Protective Malaria Vaccine in Mice Based on the Plasmodium vivax Circumsporozoite Protein Fused with the Mumps Nucleocapsid Protein

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Recombinant NLP-Fused Proteins
2.2. Transformation of P. pastoris Host Strains
2.3. Production of PvCSP
2.4. Purification of Recombinant Proteins
2.5. Transmission Electron Microscopy
2.6. Immunoblot Assay
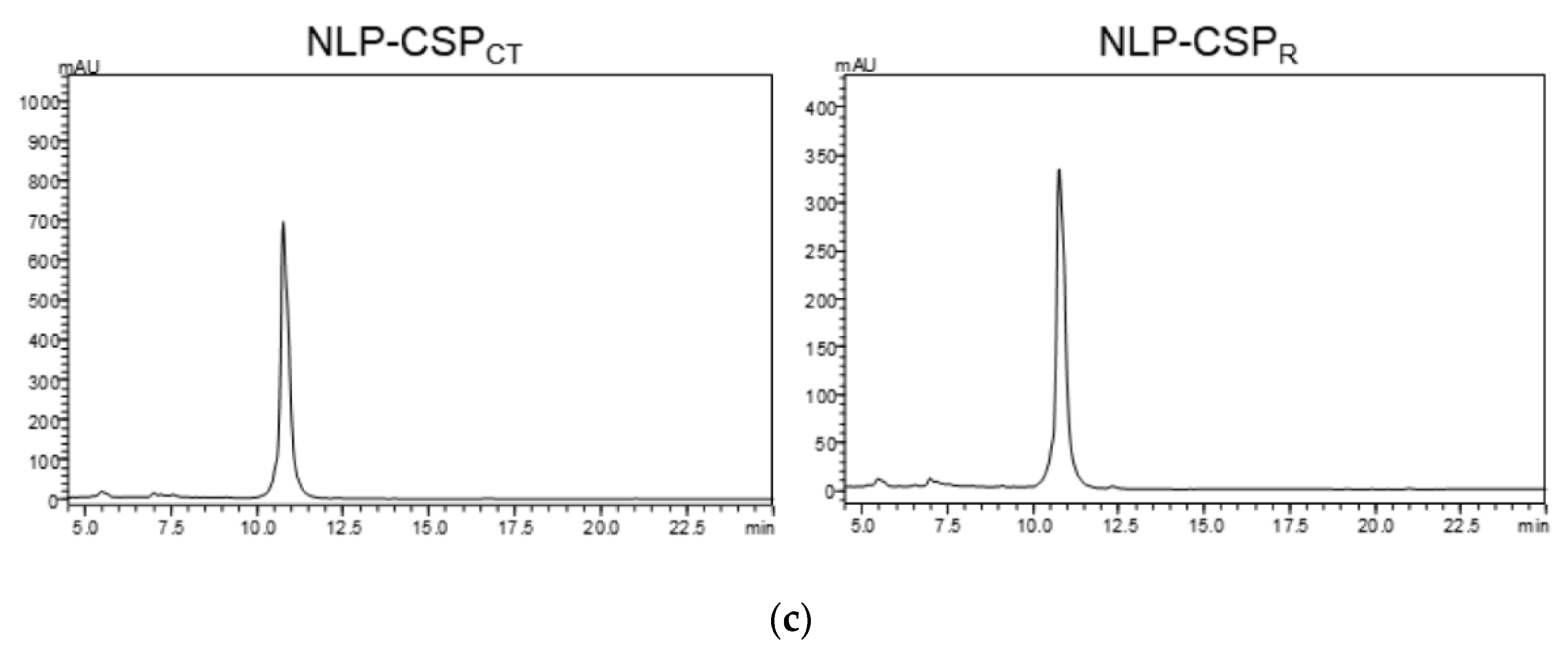
2.7. High-Performance Reverse-Phase Liquid Chromatography (RP-HPLC)
2.8. Circular Dichroism
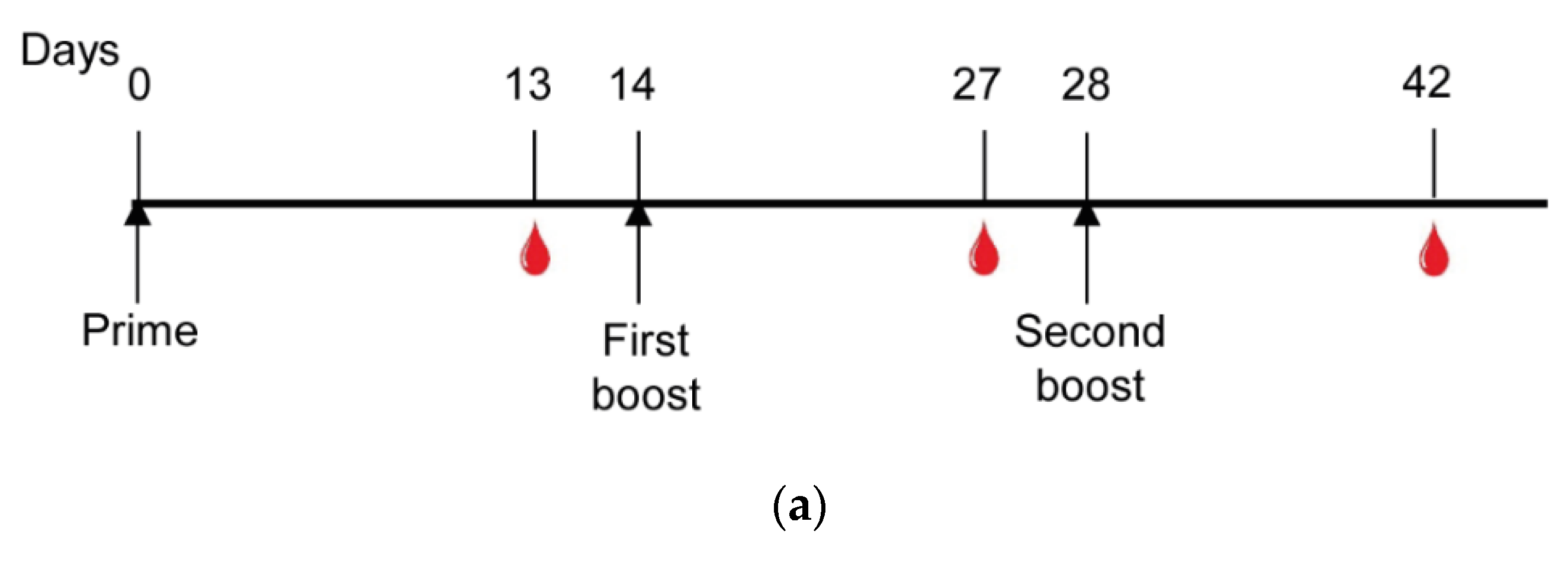
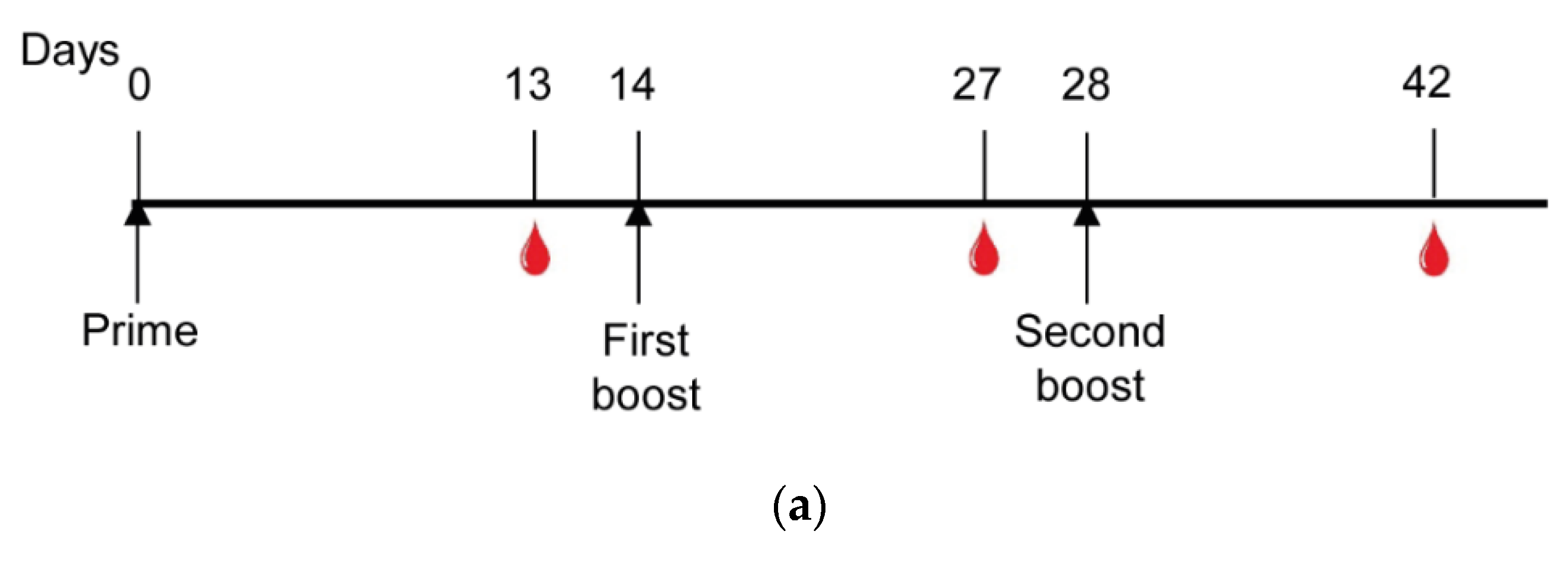
2.9. Mice Immunization
2.10. Antibody Measurement
2.11. Antibody Secreting B-Cell Measurement by ELISPOT
