Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract

1. Introduction of Hydrogels
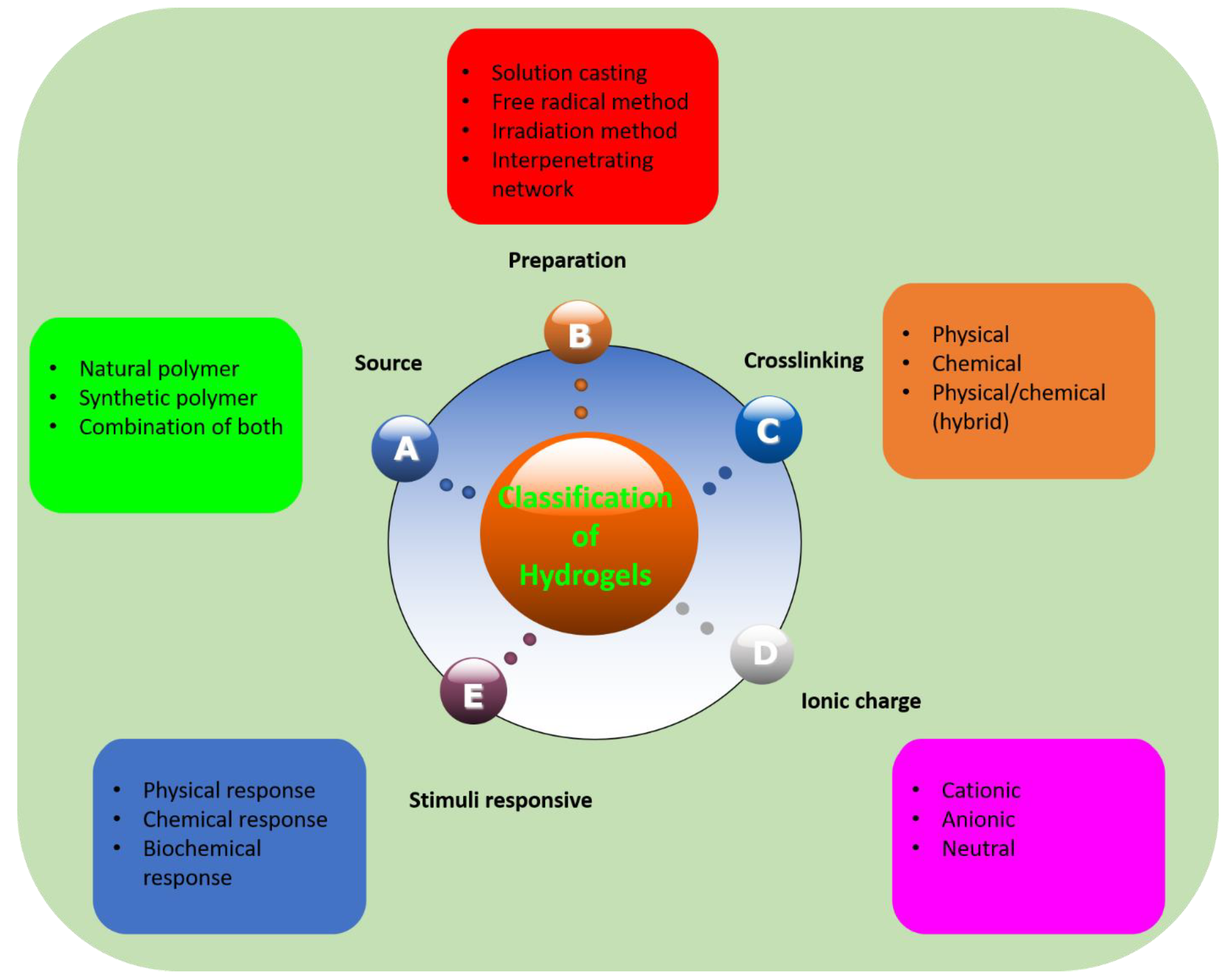
2. Classification of Hydrogels
3. Composition of Hydrogels
4. Natural Polymer Hydrogels
4.1. Polysaccharide Hydrogels
4.2. Synthetic Polymer Hydrogels
4.3. Hydrogels Based on Poly (Acrylic Acid) Derivatives
4.4. Hydrogels Based on Poly (Acrylamide) Derivatives
4.5. Natural Polymers in Combination with Synthetic Polymer Hydrogels
5. Types of Hydrogels
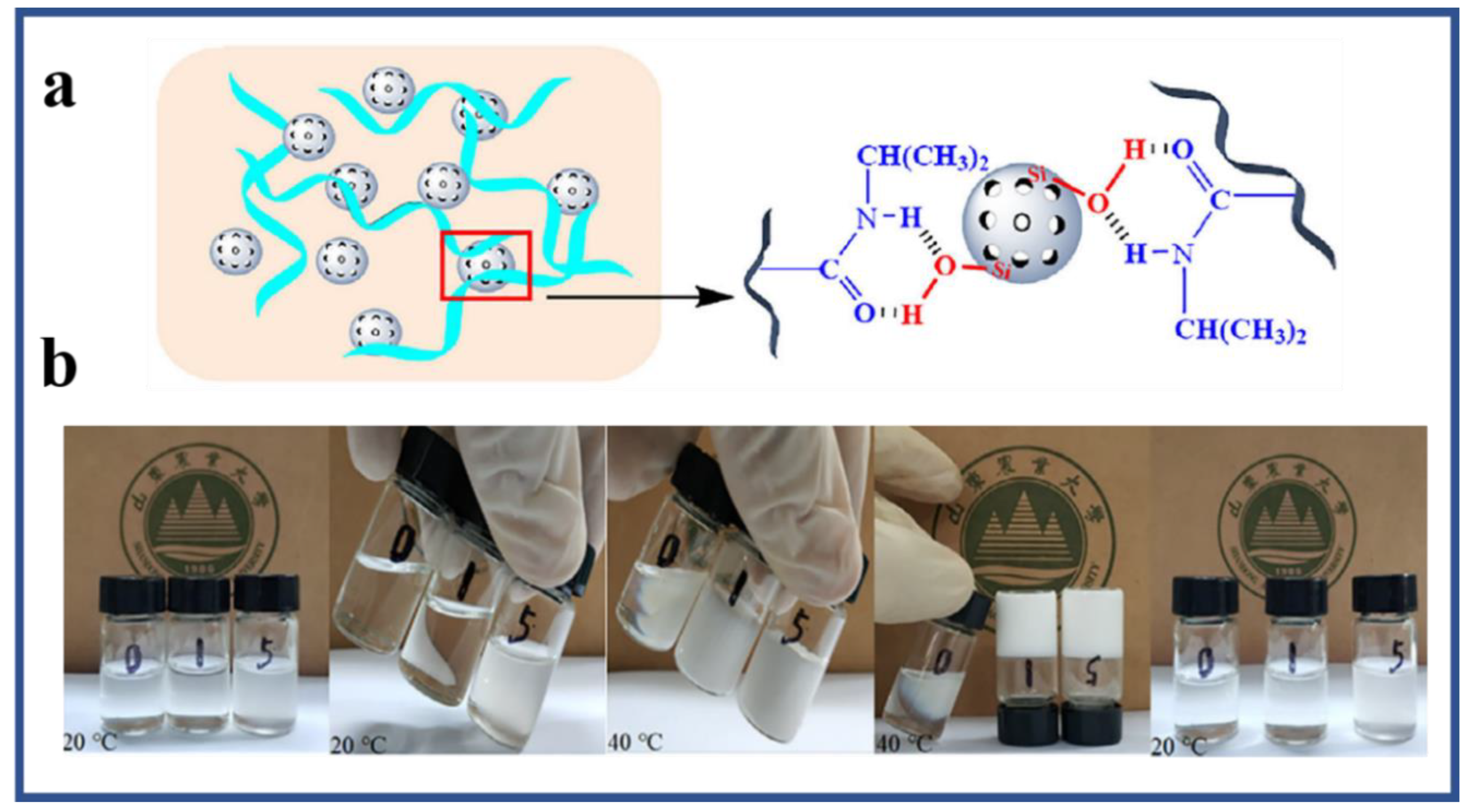
5.1. Physical Crosslinked Hydrogels
5.2. Chemically Crosslinked Hydrogels
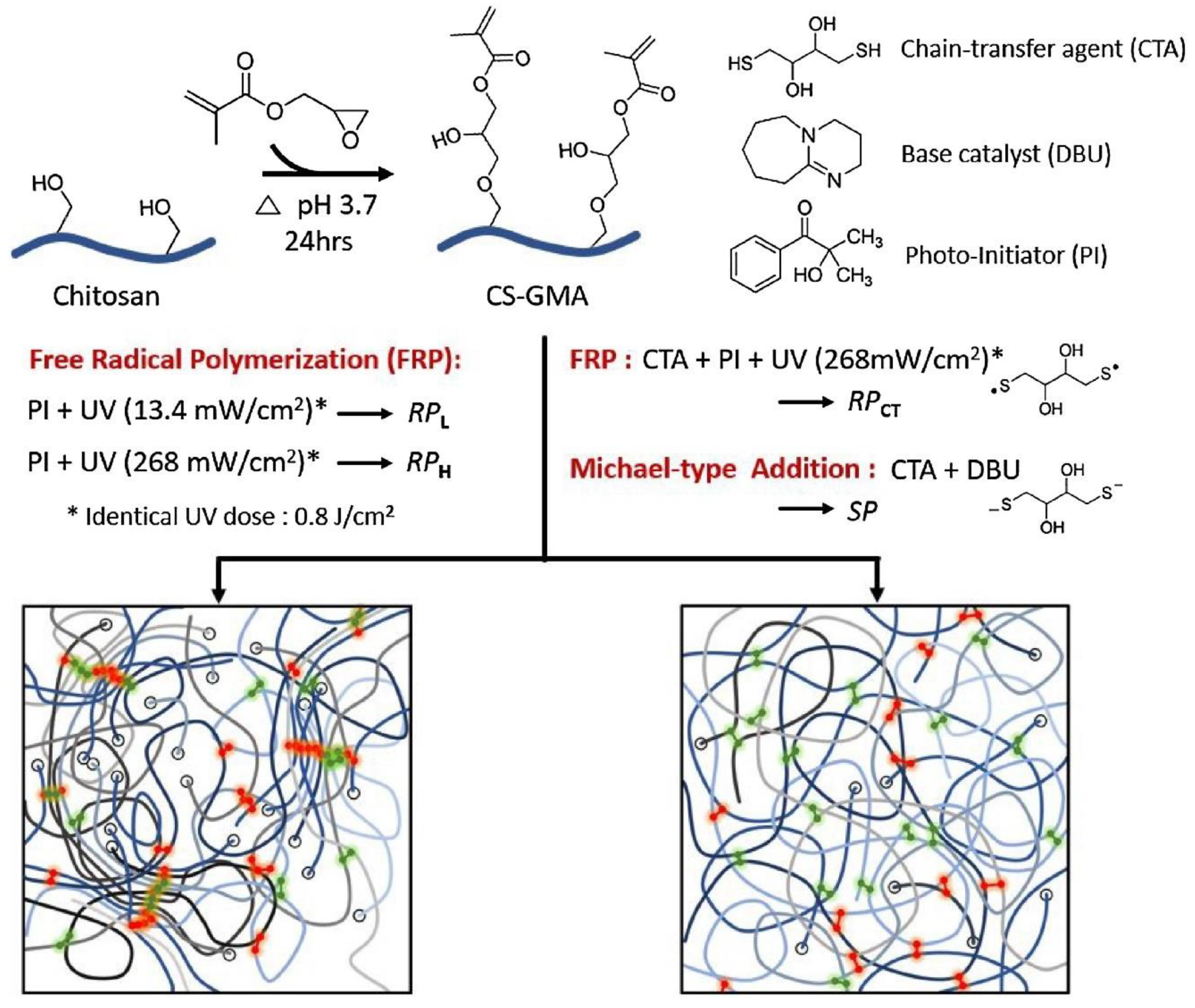
5.2.1. Crosslinking by Small Molecules
5.2.2. Crosslinking through Ionizing Radiation
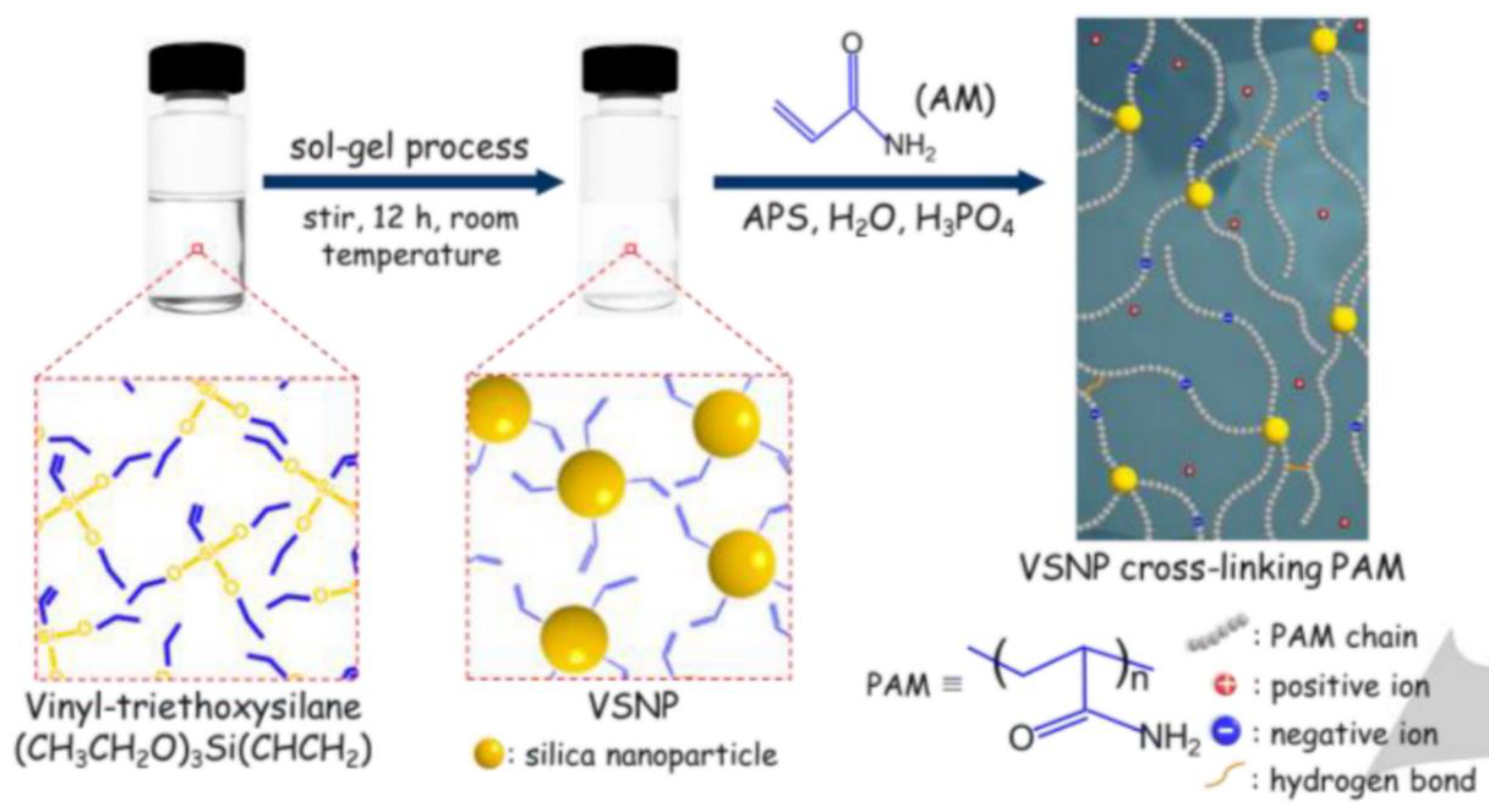
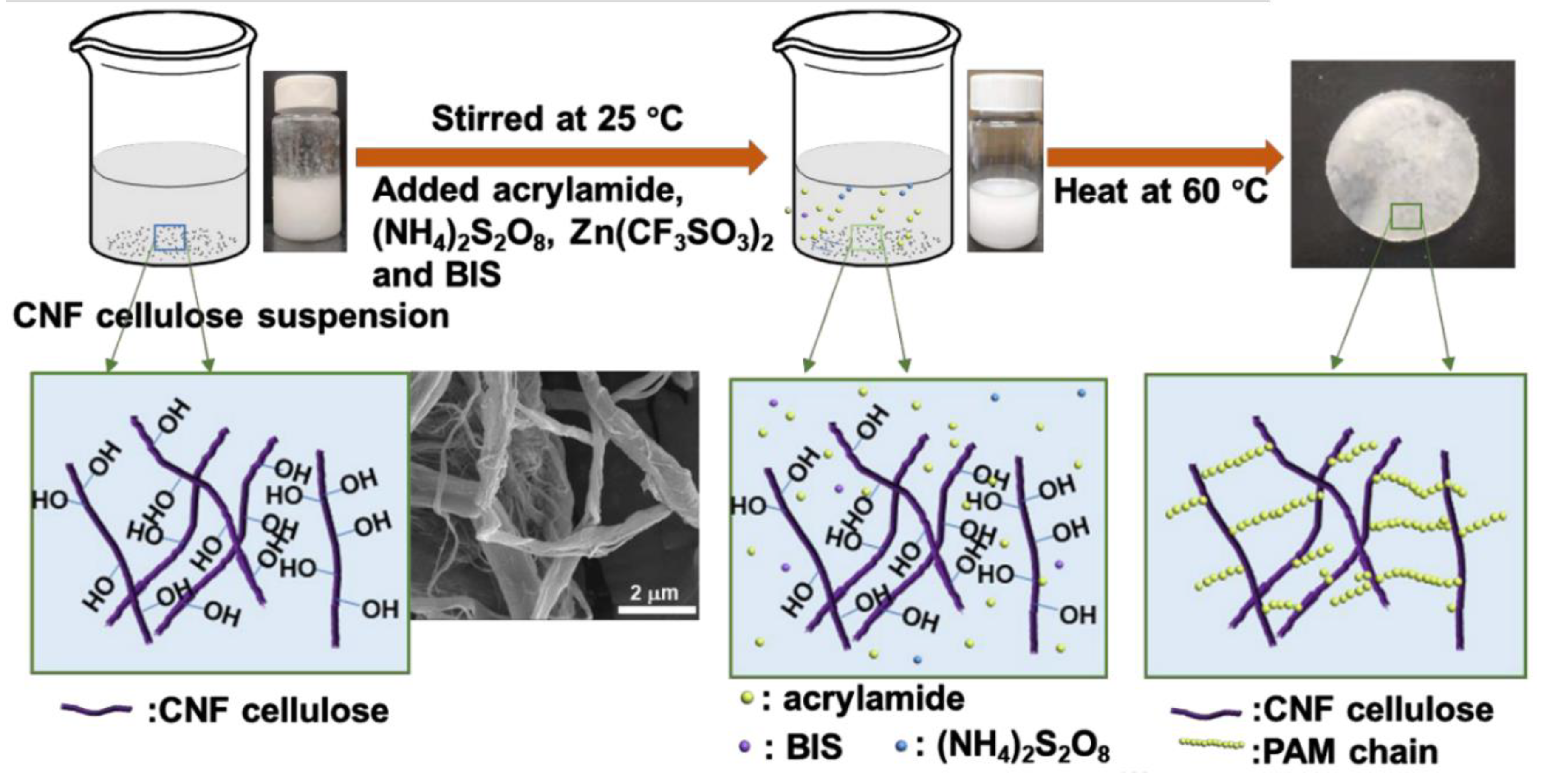
5.2.3. Crosslinking through Free Radical Mechanism
5.3. Interpenetrating Network (IPN) Hydrogel
5.3.1. Semi-Interpenetrating Network (semi-IPN) Hydrogels
5.3.2. Full-Interpenetrating Network (Full-IPN) Hydrogels
6. Properties of Hydrogels
6.1. Mechanical Properties
6.2. Biocompatibility
6.3. Biodegradability
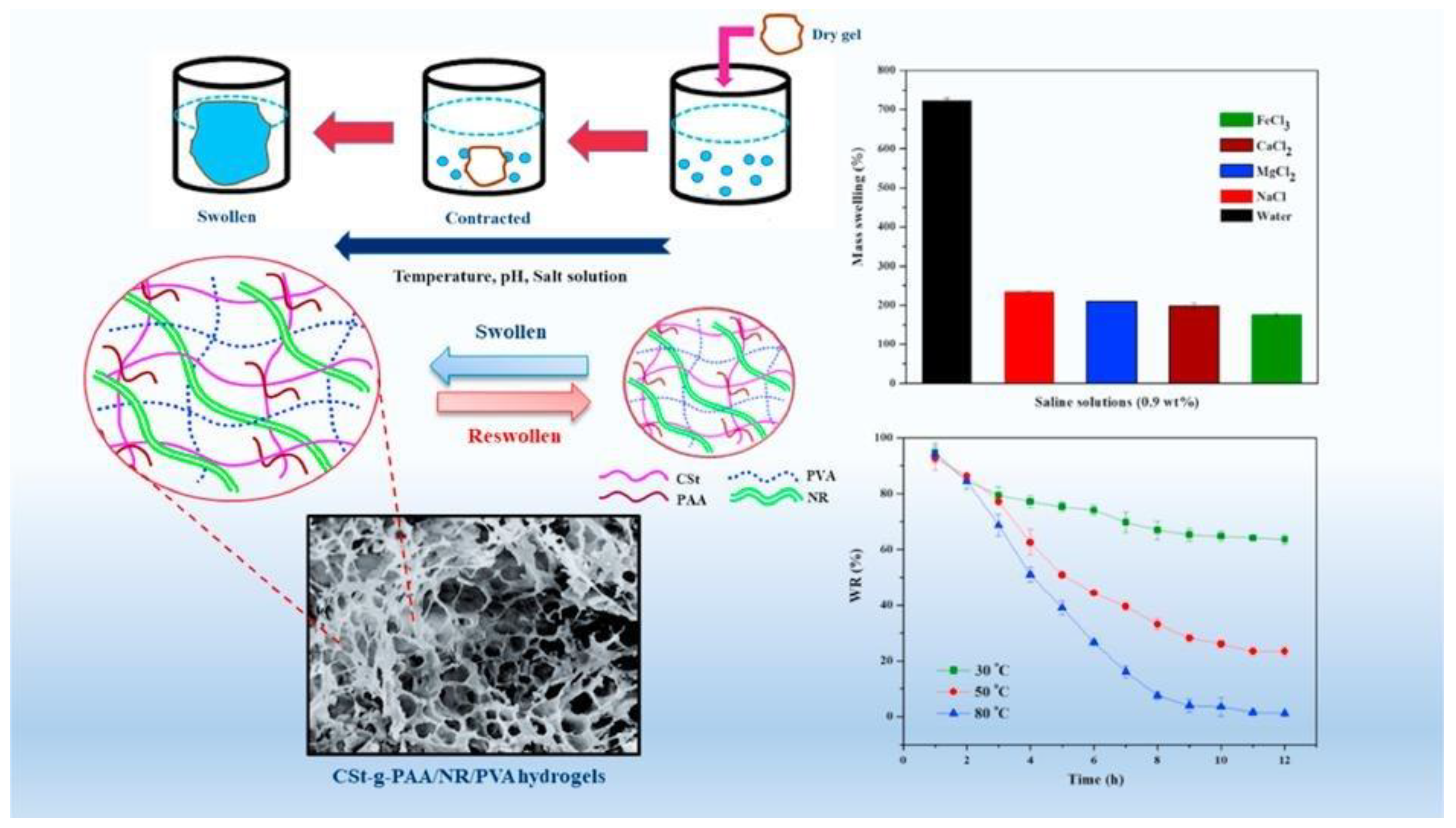
6.4. Swelling Behavior of Hydrogel
Swelling Kinetics
6.5. Stimuli Sensitive Hydrogels
6.5.1. pH-Sensitive Hydrogels
6.5.2. Thermosensitive Hydrogels
7. Electrochemical Applications of Polymer Hydrogels
7.1. Electrolytes
7.2. Quasi-Solid-State Electrolytes
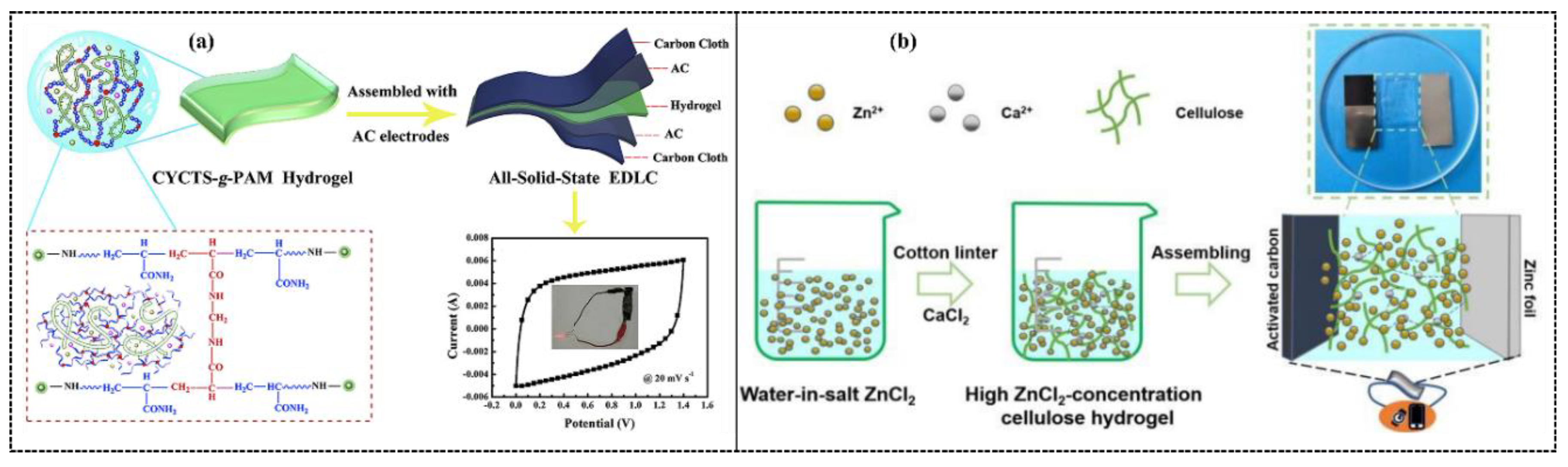
7.3. Polymer Hydrogel Electrolytes for Supercapacitors
7.4. Flexible Polymer Hydrogel Electrolytes
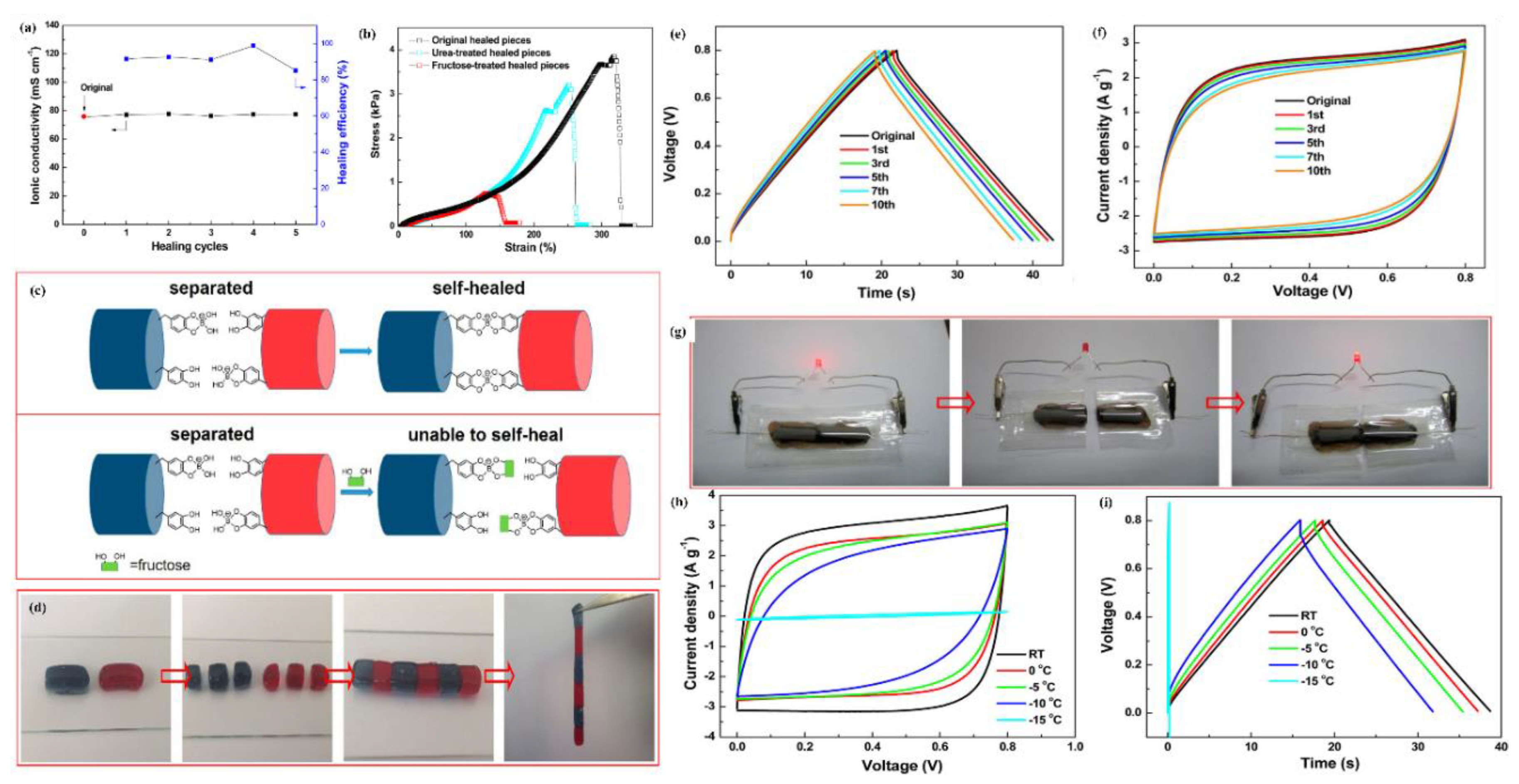
7.5. Self-Healing Properties of Polymer Hydrogel Electrolyte
Evaluation and Mechanism of Self-Healing Ability of Hydrogel Electrolytes
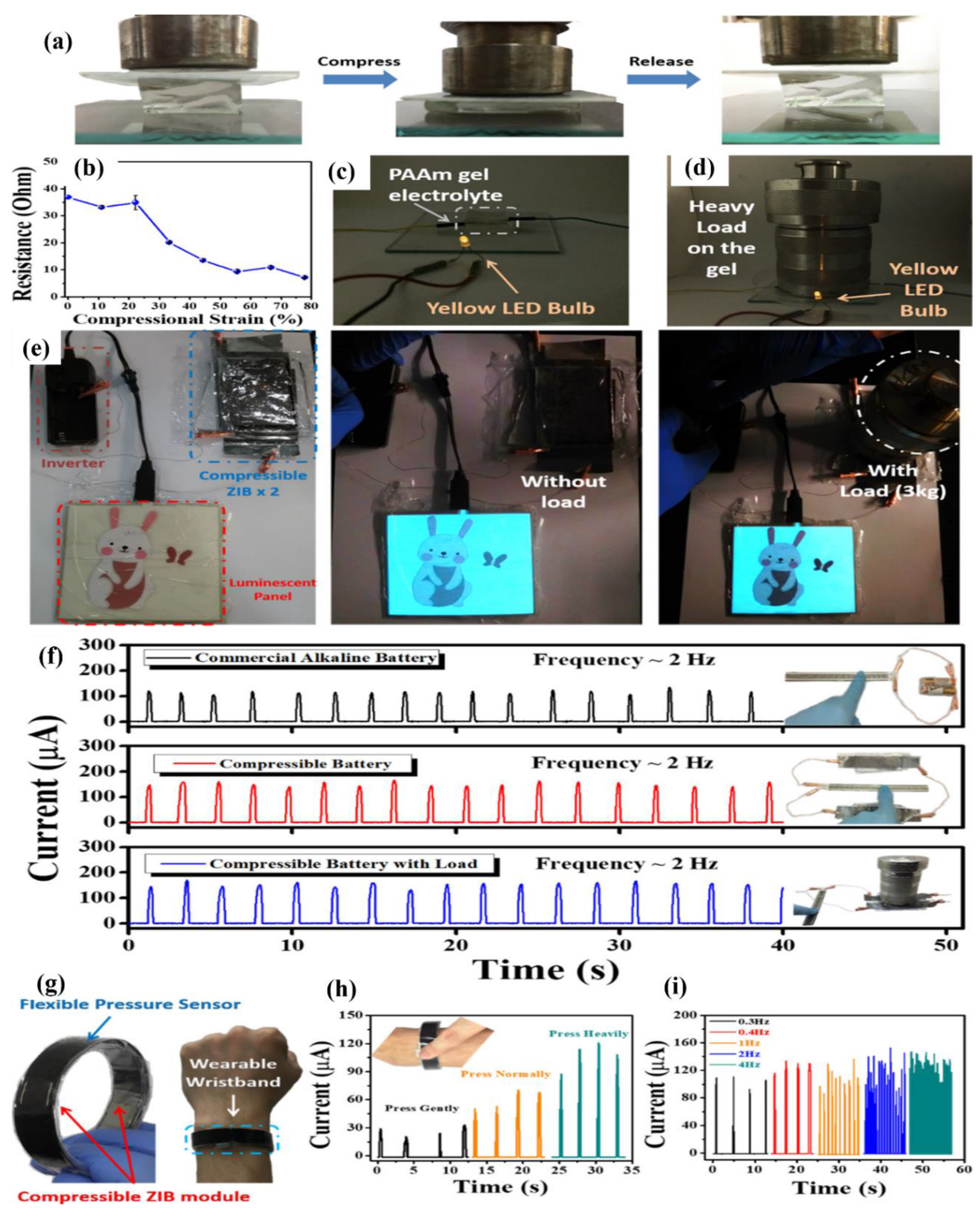
7.6. Polymer Hydrogel Electrolytes for Batteries
7.7. Polymer Hydrogel Electrolytes for Energy Conversion Devices
8. Conclusions and Future Directions
- With the passage of time, the synthesis and application of hydrogels is becoming more complex day by day. Hydrogels exposed certain limitations, such as fatigue and cracks under long-term load upon exposure in extreme environmental conditions. These limitations are attributed to their chemical composition and topology of hydrogel network. These limitations can be overcome by forming double crosslinked networks.
- Low conductivity sometimes become an unanticipated challenge in the real-time application of hydrogels. The conductivity, mechanical strength, and optoelectronic properties of hydrogels can be reinforced by incorporating conducting materials, such as nanofillers, to form hybrid hydrogel networks owing to the synergistic interaction between components and the larger surface area of the nanofillers. Nanofillers used to improve conductivity are carbon nanotubes, carbon dots, graphene, metal oxide/sulfide/phosphate, and conducting polymers.
- According to the percolation theory, flexible electrochemical devices can be developed by integrating hydrogels with certain electroactive materials, such as nanofillers. However, the balance between electroactive materials and hydrogels must be considered to attain flexible devices. In addition, more specifically, nanofillers improve the cycling stability, ionic diffusion, and connectivity of channels in the porous networks of hydrogel electrolytes.
- Hydrogels with unique properties will be appropriate candidates for electronic, sensing, cell imaging, drug-delivery, and tissue-engineering applications. Highly conducting hybrid hydrogels can be extended to produce artificial skin, soft robotics, tissue generation, 3D printing, etc.
- Ionically conductive hydrogels are applicable for limited bioelectronics. Advanced fabrication methods can introduce hydrogels in bioelectronics at a large scale and significantly support their success. Three-dimensional printing and ink-jet printing methods of ionic hydrogels are quite new developments and can be promising directions for future developments.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghorpade, V.S.; Dias, R.J.; Mali, K.K.; Mulla, S.I. Citric acid crosslinked carboxymethylcellulose-polyvinyl alcohol hydrogel films for extended release of water soluble basic drugs. J. Drug Deliv. Sci. Technol. 2019, 52, 421–430. [Google Scholar] [CrossRef]
- Dimatteo, R.; Darling, N.J.; Segura, T. In situ forming injectable hydrogels for drug delivery and wound repair. Adv. Drug Deliv. Rev. 2018, 127, 167–184. [Google Scholar] [CrossRef]
- Abad, L.V.; Relleve, L.S.; Aranilla, C.T.; Dela Rosa, A.M. Properties of radiation synthesized PVP-kappa carrageenan hydrogel blends. Radiat. Phys. Chem. 2003, 68, 901–908. [Google Scholar] [CrossRef]
- Ali, S.W.; Zaidi, S.A.R. Synthesis of copolymeric acrylamide/potassium acrylate hydrogels blended with poly (vinyl alcohol): Effect of crosslinking and the amount of poly (vinyl alcohol) on swelling behavior. J. Appl. Polym. Sci. 2005, 98, 1927–1931. [Google Scholar] [CrossRef]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef]
- Nie, J.; Pei, B.; Wang, Z.; Hu, Q. Construction of ordered structure in polysaccharide hydrogel: A review. Carbohydr. Polym. 2019, 205, 225–235. [Google Scholar] [CrossRef]
- Siepmann, J.; Siegel, R.A.; Rathbone, M.J. Fundamentals and Applications of Controlled Release Drug Delivery; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Miyata, T.; Uragami, T.; Nakamae, K. Biomolecule-sensitive hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 79–98. [Google Scholar] [CrossRef]
- Nezhad-Mokhtari, P.; Ghorbani, M.; Roshangar, L.; Rad, J.S. A review on the construction of hydrogel scaffolds by various chemically techniques for tissue engineering. Eur. Polym. J. 2019, 117, 64–76. [Google Scholar] [CrossRef]
- Huang, Y.; Zhong, M.; Shi, F.; Liu, X.; Tang, Z.; Wang, Y.; Huang, Y.; Hou, H.; Xie, X.; Zhi, C. An intrinsically stretchable and compressible supercapacitor containing a polyacrylamide hydrogel electrolyte. Angew. Chem. Int. Ed. 2017, 56, 9141–9145. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Tang, Z.; Liu, Z.; Ruan, Z.; Ma, L.; Yang, Q.; Wang, D.; Zhi, C. Hydrogel electrolytes for flexible aqueous energy storage devices. Adv. Funct. Mater. 2018, 28, 1804560. [Google Scholar] [CrossRef]
- Huang, Y.; Zhong, M.; Huang, Y.; Zhu, M.; Pei, Z.; Wang, Z.; Xue, Q.; Xie, X.; Zhi, C. A self-healable and highly stretchable supercapacitor based on a dual crosslinked polyelectrolyte. Nat. Commun. 2015, 6, 10310. [Google Scholar] [CrossRef]
- Sharma, S.; Tiwari, S. A review on biomacromolecular hydrogel classification and its applications. Int. J. Biol. Macromol. 2020, 162, 737–747. [Google Scholar] [CrossRef]
- Mahinroosta, M.; Farsangi, Z.J.; Allahverdi, A.; Shakoori, Z. Hydrogels as intelligent materials: A brief review of synthesis, properties and applications. Mater. Today Chem. 2018, 8, 42–55. [Google Scholar] [CrossRef]
- Islam, A.; Yasin, T.; Bano, I.; Riaz, M. Controlled release of aspirin from pH-sensitive chitosan/poly (vinyl alcohol) hydrogel. J. Appl. Polym. Sci. 2012, 124, 4184–4192. [Google Scholar] [CrossRef]
- Kirschner, C.M.; Anseth, K.S. Hydrogels in healthcare: From static to dynamic material microenvironments. Acta Mater. 2013, 61, 931–944. [Google Scholar] [CrossRef]
