.; Ruff, P.; Suresh, A.S.; Thomas, A.; Tjulandin, S.; Zhang, K.; et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): A randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014, 15, 569–579. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578.e517–589.e517. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Corcoran, R.B.; André, T.; Yoshino, T.; Bendell, J.C.; Atreya, C.E.; Schellens, J.H.M.; Ducreux, M.P.; McRee, A.; Siena, S.; Middleton, G.; et al. Efficacy and circulating tumor DNA (ctDNA) analysis of the BRAF inhibitor dabrafenib (D), MEK inhibitor trametinib (T), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E–mutated (BRAFm) metastatic colorectal cancer (mCRC). Ann. Oncol. 2016, 27, vi150. [Google Scholar] [CrossRef]
- Van Geel, R.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF-Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef]
- Corcoran, R.B.; André, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef]
- Hong, D.S.; Morris, V.K.; El Osta, B.; Sorokin, A.V.; Janku, F.; Fu, S.; Overman, M.J.; Piha-Paul, S.; Subbiah, V.; Kee, B.; et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016, 6, 1352–1365. [Google Scholar] [CrossRef]
- Kopetz, S.; McDonough, S.L.; Morris, V.K.; Lenz, H.-J.; Magliocco, A.M.; Atreya, C.E.; Diaz, L.A.; Allegra, C.J.; Wang, S.E.; Lieu, C.H.; et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG 1406). J. Clin. Oncol. 2017, 35, 520. [Google Scholar] [CrossRef]
- Tabernero, J.; Geel, R.V.; Guren, T.K.; Yaeger, R.D.; Spreafico, A.; Faris, J.E.; Yoshino, T.; Yamada, Y.; Kim, T.W.; Bendell, J.C.; et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J. Clin. Oncol. 2016, 34, 3544. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Passardi, A.; Nanni, O.; Tassinari, D.; Turci, D.; Cavanna, L.; Fontana, A.; Ruscelli, S.; Mucciarini, C.; Lorusso, V.; Ragazzini, A.; et al. Effectiveness of bevacizumab added to standard chemotherapy in metastatic colorectal cancer: Final results for first-line treatment from the ITACa randomized clinical trial. Ann. Oncol. 2015, 26, 1201–1207. [Google Scholar] [CrossRef]
- Simkens, L.H.; van Tinteren, H.; May, A.; Tije, T.A.J.; Creemers, G.J.; Loosveld, O.J.; de Jongh, F.E.; Erdkamp, F.L.; Erjavec, Z.; van der Torren, A.M.; et al. Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): A phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet 2015, 385, 1843–1852. [Google Scholar] [CrossRef]
- Schwartzberg, L.S.; Rivera, F.; Karthaus, M.; Fasola, G.; Canon, J.L.; Hecht, J.R.; Yu, H.; Oliner, K.S.; Go, W.Y. PEAK: A randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J. Clin. Oncol. 2014, 32, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Venook, A.P.; Niedzwiecki, D.; Lenz, H.J.; Innocenti, F.; Fruth, B.; Meyerhardt, J.A.; Schrag, D.; Greene, C.; O’Neil, B.H.; Atkins, J.N.; et al. Effect of First-Line Chemotherapy Combined With Cetuximab or Bevacizumab on Overall Survival in Patients With KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2017, 317, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson, A.B., 3rd. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Subtype | Gene Expression | Prognosis | Reference |
|---|---|---|---|
| CMS1 (MSI immune) |
| Very poor survival rate after relapse | [4] |
| CMS2 (Canonical) |
| Better survival rate after relapse in comparison to other subtypes | |
| CMS3 (Metabolic) |
| Better survival rate after relapse in comparison to other subtypes | |
| CMS4 (Mesenchymal) |
| Worse overall and relapse-free survival as compared to other subtypes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2021, 22, 130. https://doi.org/10.3390/ijms22010130
Malki A, ElRuz RA, Gupta I, Allouch A, Vranic S, Al Moustafa A-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. International Journal of Molecular Sciences. 2021; 22(1):130. https://doi.org/10.3390/ijms22010130
Chicago/Turabian StyleMalki, Ahmed, Rasha Abu ElRuz, Ishita Gupta, Asma Allouch, Semir Vranic, and Ala-Eddin Al Moustafa. 2021. "Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements" International Journal of Molecular Sciences 22, no. 1: 130. https://doi.org/10.3390/ijms22010130
APA StyleMalki, A., ElRuz, R. A., Gupta, I., Allouch, A., Vranic, S., & Al Moustafa, A.-E. (2021). Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. International Journal of Molecular Sciences, 22(1), 130. https://doi.org/10.3390/ijms22010130