2.12. Parasites, Mice, and Mosquitoes
2.13. Murine Sporozoite Challenge
2.14. Statistical Analyses
2.15. Data Availability
3. Results
3.1. Generation, purification, and recognition by mAbs of the proteins NLP-CSPCT and NLP-CSPR
3.2. Preliminary Structural Characterization of the Proteins NLP-CSPCT and NLP-CSPR
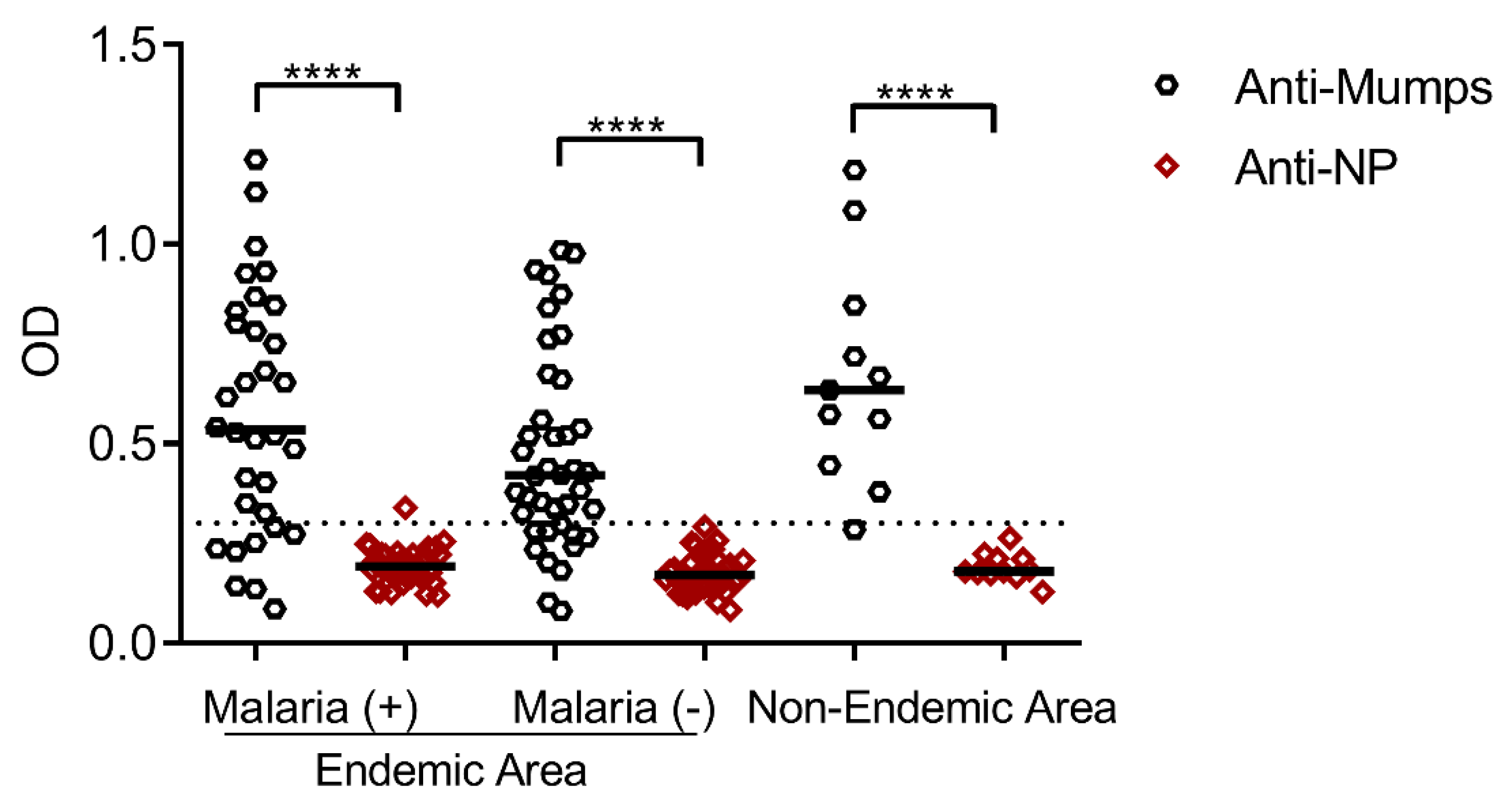
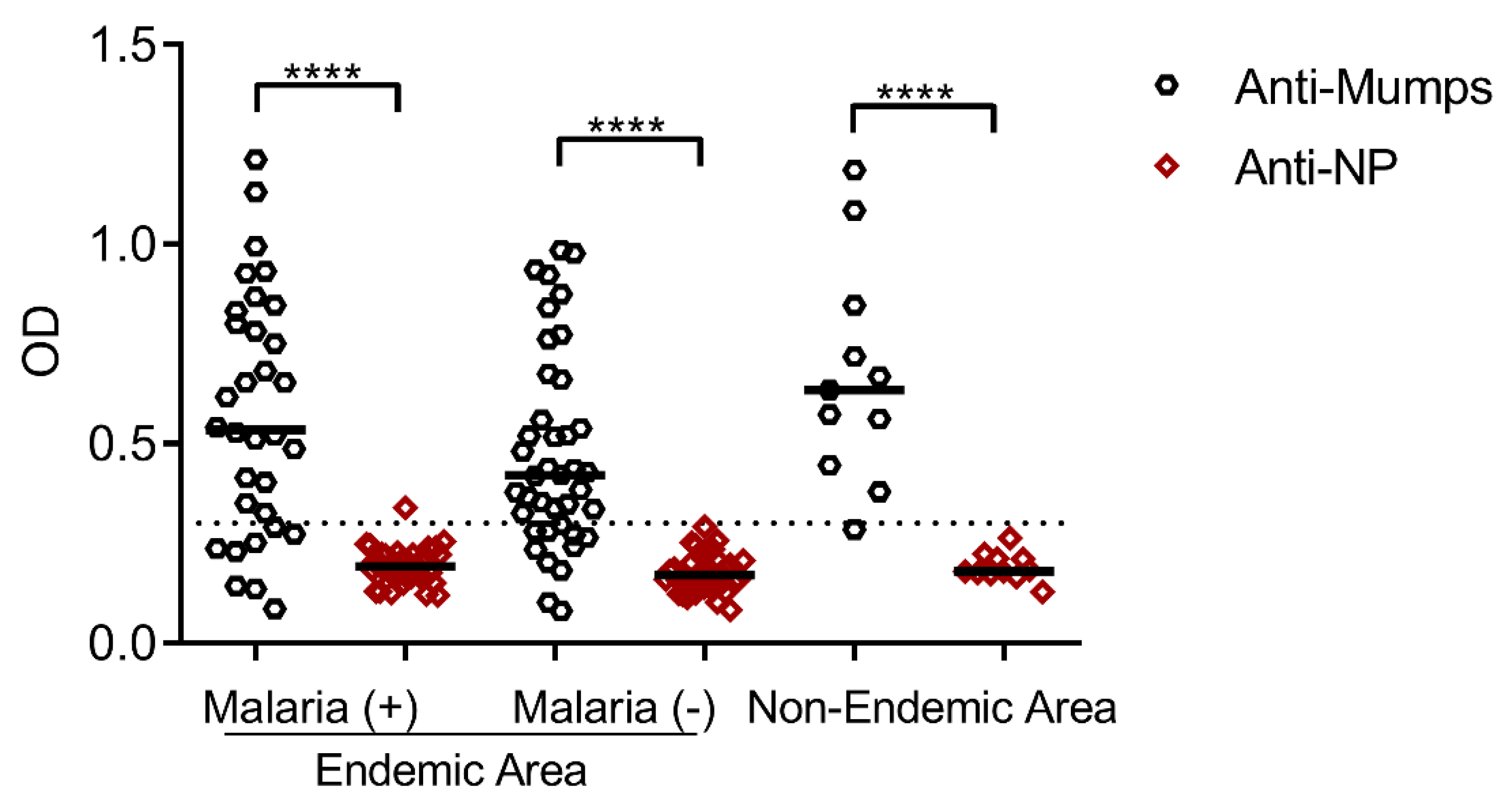
3.3. Seroprevalence of Antibodies Specific to the Mumps Virus and Its Nucleoprotein in Residents of Malaria-Endemic and Non-Endemic Areas
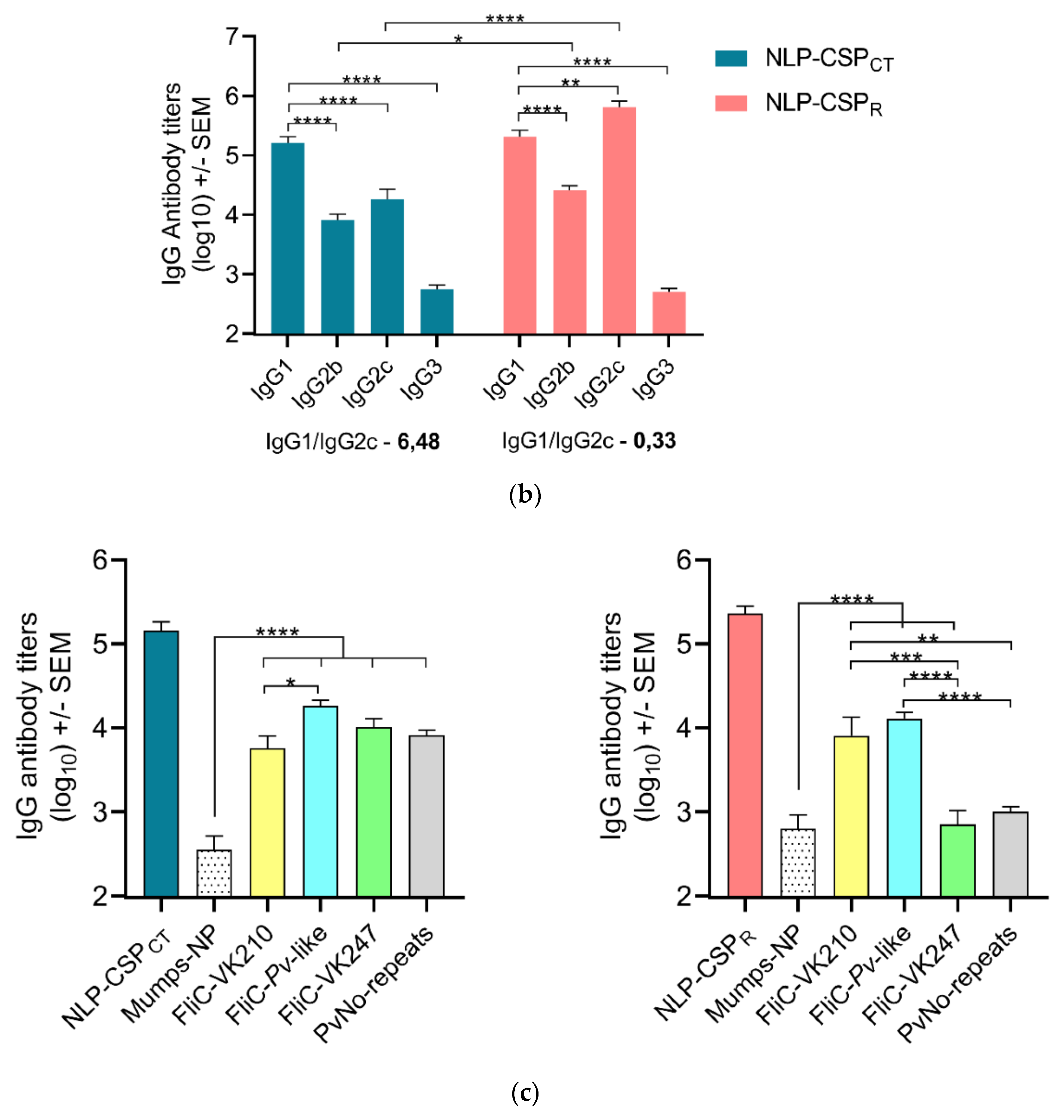
3.4. Antibody Responses in Mice Vaccinated with NLP-CSPCT and NLP-CSPR
3.5. Detection of NLP-CSPCT-Specific ASCs in Spleen and Bone Marrow of Vaccinated Mice