- Vijayavenkataraman, S.; Vialli, N.; Fuh, J.Y.; Lu, W.F. Conductive collagen/polypyrrole-b-polycaprolactone hydrogel for bioprinting of neural tissue constructs. Int. J. Bioprinting 2019, 5, 229. [Google Scholar] [CrossRef]
- Bonelli, N.; Poggi, G.; Chelazzi, D.; Giorgi, R.; Baglioni, P. Poly (vinyl alcohol)/poly (vinyl pyrrolidone) hydrogels for the cleaning of art. J. Colloid Interface Sci. 2019, 536, 339–348. [Google Scholar] [CrossRef]
- Munim, S.A.; Raza, Z.A. Poly (lactic acid) based hydrogels: Formation, characteristics and biomedical applications. J. Porous Mater. 2019, 26, 881–901. [Google Scholar] [CrossRef]
- Atta, S.; Khaliq, S.; Islam, A.; Javeria, I.; Jamil, T.; Athar, M.M.; Shafiq, M.I.; Ghaffar, A. Injectable biopolymer based hydrogels for drug delivery applications. Int. J. Biol. Macromol. 2015, 80, 240–245. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, R.; García-Carvajal, Z.; Jiménez-Palomar, I.; Jiménez-Avalos, J.; Espinosa-Andrews, H. Development of gelatin/chitosan/PVA hydrogels: Thermal stability, water state, viscoelasticity, and cytotoxicity assays. J. Appl. Polym. Sci. 2019, 136, 47149. [Google Scholar] [CrossRef]
- Bhattarai, N.; Matsen, F.A.; Zhang, M. PEG-Grafted Chitosan as an Injectable Thermoreversible Hydrogel. Macromol. Biosci. 2005, 5, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Iresha, H.; Kobayashi, T. Smart Polysaccharide Hydrogels in Drug Delivery and Release. In Advanced Biopolymeric Systems for Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2020; pp. 135–149. [Google Scholar]
- Hu, Y.; Zhang, Z.; Li, Y.; Ding, X.; Li, D.; Shen, C.; Xu, F.J. Dual-Crosslinked Amorphous Polysaccharide Hydrogels Based on Chitosan/Alginate for Wound Healing Applications. Macromol. Rapid Commun. 2018, 39, 1800069. [Google Scholar] [CrossRef] [PubMed]
- Kwiecień, I.; Kwiecień, M. Application of polysaccharide-based hydrogels as probiotic delivery systems. Gels 2018, 4, 47. [Google Scholar] [CrossRef]
- Gholamali, I. Stimuli-responsive polysaccharide hydrogels for biomedical applications: A review. Regen. Eng. Transl. Med. 2019, 1–24. [Google Scholar] [CrossRef]
- Piyakulawat, P.; Praphairaksit, N.; Chantarasiri, N.; Muangsin, N. Preparation and evaluation of chitosan/carrageenan beads for controlled release of sodium diclofenac. Aaps Pharmscitech 2007, 8, 120. [Google Scholar] [CrossRef]
- Reis, A.V.; Guilherme, M.R.; Moia, T.A.; Mattoso, L.H.; Muniz, E.C.; Tambourgi, E.B. Synthesis and characterization of a starch-modified hydrogel as potential carrier for drug delivery system. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 2567–2574. [Google Scholar] [CrossRef]
- Berger, J.; Reist, M.; Mayer, J.M.; Felt, O.; Peppas, N.; Gurny, R. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur. J. Pharm. Biopharm. 2004, 57, 19–34. [Google Scholar] [CrossRef]
- Kim, I.-S.; Oh, I.-J. Drug release from the enzyme-degradable and pH-sensitive hydrogel composed of glycidyl methacrylate dextran and poly (acrylic acid). Arch. Pharmacal Res. 2005, 28, 983–987. [Google Scholar] [CrossRef]
- Lee, A.L.S.; Ordeñez, G.; Chakraborty, S. Novel glycerol cross-linked poly (acrylic acid) hydrogel for encapsulation and release of benzocaine. Philipp. Sci. Lett. 2011, 4, 81–87. [Google Scholar]
- Amin, M.C.I.M.; Ahmad, N.; Halib, N.; Ahmad, I. Synthesis and characterization of thermo-and pH-responsive bacterial cellulose/acrylic acid hydrogels for drug delivery. Carbohydr. Polym. 2012, 88, 465–473. [Google Scholar] [CrossRef]
- Nho, Y.-C.; Park, J.-S.; Lim, Y.-M. Preparation of poly (acrylic acid) hydrogel by radiation crosslinking and its application for mucoadhesives. Polymers 2014, 6, 890–898. [Google Scholar] [CrossRef]
- Kou, J.H.; Amidon, G.L.; Lee, P.I. pH-Dependent Swelling and Solute Diffusion Characteristics of Poly (Hydroxyethyl Methacrylate–CO–Methacrylie Acid) Hydrogels. Pharm. Res. 1988, 5, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Nho, Y.C.; Lim, Y.M.; Kim, H.I. Preparation of pH-sensitive poly (vinyl alcohol-g-methacrylic acid) and poly (vinyl alcohol-g-acrylic acid) hydrogels by gamma ray irradiation and their insulin release behavior. J. Appl. Polym. Sci. 2004, 91, 636–643. [Google Scholar] [CrossRef]
- Mundargi, R.; Patil, S.; Kulkarni, P.; Mallikarjuna, N.; Aminabhavi, T. Sequential interpenetrating polymer network hydrogel microspheres of poly (methacrylic acid) and poly (vinyl alcohol) for oral controlled drug delivery to intestine. J. Microencapsul. 2008, 25, 228–240. [Google Scholar] [CrossRef]
- Khan, Y.; Bashir, S.; Hina, M.; Subramaniam, R.T.; Kasi, R.; Lahiri, I. Effect of Salt Concentration on Poly (acrylic acid) Hydrogel Electrolytes and Their Applications in Supercapacitor. J. Electrochem. Soc. 2020. [Google Scholar] [CrossRef]
- Saraydın, D.; Karadag, E.; Işıkver, Y.; Şahiner, N.; Güven, O. The influence of preparation methods on the swelling and network properties of acrylamide hydrogels with crosslinkers. J. Macromol. Sci. Part A 2004, 41, 419–431. [Google Scholar] [CrossRef]
- Xu, W.; Liu, C.; Wu, Q.; Xie, W.; Kim, W.-Y.; Lee, S.-Y.; Gwon, J. A stretchable solid-state zinc ion battery based on a cellulose nanofiber–polyacrylamide hydrogel electrolyte and a Mg0.23V2O5·1.0H2O cathode. J. Mater. Chem. A 2020, 8, 18327–18337. [Google Scholar] [CrossRef]
- Hina, M.; Bashir, S.; Kamran, K.; Ramesh, S.; Ramesh, K. Synthesis and Characterization of Self-healable Poly (acrylamide) Hydrogel Electrolytes and Their Application in Fabrication of Aqueous Supercapacitors. Polymer 2020, 210, 123020. [Google Scholar] [CrossRef]
- Nesrinne, S.; Djamel, A. Synthesis, characterization and rheological behavior of pH sensitive poly (acrylamide-co-acrylic acid) hydrogels. Arab. J. Chem. 2013. [Google Scholar] [CrossRef]
- Ao, H.; Huang, M.; Wu, J.; Lin, J.; Tang, Q.; Sun, H. Synthesis and properties of poly (acrylamide-co-acrylic acid)/polyacrylamide superporous IPN hydrogels. Polym. Adv. Technol. 2009, 20, 1044–1049. [Google Scholar] [CrossRef]
- Hirotsu, S.; Hirokawa, Y.; Tanaka, T. Volume-phase transitions of ionized N-isopropylacrylamide gels. J. Chem. Phys. 1987, 87, 1392–1395. [Google Scholar] [CrossRef]
- Matzelle, T.; Geuskens, G.; Kruse, N. Elastic properties of poly (N-isopropylacrylamide) and poly (acrylamide) hydrogels studied by scanning force microscopy. Macromolecules 2003, 36, 2926–2931. [Google Scholar] [CrossRef]
- Brazel, C.S.; Peppas, N.A. Pulsatile local delivery of thrombolytic and antithrombotic agents using poly (N-isopropylacrylamide-co-methacrylic acid) hydrogels. J. Control. Release 1996, 39, 57–64. [Google Scholar] [CrossRef]
- Fathi, M.; Alami-Milani, M.; Geranmayeh, M.H.; Barar, J.; Erfan-Niya, H.; Omidi, Y. Dual thermo-and pH-sensitive injectable hydrogels of chitosan/(poly (N-isopropylacrylamide-co-itaconic acid)) for doxorubicin delivery in breast cancer. Int. J. Biol. Macromol. 2019, 128, 957–964. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, S.; Waterhouse, G.I.; Zhang, Q.; Li, L. Poly (N-isopropylacrylamide)/mesoporous silica thermosensitive composite hydrogels for drug loading and release. J. Appl. Polym. Sci. 2020, 137, 48391. [Google Scholar] [CrossRef]
- Panayiotou, M.; Freitag, R. Synthesis and characterisation of stimuli-responsive poly (N, N′-diethylacrylamide) hydrogels. Polymer 2005, 46, 615–621. [Google Scholar] [CrossRef]
- Sohail, M.; Minhas, M.U.; Khan, S.; Hussain, Z.; de Matas, M.; Shah, S.A.; Khan, S.; Kousar, M.; Ullah, K. Natural and synthetic polymer-based smart biomaterials for management of ulcerative colitis: A review of recent developments and future prospects. Drug Deliv. Transl. Res. 2019, 9, 595–614. [Google Scholar] [CrossRef]
- Bashir, S.; Teo, Y.Y.; Ramesh, S.; Ramesh, K. Synthesis, characterization, properties of N-succinyl chitosan-g-poly (methacrylic acid) hydrogels and in vitro release of theophylline. Polymer 2016, 92, 36–49. [Google Scholar] [CrossRef]
- Chen, S.-C.; Wu, Y.-C.; Mi, F.-L.; Lin, Y.-H.; Yu, L.-C.; Sung, H.-W. A novel pH-sensitive hydrogel composed of N, O-carboxymethyl chitosan and alginate cross-linked by genipin for protein drug delivery. J. Control. Release 2004, 96, 285–300. [Google Scholar] [CrossRef]
- Chen, S.; Liu, M.; Jin, S.; Chen, Y. Synthesis and swelling properties of pH-sensitive hydrogels based on chitosan and poly (methacrylic acid) semi-interpenetrating polymer network. J. Appl. Polym. Sci. 2005, 98, 1720–1726. [Google Scholar] [CrossRef]
- Dos Santos, K.; Coelho, J.; Ferreira, P.; Pinto, I.; Lorenzetti, S.G.; Ferreira, E.; Higa, O.Z.; Gil, M. Synthesis and characterization of membranes obtained by graft copolymerization of 2-hydroxyethyl methacrylate and acrylic acid onto chitosan. Int. J. Pharm. 2006, 310, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.A.; Aminabhavi, T.M. Novel interpenetrating network chitosan-poly (ethylene oxide-g-acrylamide) hydrogel microspheres for the controlled release of capecitabine. Int. J. Pharm. 2006, 324, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.K.; Naidu, B.V.K.; Subha, M.; Sairam, M.; Aminabhavi, T. Novel chitosan-based pH-sensitive interpenetrating network microgels for the controlled release of cefadroxil. Carbohydr. Polym. 2006, 66, 333–344. [Google Scholar]
- Sokker, H.; Ghaffar, A.A.; Gad, Y.; Aly, A. Synthesis and characterization of hydrogels based on grafted chitosan for the controlled drug release. Carbohydr. Polym. 2009, 75, 222–229. [Google Scholar] [CrossRef]
- Lejardi, A.; Hernández, R.; Criado, M.; Santos, J.I.; Etxeberria, A.; Sarasua, J.; Mijangos, C. Novel hydrogels of chitosan and poly (vinyl alcohol)-g-glycolic acid copolymer with enhanced rheological properties. Carbohydr. Polym. 2014, 103, 267–273. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Ma, G.; Tan, H.; Jin, Y.; Nie, J. A pH-sensitive water-soluble N-carboxyethyl chitosan/poly (hydroxyethyl methacrylate) hydrogel as a potential drug sustained release matrix prepared by photopolymerization technique. Polym. Adv. Technol. 2008, 19, 1133–1141. [Google Scholar] [CrossRef]
- Sutar, P.B.; Mishra, R.K.; Pal, K.; Banthia, A.K. Development of pH sensitive polyacrylamide grafted pectin hydrogel for controlled drug delivery system. J. Mater. Sci. Mater. Med. 2008, 19, 2247–2253. [Google Scholar] [CrossRef]
- Karlsson, J.; Gatenholm, P. Preparation and characterization of cellulose-supported HEMA hydrogels. Polymer 1997, 38, 4727–4731. [Google Scholar] [CrossRef]
- Soppimath, K.S.; Kulkarni, A.R.; Aminabhavi, T.M. Controlled release of antihypertensive drug from the interpenetrating network poly (vinyl alcohol)–guar gum hydrogel microspheres. J. Biomater. Sci. Polym. Ed. 2000, 11, 27–43. [Google Scholar] [CrossRef]
- Soppirnath, K.S.; Aminabhavi, T.M. Water transport and drug release study from cross-linked polyacrylamide grafted guar gum hydrogel microspheres for the controlled release application. Eur. J. Pharm. Biopharm. 2002, 53, 87–98. [Google Scholar] [CrossRef]
- Schwoerer, A.D.; Harling, S.; Scheibe, K.; Menzel, H.; Daniels, R. Influence of degree of substitution of HES–HEMA on the release of incorporated drug models from corresponding hydrogels. Eur. J. Pharm. Biopharm. 2009, 73, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.; Tan, Y.; Li, Y.; Yang, Y. Effect of glutaraldehyde crosslinking conditions on the strength and water stability of wheat gluten fibers. Macromol. Mater. Eng. 2008, 293, 614–620. [Google Scholar] [CrossRef]
- Magami, S.M.; Oldring, P.K.T.; Castle, L.; Guthrie, J.T. The physical–chemical behaviour of amino cross-linkers and the effect of their chemistry on selected epoxy can coatings’ hydrolysis to melamine and to formaldehyde into aqueous food simulants. Prog. Org. Coat. 2015, 78, 325–333. [Google Scholar] [CrossRef]
- Costa-Júnior, E.S.; Barbosa-Stancioli, E.F.; Mansur, A.A.P.; Vasconcelos, W.L.; Mansur, H.S. Preparation and characterization of chitosan/poly(vinyl alcohol) chemically crosslinked blends for biomedical applications. Carbohydr. Polym. 2009, 76, 472–481. [Google Scholar] [CrossRef]
- Lin, J.; Zheng, S.Y.; Xiao, R.; Yin, J.; Wu, Z.L.; Zheng, Q.; Qian, J. Constitutive behaviors of tough physical hydrogels with dynamic metal-coordinated bonds. J. Mech. Phys. Solids 2020, 139, 103935. [Google Scholar] [CrossRef]
- Brack, H.; Tirmizi, S.; Risen, W. A spectroscopic and viscometric study of the metal ion-induced gelation of the biopolymer chitosan. Polymer 1997, 38, 2351–2362. [Google Scholar] [CrossRef]
- Fan, L.; Yu, L.; Xu, Y.; Yi, C.; Cai, J.; Li, M.; Huang, J. The novel alginate/N-succinyl-chitosan antibacterial blend fibers. J. Appl. Polym. Sci. 2010, 116, 2151–2156. [Google Scholar] [CrossRef]
- Gacesa, P. Alginates. Carbohydr. Polym. 1988, 8, 161–182. [Google Scholar] [CrossRef]
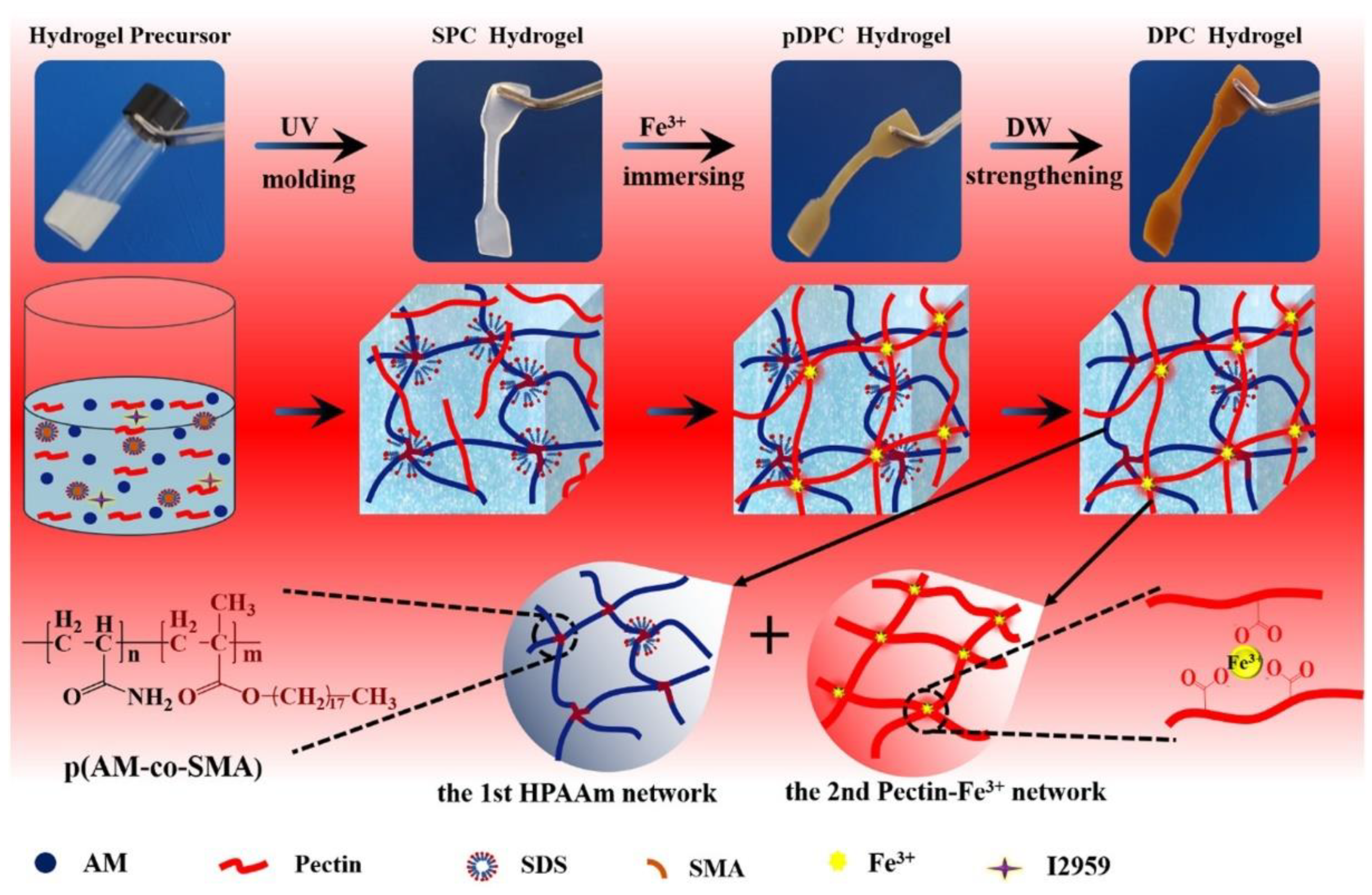
- Niu, R.; Qin, Z.; Ji, F.; Xu, M.; Tian, X.; Li, J.; Yao, F. Hybrid pectin–Fe3+/polyacrylamide double network hydrogels with excellent strength, high stiffness, superior toughness and notch-insensitivity. Soft Matter 2017, 13, 9237–9245. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Sun, H.; Qin, Z.; Che, P.; Yi, X.; Yu, Q.; Zhang, H.; Sun, X.; Yao, F.; Li, J. Fully physically crosslinked pectin-based hydrogel with high stretchability and toughness for biomedical application. Int. J. Biol. Macromol. 2020, 149, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Oderinde, O.; Liu, S.; Huang, Q.; Ma, W.; Yao, F.; Fu, G. Characterization of Xanthan gum-based hydrogel with Fe3+ ions coordination and its reversible sol-gel conversion. Carbohydr. Polym. 2019, 203, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhao, Y.; Li, Y.; Xu, J.; Fu, X.; Gao, X.; Mao, X.; Li, Z. UV-shielding alginate films crosslinked with Fe3+ containing EDTA. Carbohydr. Polym. 2020, 239, 115480. [Google Scholar] [CrossRef] [PubMed]
- Hossain, K.S.; Miyanaga, K.; Maeda, H.; Nemoto, N. Sol-gel transition behavior of pure ι-carrageenan in both salt-free and added salt states. Biomacromolecules 2001, 2, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.A.; Fresneau, M.P.; Marazuela, A.; Fabra, A.; Alonso, M.A.J. Chitosan nanoparticles as delivery systems for doxorubicin. J. Control. Release 2001, 73, 255–267. [Google Scholar] [CrossRef]
- Yokoyama, F.; Masada, I.; Shimamura, K.; Ikawa, T.; Monobe, K. Morphology and structure of highly elastic poly (vinyl alcohol) hydrogel prepared by repeated freezing-and-melting. Colloid Polym. Sci. 1986, 264, 595–601. [Google Scholar] [CrossRef]
- Takamura, A.; Ishii, F.; Hidaka, H. Drug release from poly (vinyl alcohol) gel prepared by freeze-thaw procedure. J. Control. Release 1992, 20, 21–27. [Google Scholar] [CrossRef]
- Cascone, M.; Maltinti, S.; Barbani, N.; Laus, M. Effect of chitosan and dextran on the properties of poly (vinyl alcohol) hydrogels. J. Mater. Sci. Mater. Med. 1999, 10, 431–435. [Google Scholar] [CrossRef]