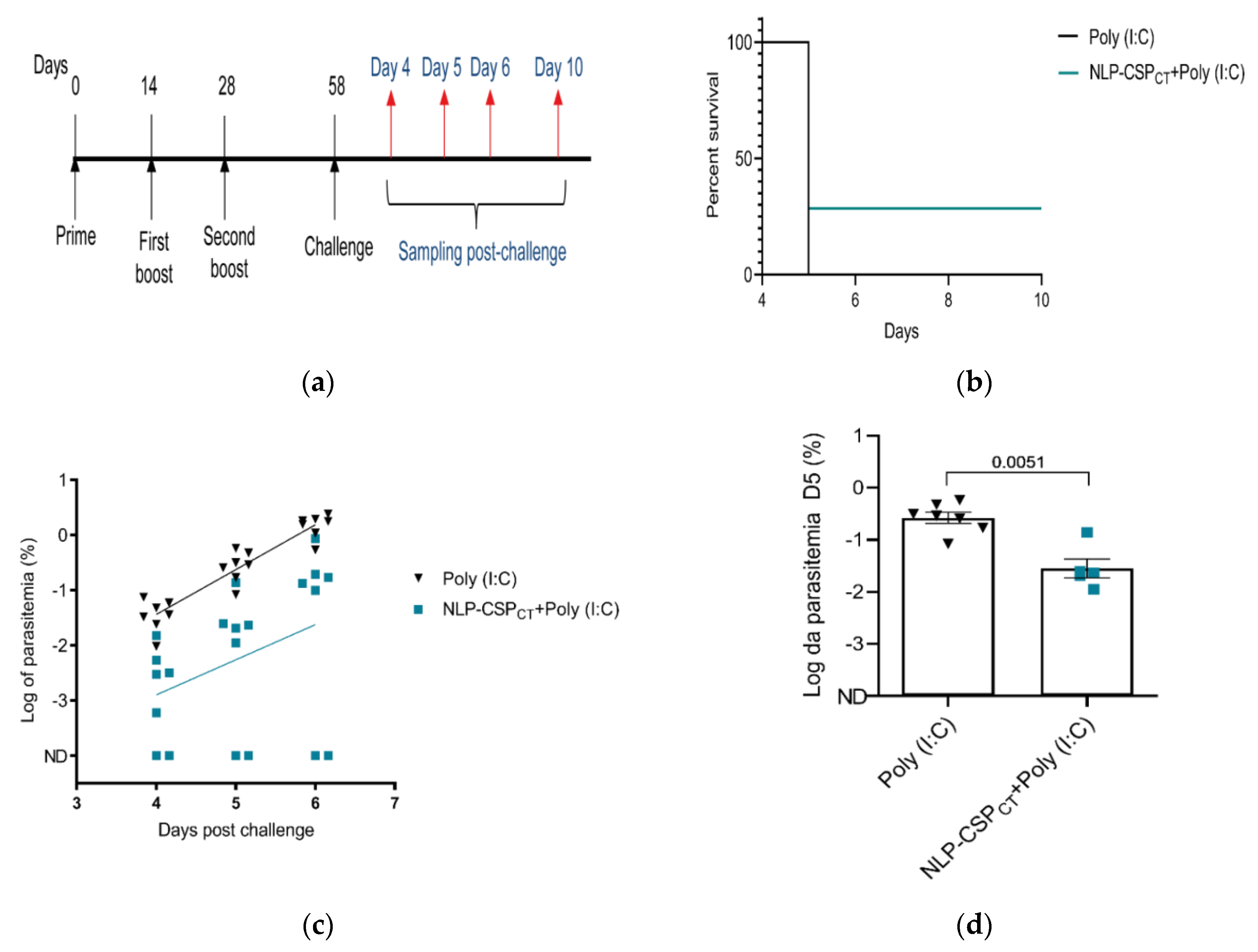
3.6. Assessment of Vaccine Efficacy
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Price, R.N.; von Seidlein, L.; Valecha, N.; Nosten, F.; Baird, J.K.; White, N.J. Global extent of chloroquine-resistant Plasmodium vivax: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 982–991. [Google Scholar] [CrossRef] [Green Version]
- Villamil-Gómez, W.E.; Eyes-Escalante, M.; Franco-Paredes, C. Severe and Complicated Malaria Due to Plasmodium Vivax. In Current Topics in Malaria; InTech: London, UK, 2016. [Google Scholar] [CrossRef] [Green Version]
- malERA. Consultative Group on Vaccines. A research agenda for malaria eradication: Vaccines. PLoS Med. 2011, 8, e1000398. [Google Scholar] [CrossRef]