- Weng, L.; Chen, X.; Chen, W. Rheological characterization of in situ crosslinkable hydrogels formulated from oxidized dextran and N-carboxyethyl chitosan. Biomacromolecules 2007, 8, 1109–1115. [Google Scholar] [CrossRef]
- Tan, H.; Chu, C.R.; Payne, K.A.; Marra, K.G. Injectable in situ forming biodegradable chitosan–hyaluronic acid based hydrogels for cartilage tissue engineering. Biomaterials 2009, 30, 2499–2506. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yeo, Y.; Highley, C.B.; Bellas, E.; Kohane, D.S. Dextran-based in situ cross-linked injectable hydrogels to prevent peritoneal adhesions. Biomaterials 2007, 28, 3418–3426. [Google Scholar] [CrossRef]
- Lee, K.Y.; Alsberg, E.; Mooney, D.J. Degradable and injectable poly (aldehyde guluronate) hydrogels for bone tissue engineering. J. Biomed. Mater. Res. 2001, 56, 228–233. [Google Scholar] [CrossRef]
- Ono, K.; Saito, Y.; Yura, H.; Ishikawa, K.; Kurita, A.; Akaike, T.; Ishihara, M. Photocrosslinkable chitosan as a biological adhesive. J. Biomed. Mater. Res. 2000, 49, 289–295. [Google Scholar] [CrossRef]
- Yoo, H.S. Photo-cross-linkable and thermo-responsive hydrogels containing chitosan and Pluronic for sustained release of human growth hormone (hGH). J. Biomater. Sci. Polym. Ed. 2007, 18, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Ryu, J.; Song, G.; Whang, M.; Kim, J. Network structure and enzymatic degradation of chitosan hydrogels determined by crosslinking methods. Carbohydr. Polym. 2019, 217, 160–167. [Google Scholar] [CrossRef]
- Pourjavadi, A.; Mahdavinia, G.R. Superabsorbency, pH-sensitivity and swelling kinetics of partially hydrolyzed chitosan-g-poly (acrylamide) hydrogels. Turk. J. Chem. 2006, 30, 595–608. [Google Scholar]
- Mahdavinia, G.; Pourjavadi, A.; Hosseinzadeh, H.; Zohuriaan, M. Modified chitosan 4. Superabsorbent hydrogels from poly (acrylic acid-co-acrylamide) grafted chitosan with salt-and pH-responsiveness properties. Eur. Polym. J. 2004, 40, 1399–1407. [Google Scholar] [CrossRef]
- Yasin, T.; Rasool, N.; Akhter, Z. Synthesis of carboxymethyl-chitosan/acrylic acid hydrogel using silane crosslinker. e-Polymers 2008, 8, 1636–1646. [Google Scholar] [CrossRef]
- Pourjavadi, A.; Harzandi, A.; Hosseinzadeh, H. Modified carrageenan 3. Synthesis of a novel polysaccharide-based superabsorbent hydrogel via graft copolymerization of acrylic acid onto kappa-carrageenan in air. Eur. Polym. J. 2004, 40, 1363–1370. [Google Scholar] [CrossRef]
- Rasool, N.; Yasin, T.; Heng, J.Y.; Akhter, Z. Synthesis and characterization of novel pH-, ionic strength and temperature-sensitive hydrogel for insulin delivery. Polymer 2010, 51, 1687–1693. [Google Scholar] [CrossRef]
- Elvira, C.; Mano, J.; San Roman, J.; Reis, R. Starch-based biodegradable hydrogels with potential biomedical applications as drug delivery systems. Biomaterials 2002, 23, 1955–1966. [Google Scholar] [CrossRef]
- Bao, Y.; Ma, J.; Li, N. Synthesis and swelling behaviors of sodium carboxymethyl cellulose-g-poly (AA-co-AM-co-AMPS)/MMT superabsorbent hydrogel. Carbohydr. Polym. 2011, 84, 76–82. [Google Scholar] [CrossRef]
- Dragan, E.S. Design and applications of interpenetrating polymer network hydrogels. A review. Chem. Eng. J. 2014, 243, 572–590. [Google Scholar] [CrossRef]
- Cai, Z.; Kim, J. Cellulose–chitosan interpenetrating polymer network for electro-active paper actuator. J. Appl. Polym. Sci. 2009, 114, 288–297. [Google Scholar] [CrossRef]
- Rokhade, A.P.; Shelke, N.B.; Patil, S.A.; Aminabhavi, T.M. Novel interpenetrating polymer network microspheres of chitosan and methylcellulose for controlled release of theophylline. Carbohydr. Polym. 2007, 69, 678–687. [Google Scholar] [CrossRef]
- Ahmed, A.A.-K.; Naik, H.B.; Sherigara, B. Synthesis and characterization of chitosan-based pH-sensitive semi-interpenetrating network microspheres for controlled release of diclofenac sodium. Carbohydr. Res. 2009, 344, 699–706. [Google Scholar] [CrossRef]
- Angadi, S.C.; Manjeshwar, L.S.; Aminabhavi, T.M. Interpenetrating polymer network blend microspheres of chitosan and hydroxyethyl cellulose for controlled release of isoniazid. Int. J. Biol. Macromol. 2010, 47, 171–179. [Google Scholar] [CrossRef]
- Rokhade, A.P.; Patil, S.A.; Aminabhavi, T.M. Synthesis and characterization of semi-interpenetrating polymer network microspheres of acrylamide grafted dextran and chitosan for controlled release of acyclovir. Carbohydr. Polym. 2007, 67, 605–613. [Google Scholar] [CrossRef]
- Yang, J.; Chen, J.; Pan, D.; Wan, Y.; Wang, Z. pH-sensitive interpenetrating network hydrogels based on chitosan derivatives and alginate for oral drug delivery. Carbohydr. Polym. 2013, 92, 719–725. [Google Scholar] [CrossRef]
- Povea, M.B.; Monal, W.A.; Cauich-Rodríguez, J.V.; Pat, A.M.; Rivero, N.B.; Covas, C.P. Interpenetrated chitosan-poly (acrylic acid-co-acrylamide) hydrogels. Synthesis, characterization and sustained protein release studies. Mater. Sci. Appl. 2011, 2, 509. [Google Scholar] [CrossRef]
- Pulat, M.; Asıl, D. Fluconazole release through semi-interpenetrating polymer network hydrogels based on chitosan, acrylic acid, and citraconic acid. J. Appl. Polym. Sci. 2009, 113, 2613–2619. [Google Scholar] [CrossRef]
- Yin, L.; Zhao, Z.; Hu, Y.; Ding, J.; Cui, F.; Tang, C.; Yin, C. Polymer–protein interaction, water retention, and biocompatibility of a stimuli-sensitive superporous hydrogel containing interpenetrating polymer networks. J. Appl. Polym. Sci. 2008, 108, 1238–1248. [Google Scholar] [CrossRef]
- Milosavljević, N.B.; Milašinović, N.Z.; Popović, I.G.; Filipović, J.M.; Kalagasidis Krušić, M.T. Preparation and characterization of pH-sensitive hydrogels based on chitosan, itaconic acid and methacrylic acid. Polym. Int. 2011, 60, 443–452. [Google Scholar] [CrossRef]
- Sharma, C.P. Interpolymer complex microparticles based on polymethacrylic acid-chitosan for oral insulin delivery. J. Appl. Polym. Sci. 2006, 99, 506–512. [Google Scholar]
- El-Sherbiny, I.; Lins, R.; Abdel-Bary, E.; Harding, D. Preparation, characterization, swelling and in vitro drug release behaviour of poly [N-acryloylglycine-chitosan] interpolymeric pH and thermally-responsive hydrogels. Eur. Polym. J. 2005, 41, 2584–2591. [Google Scholar] [CrossRef]
- Kim, S.J.; Yoon, S.G.; Kim, I.Y.; Kim, S.I. Swelling characterization of the semiinterpenetrating polymer network hydrogels composed of chitosan and poly (diallyldimethylammonium chloride). J. Appl. Polym. Sci. 2004, 91, 2876–2880. [Google Scholar] [CrossRef]
- Kim, S.J.; Shin, S.R.; Spinks, G.M.; Kim, I.Y.; Kim, S.I. Synthesis and characteristics of a semi-interpenetrating polymer network based on chitosan/polyaniline under different pH conditions. J. Appl. Polym. Sci. 2005, 96, 867–873. [Google Scholar] [CrossRef][Green Version]
- Guo, B.; Yuan, J.; Yao, L.; Gao, Q. Preparation and release profiles of pH/temperature-responsive carboxymethyl chitosan/P (2-(dimethylamino) ethyl methacrylate) semi-IPN amphoteric hydrogel. Colloid Polym. Sci. 2007, 285, 665–671. [Google Scholar] [CrossRef]
- Martinez-Ruvalcaba, A.; Sanchez-Diaz, J.; Becerra, F.; Cruz-Barba, L.; Gonzalez-Alvarez, A. Swelling characterization and drug delivery kinetics of polyacrylamide-co-itaconic acid/chitosan hydrogels. Express Polym. Lett. 2009, 3, 25–32. [Google Scholar] [CrossRef]
- Xia, Y.-Q.; Guo, T.-Y.; Song, M.-D.; Zhang, B.-H.; Zhang, B.-L. Hemoglobin recognition by imprinting in semi-interpenetrating polymer network hydrogel based on polyacrylamide and chitosan. Biomacromolecules 2005, 6, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Babu, V.R.; Hosamani, K.; Aminabhavi, T. Preparation and in-vitro release of chlorothiazide novel pH-sensitive chitosan-N, N′-dimethylacrylamide semi-interpenetrating network microspheres. Carbohydr. Polym. 2008, 71, 208–217. [Google Scholar] [CrossRef]
- Han, Y.A.; Lee, E.M.; Ji, B.C. Mechanical properties of semi-interpenetrating polymer network hydrogels based on poly (2-hydroxyethyl methacrylate) copolymer and chitosan. Fibers Polym. 2008, 9, 393–399. [Google Scholar] [CrossRef]
- Kim, S.J.; Shin, S.R.; Lee, S.M.; Kim, I.Y.; Kim, S.I. Electromechanical properties of hydrogels based on chitosan and poly (hydroxyethyl methacrylate) in NaCl solution. Smart Mater. Struct. 2004, 13, 1036. [Google Scholar] [CrossRef]
- Kim, S.J.; Yoon, S.G.; Lee, Y.H.; Kim, S.I. Bending behavior of hydrogels composed of poly (methacrylic acid) and alginate by electrical stimulus. Polym. Int. 2004, 53, 1456–1460. [Google Scholar] [CrossRef]
- Li, X.; Xu, S.; Wang, J.; Chen, X.; Feng, S. Structure and characterization of amphoteric semi-IPN hydrogel based on cationic starch. Carbohydr. Polym. 2009, 75, 688–693. [Google Scholar] [CrossRef]
- Chen, J.; Liu, M.; Jin, S.; Liu, H. Synthesis and characterization of κ-carrageenan/poly (N, N-diethylacrylamide) semi-interpenetrating polymer network hydrogels with rapid response to temperature. Polym. Adv. Technol. 2008, 19, 1656–1663. [Google Scholar] [CrossRef]
- Wang, M.; Fang, Y.; Hu, D. Preparation and properties of chitosan-poly (N-isopropylacrylamide) full-IPN hydrogels. React. Funct. Polym. 2001, 48, 215–221. [Google Scholar] [CrossRef]
- Kim, S.J.; Park, S.J.; Kim, S.I. Swelling behavior of interpenetrating polymer network hydrogels composed of poly (vinyl alcohol) and chitosan. React. Funct. Polym. 2003, 55, 53–59. [Google Scholar] [CrossRef]
- Yin, L.; Fei, L.; Cui, F.; Tang, C.; Yin, C. Superporous hydrogels containing poly (acrylic acid-co-acrylamide)/O-carboxymethyl chitosan interpenetrating polymer networks. Biomaterials 2007, 28, 1258–1266. [Google Scholar] [CrossRef]
- Dragan, E.S.; Perju, M.M.; Dinu, M.V. Preparation and characterization of IPN composite hydrogels based on polyacrylamide and chitosan and their interaction with ionic dyes. Carbohydr. Polym. 2012, 88, 270–281. [Google Scholar] [CrossRef]
- Yin, L.; Fei, L.; Tang, C.; Yin, C. Synthesis, characterization, mechanical properties and biocompatibility of interpenetrating polymer network–super-porous hydrogel containing sodium alginate. Polym. Int. 2007, 56, 1563–1571. [Google Scholar] [CrossRef]
- Lee, S.B.; Park, E.K.; Lim, Y.M.; Cho, S.K.; Kim, S.Y.; Lee, Y.M.; Nho, Y.C. Preparation of alginate/poly (N-isopropylacrylamide) semi-interpenetrating and fully interpenetrating polymer network hydrogels with γ-ray irradiation and their swelling behaviors. J. Appl. Polym. Sci. 2006, 100, 4439–4446. [Google Scholar] [CrossRef]
- Zadražil, A.; Štěpánek, F. Investigation of thermo-responsive optical properties of a composite hydrogel. Colloids Surf. A Physicochem. Eng. Asp. 2010, 372, 115–119. [Google Scholar]
- Buenger, D.; Topuz, F.; Groll, J. Hydrogels in sensing applications. Prog. Polym. Sci. 2012, 37, 1678–1719. [Google Scholar] [CrossRef]
- Guo, W.; Lu, C.H.; Orbach, R.; Wang, F.; Qi, X.J.; Cecconello, A.; Seliktar, D.; Willner, I. pH-Stimulated DNA Hydrogels Exhibiting Shape-Memory Properties. Adv. Mater. 2015, 27, 73–78. [Google Scholar] [CrossRef]
- Sun, J.; Jiang, G.; Wang, Y.; Ding, F. Thermosensitive chitosan hydrogel for implantable drug delivery: Blending PVA to mitigate body response and promote bioavailability. J. Appl. Polym. Sci. 2012, 125, 2092–2101. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, K.J.; Kim, I.Y.; Kim, S.I. Swelling Kinetics of Interpenetrating Polymer Hydrogels Composed of Poly(Vinyl Alcohol)/Chitosan. J. Macromol. Sci. Part A 2003, 40, 501–510. [Google Scholar] [CrossRef]
- Wu, C.-J.; Gaharwar, A.K.; Schexnailder, P.J.; Schmidt, G. Development of Biomedical Polymer-Silicate Nanocomposites: A Materials Science Perspective. Materials 2010, 3, 2986–3005. [Google Scholar] [CrossRef]
- Bashir, S.; Teo, Y.Y.; Ramesh, S.; Ramesh, K.; Mushtaq, M.W. Rheological behavior of biodegradable N-succinyl chitosan-g-poly (acrylic acid) hydrogels and their applications as drug carrier and in vitro theophylline release. Int. J. Biol. Macromol. 2018, 117, 454–466. [Google Scholar] [CrossRef]
- Das, N. Preparation methods and properties of hydrogel: A review. Int. J. Pharm. Pharm. Sci. 2013, 5, 112–117. [Google Scholar]
- Oyen, M. Mechanical characterisation of hydrogel materials. Int. Mater. Rev. 2014, 59, 44–59. [Google Scholar] [CrossRef]
- Goodwin, J.W.; Hughes, R.W. Rheology for Chemists: An Introduction; Royal Society of Chemistry: Cambridge, UK, 2008. [Google Scholar]
- Barnes, H.A.; Hutton, J.F.; Walters, K. An Introduction to Rheology; Elsevier: Amsterdam, The Netherlands, 1989; Volume 3. [Google Scholar]
- Kang, E.Y.; Moon, H.J.; Joo, M.K.; Jeong, B. Thermogelling chitosan-g-(PAF-PEG) aqueous solution as an injectable scaffold. Biomacromolecules 2012, 13, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Moura, M.J.; Figueiredo, M.M.; Gil, M.H. Rheological study of genipin cross-linked chitosan hydrogels. Biomacromolecules 2007, 8, 3823–3829. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Wang, Q.; Sun, S.; Zhu, W.; Wu, P. A bioinspired mineral hydrogel as a self-healable, mechanically adaptable ionic skin for highly sensitive pressure sensing. Adv. Mater. 2017, 29, 1700321. [Google Scholar] [CrossRef]
- Khan, Y.; Bashir, S.; Hina, M.; Ramesh, S.; Ramesh, K.; Mujtaba, M.; Lahiri, I. Effect of Charge Density on the Mechanical and Electrochemical Properties of Poly (acrylic acid) Hydrogel Electrolytes Based Flexible Supercapacitors. Mater. Today Commun. 2020, 25, 101558. [Google Scholar] [CrossRef]
- Yang, Z.; Han, B.; Fu, D.; Liu, W. Acute toxicity of high dosage carboxymethyl chitosan and its effect on the blood parameters in rats. J. Mater. Sci. Mater. Med. 2012, 23, 457–462. [Google Scholar] [CrossRef]
- Chan, P.; Kurisawa, M.; Chung, J.E.; Yang, Y.-Y. Synthesis and characterization of chitosan-g-poly (ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene delivery. Biomaterials 2007, 28, 540–549. [Google Scholar] [CrossRef]
- Anitha, A.; Rani, V.D.; Krishna, R.; Sreeja, V.; Selvamurugan, N.; Nair, S.; Tamura, H.; Jayakumar, R. Synthesis, characterization, cytotoxicity and antibacterial studies of chitosan, O-carboxymethyl and N, O-carboxymethyl chitosan nanoparticles. Carbohydr. Polym. 2009, 78, 672–677. [Google Scholar] [CrossRef]
- Lü, S.; Liu, M.; Ni, B. An injectable oxidized carboxymethylcellulose/N-succinyl-chitosan hydrogel system for protein delivery. Chem. Eng. J. 2010, 160, 779–787. [Google Scholar] [CrossRef]