- Tham, W.H.; Beeson, J.G.; Rayner, J.C. Plasmodium vivax vaccine research—we’ve only just begun. Int. J. Parasitol. 2017, 47, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olotu, A.; Fegan, G.; Wambua, J.; Nyangweso, G.; Awuondo, K.O.; Leach, A.; Lievens, M.; Leboulleux, D.; Njuguna, P.; Peshu, N.; et al. Four-year efficacy of RTS,S/AS01E and its interaction with malaria exposure. N. Engl. J. Med. 2013, 368, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- RTS, S.C.T.P. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 2015, 386, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Dobano, C.; Sanz, H.; Sorgho, H.; Dosoo, D.; Mpina, M.; Ubillos, I.; Aguilar, R.; Ford, T.; Diez-Padrisa, N.; Williams, N.A.; et al. Concentration and avidity of antibodies to different circumsporozoite epitopes correlate with RTS,S/AS01E malaria vaccine efficacy. Nat. Commun. 2019, 10, 2174. [Google Scholar] [CrossRef]
- Arnot, D.E.; Barnwell, J.W.; Tam, J.P.; Nussenzweig, V.; Nussenzweig, R.S.; Enea, V. Circumsporozoite protein of Plasmodium vivax: Gene cloning and characterization of the immunodominant epitope. Science 1985, 230, 815–818. [Google Scholar] [CrossRef]
- Rosenberg, R.; Wirtz, R.A.; Lanar, D.E.; Sattabongkot, J.; Hall, T.; Waters, A.P.; Prasittisuk, C. Circumsporozoite protein heterogeneity in the human malaria parasite Plasmodium vivax. Science 1989, 245, 973–976. [Google Scholar] [CrossRef]