- Lu, B.; Wu, D.-Q.; Zheng, H.; Quan, C.-Y.; Zhang, X.-Z.; Zhuo, R.-X. Galactosyl conjugated N-succinyl-chitosan-graft-polyethylenimine for targeting gene transfer. Mol. Biosyst. 2010, 6, 2529–2538. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Sun, Y.-X.; Li, Y.-Q.; Zhang, X.-Z.; Zhuo, R.-X. N-Succinyl-chitosan grafted with low molecular weight polyethylenimine as a serum-resistant gene vector. Mol. Biosyst. 2009, 5, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Aiping, Z.; Tian, C.; Lanhua, Y.; Hao, W.; Ping, L. Synthesis and characterization of N-succinyl-chitosan and its self-assembly of nanospheres. Carbohydr. Polym. 2006, 66, 274–279. [Google Scholar] [CrossRef]
- Mishra, R.K.; Datt, M.; Banthia, A.K. Synthesis and characterization of pectin/PVP hydrogel membranes for drug delivery system. AAPS PharmSciTech 2008, 9, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Xu, X.; Wang, Y.; Yan, X.; Guo, G.; Huang, M.; Luo, F.; Zhao, X.; Wei, Y.; Qian, Z. Synthesis and characterization of poly (methoxyl ethylene glycol-caprolactone-co-methacrylic acid-co-poly (ethylene glycol) methyl ether methacrylate) pH-sensitive hydrogel for delivery of dexamethasone. Int. J. Pharm. 2010, 389, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fan, L.; Yi, X.; Zhou, Z.; Liu, C.; Fu, R.; Dai, C.; Wang, Z.; Chen, X.; Yu, P. Soft conducting polymer hydrogels cross-linked and doped by tannic acid for spinal cord injury repair. ACS Nano 2018, 12, 10957–10967. [Google Scholar] [CrossRef]
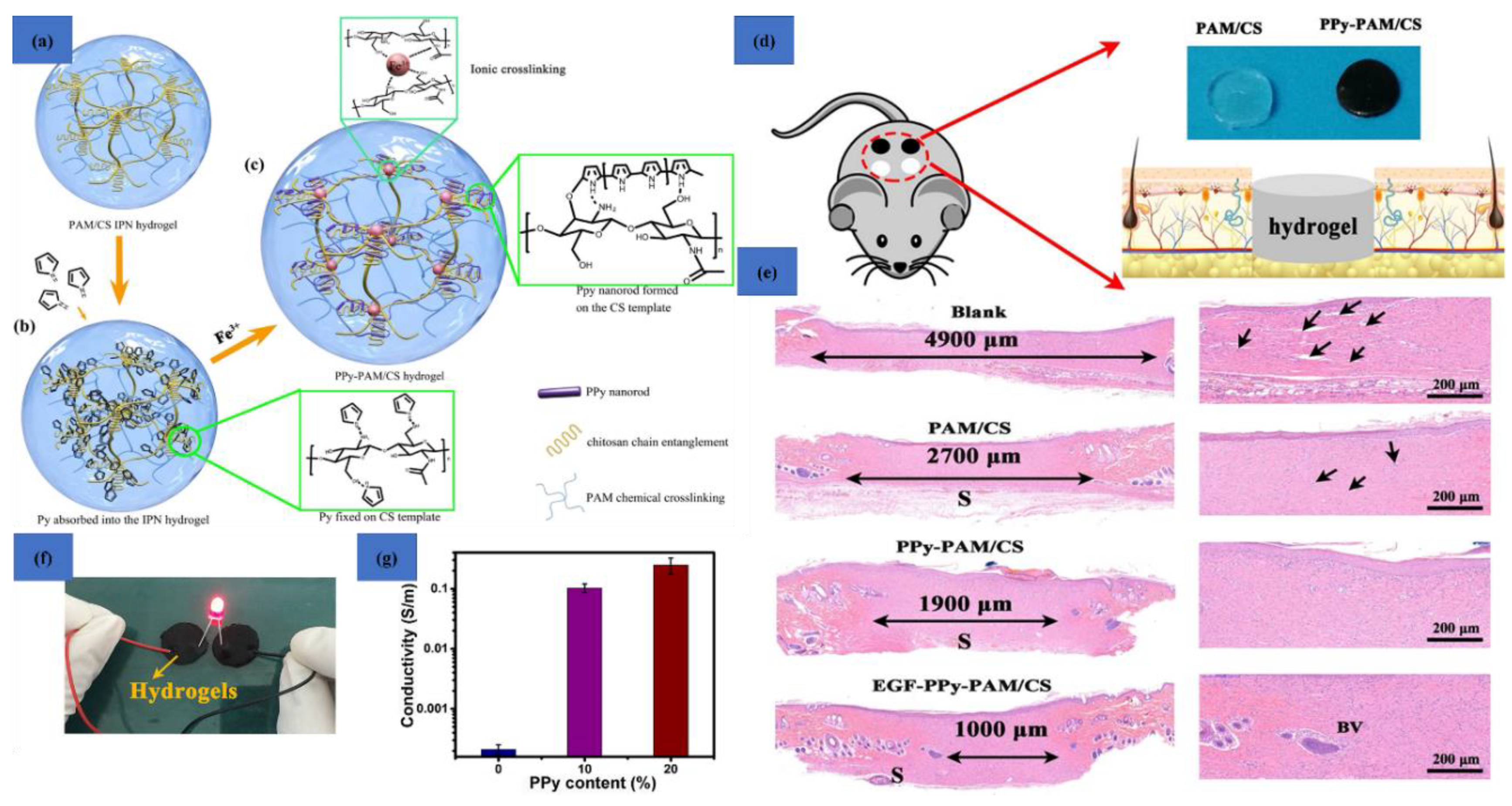
- Gan, D.; Han, L.; Wang, M.; Xing, W.; Xu, T.; Zhang, H.; Wang, K.; Fang, L.; Lu, X. Conductive and tough hydrogels based on biopolymer molecular templates for controlling in situ formation of polypyrrole nanorods. ACS Appl. Mater. Interfaces 2018, 10, 36218–36228. [Google Scholar] [CrossRef]
- Onishi, H.; Machida, Y. Biodegradation and distribution of water-soluble chitosan in mice. Biomaterials 1999, 20, 175–182. [Google Scholar] [CrossRef]
- Verheul, R.J.; Amidi, M.; van Steenbergen, M.J.; van Riet, E.; Jiskoot, W.; Hennink, W.E. Influence of the degree of acetylation on the enzymatic degradation and in vitro biological properties of trimethylated chitosans. Biomaterials 2009, 30, 3129–3135. [Google Scholar] [CrossRef]
- Ito, T.; Yeo, Y.; Highley, C.B.; Bellas, E.; Benitez, C.A.; Kohane, D.S. The prevention of peritoneal adhesions by in situ cross-linking hydrogels of hyaluronic acid and cellulose derivatives. Biomaterials 2007, 28, 975–983. [Google Scholar] [CrossRef]
- Das, R.; Panda, A.; Pal, S. Synthesis and characterization of a novel polymeric hydrogel based on hydroxypropyl methyl cellulose grafted with polyacrylamide. Cellulose 2012, 19, 933–945. [Google Scholar] [CrossRef]
- van Dijk-Wolthuis, W.; Hoogeboom, J.; Van Steenbergen, M.; Tsang, S.; Hennink, W. Degradation and release behavior of dextran-based hydrogels. Macromolecules 1997, 30, 4639–4645. [Google Scholar] [CrossRef]
- Majzoob, S.; Atyabi, F.; Dorkoosh, F.; Kafedjiiski, K.; Loretz, B.; Bernkop-Schnürch, A. Pectin-cysteine conjugate: Synthesis and in-vitro evaluation of its potential for drug delivery. J. Pharm. Pharmacol. 2006, 58, 1601–1610. [Google Scholar] [CrossRef]
- Rana, V.; Rai, P.; Tiwary, A.K.; Singh, R.S.; Kennedy, J.F.; Knill, C.J. Modified gums: Approaches and applications in drug delivery. Carbohydr. Polym. 2011, 83, 1031–1047. [Google Scholar] [CrossRef]
- Tan, H.; Marra, K.G. Injectable, biodegradable hydrogels for tissue engineering applications. Materials 2010, 3, 1746–1767. [Google Scholar] [CrossRef]
- Gao, X.; He, C.; Xiao, C.; Zhuang, X.; Chen, X. Biodegradable pH-responsive polyacrylic acid derivative hydrogels with tunable swelling behavior for oral delivery of insulin. Polymer 2013, 54, 1786–1793. [Google Scholar] [CrossRef]
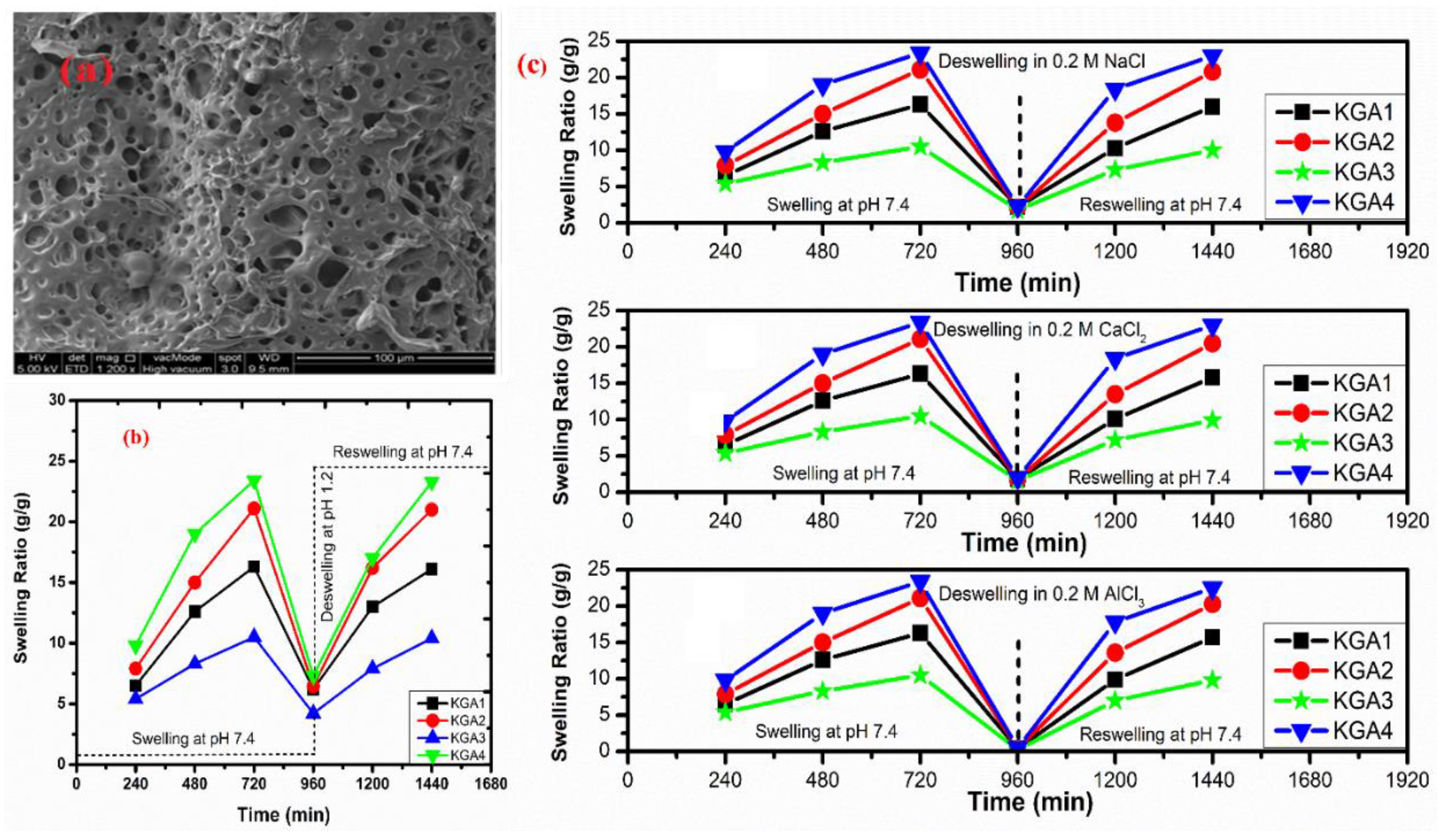
- Bashir, S.; Teo, Y.Y.; Ramesh, S.; Ramesh, K. Synthesis and characterization of karaya gum-g-poly (acrylic acid) hydrogels and in vitro release of hydrophobic quercetin. Polymer 2018, 147, 108–120. [Google Scholar] [CrossRef]
- Tanan, W.; Panichpakdee, J.; Saengsuwan, S. Novel biodegradable hydrogel based on natural polymers: Synthesis, characterization, swelling/reswelling and biodegradability. Eur. Polym. J. 2019, 112, 678–687. [Google Scholar] [CrossRef]
- Qureshi, D.; Nayak, S.K.; Maji, S.; Anis, A.; Kim, D.; Pal, K. Environment sensitive hydrogels for drug delivery applications. Eur. Polym. J. 2019, 120, 109220. [Google Scholar] [CrossRef]
- Siegel, R.A.; Falamarzian, M.; Firestone, B.A.; Moxley, B.C. pH-controlled release from hydrophobic/polyelectrolyte copolymer hydrogels. J. Control. Release 1988, 8, 179–182. [Google Scholar] [CrossRef]
- Patel, V.R.; Amiji, M.M. Preparation and characterization of freeze-dried chitosan-poly (ethylene oxide) hydrogels for site-specific antibiotic delivery in the stomach. Pharm. Res. 1996, 13, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Bilia, A.; Carelli, V.; Di Colo, G.; Nannipieri, E. In vitro evaluation of a pH-sensitive hydrogel for control of GI drug delivery from silicone-based matrices. Int. J. Pharm. 1996, 130, 83–92. [Google Scholar] [CrossRef]
- Vaghani, S.S.; Patel, M.M.; Satish, C. Synthesis and characterization of pH-sensitive hydrogel composed of carboxymethyl chitosan for colon targeted delivery of ornidazole. Carbohydr. Res. 2012, 347, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Mura, C.; Manconi, M.; Valenti, D.; Manca, M.L.; Díez-Sales, O.; Loy, G.; Fadda, A.M. In vitro study of N-succinyl chitosan for targeted delivery of 5-aminosalicylic acid to colon. Carbohydr. Polym. 2011, 85, 578–583. [Google Scholar] [CrossRef]
- Chen, H.; Gu, Y.; Hu, Y.; Qian, Z. Characterization of pH-and temperature-sensitive hydrogel nanoparticles for controlled drug release. PDA J. Pharm. Sci. Technol. 2007, 61, 303–313. [Google Scholar]
- Katono, H.; Maruyama, A.; Sanui, K.; Ogata, N.; Okano, T.; Sakurai, Y. Thermo-responsive swelling and drug release switching of interpenetrating polymer networks composed of poly (acrylamide-co-butyl methacrylate) and poly (acrylic acid). J. Control. Release 1991, 16, 215–227. [Google Scholar] [CrossRef]
- Schmolka, I.R. Artificial skin I. Preparation and properties of pluronic F-127 gels for treatment of burns. J. Biomed. Mater. Res. 1972, 6, 571–582. [Google Scholar] [CrossRef]
- Hoffman, A.; Chen, G.; Wu, X.; Ding, Z. Graft copolymers of PEO-PPO-PEO triblock polyethers on bioadhesive polymer backbones: Synthesis and properties. In Abstracts of Papers of the American Chemical Society; American Chemical Society: Washington, DC, USA, 1997. [Google Scholar]
- Wang, G.; Zhang, L.; Zhang, J. A review of electrode materials for electrochemical supercapacitors. Chem. Soc. Rev. 2012, 41, 797–828. [Google Scholar] [CrossRef]
- Pal, B.; Yang, S.; Ramesh, S.; Thangadurai, V.; Jose, R. Electrolyte selection for supercapacitive devices: A critical review. Nanoscale Adv. 2019, 1, 3807–3835. [Google Scholar] [CrossRef]
- Choudhury, N.; Sampath, S.; Shukla, A. Hydrogel-polymer electrolytes for electrochemical capacitors: An overview. Energy Environ. Sci. 2009, 2, 55–67. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y. Materials for electrochemical capacitors. Nat Mater 2008, 7, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Omar, F.S.; Hina, M.; Numan, A.; Iqbal, J.; Ramesh, S.; Ramesh, K. Synthesis and characterization of hybrid poly (N, N-dimethylacrylamide) composite hydrogel electrolytes and their performance in supercapacitor. Electrochim. Acta 2020, 332, 135438. [Google Scholar] [CrossRef]
- Yang, H.; Ji, X.; Tan, Y.; Liu, Y.; Ran, F. Modified supramolecular carboxylated chitosan as hydrogel electrolyte for quasi-solid-state supercapacitors. J. Power Sources 2019, 441, 227174. [Google Scholar] [CrossRef]
- Han, L.; Huang, H.; Fu, X.; Li, J.; Yang, Z.; Liu, X.; Pan, L.; Xu, M. A flexible, high-voltage and safe zwitterionic natural polymer hydrogel electrolyte for high-energy-density zinc-ion hybrid supercapacitor. Chem. Eng. J. 2020, 392, 123733. [Google Scholar] [CrossRef]
- Yang, L.; Song, L.; Feng, Y.; Cao, M.; Zhang, P.; Zhang, X.; Yao, J. Zinc Ion Trapping in Cellulose Hydrogel as Solid Electrolyte for Safe and Flexible Supercapacitor. J. Mater. Chem. A 2020, 8, 12314–12318. [Google Scholar] [CrossRef]
- Willfahrt, A.; Steiner, E.; Hötzel, J.; Crispin, X. Printable acid-modified corn starch as non-toxic, disposable hydrogel-polymer electrolyte in supercapacitors. Appl. Phys. A 2019, 125, 474. [Google Scholar] [CrossRef]
- Alipoori, S.; Mazinani, S.; Aboutalebi, S.H.; Sharif, F. Review of PVA-based gel polymer electrolytes in flexible solid-state supercapacitors: Opportunities and challenges. J. Energy Storage 2020, 27, 101072. [Google Scholar] [CrossRef]
- Zhang, H.; Niu, W.; Zhang, S. Extremely stretchable, sticky and conductive double-network ionic hydrogel for ultra-stretchable and compressible supercapacitors. Chem. Eng. J. 2020, 387, 124105. [Google Scholar] [CrossRef]
- Guo, L.; Ma, W.-B.; Wang, Y.; Song, X.-Z.; Ma, J.; Han, X.-D.; Tao, X.-Y.; Guo, L.-T.; Fan, H.-L.; Liu, Z.-S. A chemically crosslinked hydrogel electrolyte based all-in-one flexible supercapacitor with superior performance. J. Alloys Compd. 2020, 843, 155895. [Google Scholar] [CrossRef]
- Fang, L.; Cai, Z.; Ding, Z.; Chen, T.; Zhang, J.; Chen, F.; Shen, J.; Chen, F.; Li, R.; Zhou, X. Skin-Inspired Surface-Microstructured Tough Hydrogel Electrolytes for Stretchable Supercapacitors. ACS Appl. Mater. Interfaces 2019, 11, 21895–21903. [Google Scholar] [CrossRef]
- Saborío, M.G.; Svelic, P.; Casanovas, J.; Ruano, G.; Pérez-Madrigal, M.M.; Franco, L.; Torras, J.; Estrany, F.; Alemán, C. Hydrogels for flexible and compressible free standing cellulose supercapacitors. Eur. Polym. J. 2019, 118, 347–357. [Google Scholar] [CrossRef]
- Chen, Q.; Li, X.; Zang, X.; Cao, Y.; He, Y.; Li, P.; Wang, K.; Wei, J.; Wu, D.; Zhu, H. Effect of different gel electrolytes on graphene-based solid-state supercapacitors. RSC Adv. 2014, 4, 36253–36256. [Google Scholar] [CrossRef]
- Wang, H.; Wu, J.; Qiu, J.; Zhang, K.; Shao, J.; Yan, L. In situ formation of a renewable cellulose hydrogel electrolyte for high-performance flexible all-solid-state asymmetric supercapacitors. Sustain. Energy Fuels 2019, 3, 3109–3115. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, X.; Li, C.; Sun, X.; Meng, Q.; Ma, Y.; Wei, Z. Chemically crosslinked hydrogel film leads to integrated flexible supercapacitors with superior performance. Adv. Mater. 2015, 27, 7451–7457. [Google Scholar] [CrossRef]
- Nohara, S.; Wada, H.; Furukawa, N.; Inoue, H.; Morita, M.; Iwakura, C. Electrochemical characterization of new electric double layer capacitor with polymer hydrogel electrolyte. Electrochim. Acta 2003, 48, 749–753. [Google Scholar] [CrossRef]
- Lopez, J.; Sun, Y.; Mackanic, D.G.; Lee, M.; Foudeh, A.M.; Song, M.S.; Cui, Y.; Bao, Z. A Dual-Crosslinking Design for Resilient Lithium-Ion Conductors. Adv. Mater. 2018, 30, 1804142. [Google Scholar] [CrossRef]
- Na, R.; Liu, Y.; Lu, N.; Zhang, S.; Liu, F.; Wang, G. Mechanically robust hydrophobic association hydrogel electrolyte with efficient ionic transport for flexible supercapacitors. Chem. Eng. J. 2019, 374, 738–747. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, H.; Ma, P.X.; Guo, B. Self-healing conductive hydrogels: Preparation, properties and applications. Nanoscale 2020, 12, 1224–1246. [Google Scholar] [CrossRef]
- Taylor, D.L.; in het Panhuis, M. Self-healing hydrogels. Adv. Mater. 2016, 28, 9060–9093. [Google Scholar] [CrossRef]
- Wang, Z.; Tao, F.; Pan, Q. A self-healable polyvinyl alcohol-based hydrogel electrolyte for smart electrochemical capacitors. J. Mater. Chem. A 2016, 4, 17732–17739. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, X.; Tang, Q.; Bao, H.; Wang, G.; Saha, P. A self-healable and easily recyclable supramolecular hydrogel electrolyte for flexible supercapacitors. J. Mater. Chem. A 2016, 4, 8769–8776. [Google Scholar] [CrossRef]
- Tao, F.; Qin, L.; Wang, Z.; Pan, Q. Self-healable and cold-resistant supercapacitor based on a multifunctional hydrogel electrolyte. ACS Appl. Mater. Interfaces 2017, 9, 15541–15548. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Qi, P.; Zhang, X.; Wang, L.; Tan, Y.; Luan, Z.; Xia, Y.; Li, Y.; Sui, K. Multiple weak H-bonds lead to highly sensitive, stretchable, self-adhesive, and self-healing ionic sensors. ACS Appl. Mater. Interfaces 2019, 11, 7755–7763. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.K.; Guo, H.L.; Sun, T.L.; Ihsan, A.B.; Kurokawa, T.; Takahata, M.; Nonoyama, T.; Nakajima, T.; Gong, J.P. Self-Adjustable Adhesion of Polyampholyte Hydrogels. Adv. Mater. 2015, 27, 7344–7348. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Sun, T.L.; Nakajima, T.; Kurokawa, T.; Zhao, Y.; Sato, K.; Ihsan, A.B.; Li, X.; Guo, H.; Gong, J.P. Oppositely charged polyelectrolytes form tough, self-healing, and rebuildable hydrogels. Adv. Mater. 2015, 27, 2722–2727. [Google Scholar] [CrossRef]
- Yuan, N.; Xu, L.; Xu, B.; Zhao, J.; Rong, J. Chitosan derivative-based self-healable hydrogels with enhanced mechanical properties by high-density dynamic ionic interactions. Carbohydr. Polym. 2018, 193, 259–267. [Google Scholar] [CrossRef]
- Peng, H.; Lv, Y.; Wei, G.; Zhou, J.; Gao, X.; Sun, K.; Ma, G.; Lei, Z. A flexible and self-healing hydrogel electrolyte for smart supercapacitor. J. Power Sources 2019, 431, 210–219. [Google Scholar] [CrossRef]
- Han, J.; Wang, H.; Yue, Y.; Mei, C.; Chen, J.; Huang, C.; Wu, Q.; Xu, X. A self-healable and highly flexible supercapacitor integrated by dynamically cross-linked electro-conductive hydrogels based on nanocellulose-templated carbon nanotubes embedded in a viscoelastic polymer network. Carbon 2019, 149, 1–18. [Google Scholar] [CrossRef]
- Du, Z.; Su, Y.; Qu, Y.; Zhao, L.; Jia, X.; Mo, Y.; Yu, F.; Du, J.; Chen, Y. A mechanically robust, biodegradable and high performance cellulose gel membrane as gel polymer electrolyte of lithium-ion battery. Electrochim. Acta 2019, 299, 19–26. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Zhu, M.; Huang, Y.; Tang, Z.; Pei, Z.; Wang, Z.; Shi, Z.; Liu, J.; Huang, Y. Towards wearable electronic devices: A quasi-solid-state aqueous lithium-ion battery with outstanding stability, flexibility, safety and breathability. Nano Energy 2018, 44, 164–173. [Google Scholar] [CrossRef]
- Zhong, L.; Lu, Y.; Li, H.; Tao, Z.; Chen, J. High-performance aqueous sodium-ion batteries with hydrogel electrolyte and alloxazine/CMK-3 anode. ACS Sustain. Chem. Eng. 2018, 6, 7761–7768. [Google Scholar] [CrossRef]
- Han, Q.; Chi, X.; Zhang, S.; Liu, Y.; Zhou, B.; Yang, J.; Liu, Y. Durable, flexible self-standing hydrogel electrolytes enabling high-safety rechargeable solid-state zinc metal batteries. J. Mater. Chem. A 2018, 6, 23046–23054. [Google Scholar] [CrossRef]
- Wang, Z.; Mo, F.; Ma, L.; Yang, Q.; Liang, G.; Liu, Z.; Li, H.; Li, N.; Zhang, H.; Zhi, C. Highly Compressible Cross-Linked Polyacrylamide Hydrogel-Enabled Compressible Zn–MnO2 Battery and a Flexible Battery–Sensor System. ACS Appl. Mater. Interfaces 2018, 10, 44527–44534. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, C.; Huang, Y.; Huang, Y.; Zhu, M.; Pei, Z.; Xue, Q.; Wang, Z.; Liu, Z.; Tang, Z. An extremely safe and wearable solid-state zinc ion battery based on a hierarchical structured polymer electrolyte. Energy Environ. Sci. 2018, 11, 941–951. [Google Scholar] [CrossRef]
- Ma, L.; Chen, S.; Wang, D.; Yang, Q.; Mo, F.; Liang, G.; Li, N.; Zhang, H.; Zapien, J.A.; Zhi, C. Super-Stretchable Zinc–Air Batteries Based on an Alkaline-Tolerant Dual-Network Hydrogel Electrolyte. Adv. Energy Mater. 2019, 9, 1803046. [Google Scholar] [CrossRef]
- Leng, K.; Li, G.; Guo, J.; Zhang, X.; Wang, A.; Liu, X.; Luo, J. A Safe Polyzwitterionic Hydrogel Electrolyte for Long-Life Quasi-Solid State Zinc Metal Batteries. Adv. Funct. Mater. 2020, 30, 2001317. [Google Scholar] [CrossRef]
- Chen, M.; Zhou, W.; Wang, A.; Huang, A.; Chen, J.; Xu, J.; Wong, C.-P. Anti-freezing flexible aqueous Zn–MnO2 batteries working at −35° C enabled by a borax-crosslinked polyvinyl alcohol/glycerol gel electrolyte. J. Mater. Chem. A 2020, 8, 6828–6841. [Google Scholar] [CrossRef]
- Duan, J.; Xie, W.; Yang, P.; Li, J.; Xue, G.; Chen, Q.; Yu, B.; Liu, R.; Zhou, J. Tough hydrogel diodes with tunable interfacial adhesion for safe and durable wearable batteries. Nano Energy 2018, 48, 569–574. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, X.; Tang, H.; Wang, J.; Hao, Q.; Liu, L.; Li, Y.; Zhang, K.; Schmidt, O.G. Antifreezing hydrogel with high zinc reversibility for flexible and durable aqueous batteries by cooperative hydrated cations. Adv. Funct. Mater. 2020, 30, 1907218. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Z.; Pei, Z.; Liu, Z.; Li, H.; Zhu, M.; Fan, J.; Dai, Q.; Zhang, M.; Dai, L. Solid-State Rechargeable Zn//NiCo and Zn–Air Batteries with Ultralong Lifetime and High Capacity: The Role of a Sodium Polyacrylate Hydrogel Electrolyte. Adv. Energy Mater. 2018, 8, 1802288. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, Y.; Ren, J.; Zhang, Y.; Peng, H. An all-solid-state fiber-shaped aluminum–air battery with flexibility, stretchability, and high electrochemical performance. Angew. Chem. 2016, 128, 8111–8114. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, J.; Wang, J.; Hu, M.; Mo, F.; Liang, G.; Zhi, C. An Intrinsically Self-Healing NiCo|| Zn Rechargeable Battery with a Self-Healable Ferric-Ion-Crosslinking Sodium Polyacrylate Hydrogel Electrolyte. Angew. Chem. Int. Ed. 2018, 57, 9810–9813. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, J.; Hou, C.; Zhang, Q.; Li, Y.; Wang, H. Stable Hydrogel Electrolytes for Flexible and Submarine-Use Zn-Ion Batteries. ACS Appl. Mater. Interfaces 2020, 12, 46005–46014. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chakraborty, P.; Shit, A.; Mondal, S.; Nandi, A.K. Robust hybrid hydrogels with good rectification properties and their application as active materials for dye-sensitized solar cells: Insights from AC impedance spectroscopy. J. Mater. Chem. A 2016, 4, 4194–4210. [Google Scholar] [CrossRef]
- Galliano, S.; Bella, F.; Bonomo, M.; Viscardi, G.; Gerbaldi, C.; Boschloo, G.; Barolo, C. Hydrogel Electrolytes Based on Xanthan Gum: Green Route towards Stable Dye-Sensitized Solar Cells. Nanomaterials 2020, 10, 1585. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, Q.; Qin, D.; Luo, Y.; Li, D.; Shen, Q.; Toyoda, T.; Meng, Q. Highly efficient quasi-solid-state quantum-dot-sensitized solar cell based on hydrogel electrolytes. Electrochem. Commun. 2010, 12, 1776–1779. [Google Scholar] [CrossRef]
- Yang, Q.; Yang, W.; Duan, J.; Yang, P. A series of conducting gel electrolytes for quasi-solid-state quantum dot-sensitized solar cells with boosted electron transfer processes. J. Energy Chem. 2018, 27, 335–341. [Google Scholar] [CrossRef]
- Jin, X.; Chang, C.; Chen, Z.; Li, Q. Graphene tailored gel electrolytes for quasi-solid-state quantum dot-sensitized solar cells. Electrochim. Acta 2018, 283, 597–602. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Lin, L.; Yu, X.-Y.; Qiu, K.-Q.; Lü, X.-Y.; Kuang, D.-B.; Su, C.-Y. Dextran based highly conductive hydrogel polysulfide electrolyte for efficient quasi-solid-state quantum dot-sensitized solar cells. Electrochim. Acta 2013, 92, 117–123. [Google Scholar] [CrossRef]
- Huo, Z.; Tao, L.; Wang, S.; Wei, J.; Zhu, J.; Dong, W.; Liu, F.; Chen, S.; Zhang, B.; Dai, S. A novel polysulfide hydrogel electrolyte based on low molecular mass organogelator for quasi-solid-state quantum dot-sensitized solar cells. J. Power Sources 2015, 284, 582–587. [Google Scholar] [CrossRef]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Target Functional Group | Reaction Conditions | Crosslinkage | Comments | |
|---|---|---|---|---|---|
| Small molecules |  Formaldehyde | Primary amines and aldehydes | Reaction favors basic and neutral pH |  | Reaction takes one hour to complete. Difficult to remove trace formaldehyde. |
 Glutaraldehyde | Primary amines and aldehydes | Reaction favors basic and neutral pH |  | Reaction takes one hour to complete. | |
 Genipin | Primary amines and aldehydes | Independent of pH |  | Non-toxic crosslinker and can undergo self-polymerization | |
 EGDMA | Primary amines and oxiranes | Basic pH and at high temperature |  | Weak base, reaction takes one hour. Hydrogels beads form. |
| Reaction | Reaction Conditions | Reactive Polymer Groups | Crosslinkage | Comments |
|---|---|---|---|---|
| Schiff base mechanism | Neutral pH |  |  | Good candidate for in situ gel formation. Reaction takes minimum 10 min. |
| Disulfide bonding | Neutral pH |  |  | Good candidate for in situ gel formation and hydrogels have good mucoadhesivity. |
| Michael addition | Weak base and in presence of catalyst |  |  | Good candidate for in situ gel formation and hydrogels have good mucoadhesivity. |
| Agent | Target Functional Group | Reaction Conditions | Crosslinkage | Comments | |
|---|---|---|---|---|---|
| Light sensitive groups |  Functional azides | Primary amines | pH-Independent |  | Multi step crosslinker. Suitable for injectable hydrogel synthesis. |
 Functional acrylates | Other acrylic acids | pH-Independent |  | Multi step crosslinker. Suitable for injectable hydrogel synthesis. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bashir, S.; Hina, M.; Iqbal, J.; Rajpar, A.H.; Mujtaba, M.A.; Alghamdi, N.A.; Wageh, S.; Ramesh, K.; Ramesh, S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12, 2702. https://doi.org/10.3390/polym12112702
Bashir S, Hina M, Iqbal J, Rajpar AH, Mujtaba MA, Alghamdi NA, Wageh S, Ramesh K, Ramesh S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers. 2020; 12(11):2702. https://doi.org/10.3390/polym12112702
Chicago/Turabian StyleBashir, Shahid, Maryam Hina, Javed Iqbal, A. H. Rajpar, M. A. Mujtaba, N. A. Alghamdi, S. Wageh, K. Ramesh, and S. Ramesh. 2020. "Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications" Polymers 12, no. 11: 2702. https://doi.org/10.3390/polym12112702
APA StyleBashir, S., Hina, M., Iqbal, J., Rajpar, A. H., Mujtaba, M. A., Alghamdi, N. A., Wageh, S., Ramesh, K., & Ramesh, S. (2020). Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers, 12(11), 2702. https://doi.org/10.3390/polym12112702