- Qari, S.H.; Shi, Y.P.; Goldman, I.F.; Udhayakumar, V.; Alpers, M.P.; Collins, W.E.; Lal, A.A. Identification of Plasmodium vivax-like human malaria parasite. Lancet 1993, 341, 780–783. [Google Scholar] [CrossRef]
- De Souza-Neiras, W.C.; de Melo, L.M.; Machado, R.L. The genetic diversity of Plasmodium vivax—A review. Mem Inst. Oswaldo Cruz 2007, 102, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Raza, A.; Ghanchi, N.K.; Thaver, A.M.; Jafri, S.; Beg, M.A. Genetic diversity of Plasmodium vivax clinical isolates from southern Pakistan using pvcsp and pvmsp1 genetic markers. Malaria J. 2013, 12, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, V.A.; Sanchez-Arcila, J.C.; Vasconcelos, M.P.A.; Ferreira, A.R.; de Souza Videira, L.; Teva, A.; Perce-da-Silva, D.; Marques, M.T.Q.; de Carvalho, L.H.; Banic, D.M.; et al. Evaluating seroprevalence to circumsporozoite protein to estimate exposure to three species of Plasmodium in the Brazilian Amazon. Infect. Dis. Poverty 2018, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Arruda, M.E.; Zimmerman, R.H.; Souza, R.M.; Oliveira-Ferreira, J. Prevalence and level of antibodies to the circumsporozoite protein of human malaria parasites in five states of the Amazon region of Brazil. Mem. Inst. Oswaldo Cruz 2007, 102, 367–371. [Google Scholar] [CrossRef] [PubMed]
- White, M.T.; Verity, R.; Griffin, J.T.; Asante, K.P.; Owusu-Agyei, S.; Greenwood, B.; Drakeley, C.; Gesase, S.; Lusingu, J.; Ansong, D.; et al. Immunogenicity of the RTS,S/AS01 malaria vaccine and implications for duration of vaccine efficacy: Secondary analysis of data from a phase 3 randomised controlled trial. Lancet Infect. Dis. 2015, 15, 1450–1458. [Google Scholar] [CrossRef] [Green Version]
- Salman, A.M.; Montoya-Diaz, E.; West, H.; Lall, A.; Atcheson, E.; Lopez-Camacho, C.; Ramesar, J.; Bauza, K.; Collins, K.A.; Brod, F.; et al. Rational development of a protective P. vivax vaccine evaluated with transgenic rodent parasite challenge models. Sci. Rep. 2017, 7, 46482. [Google Scholar] [CrossRef]
- Atcheson, E.; Bauza, K.; Salman, A.M.; Alves, E.; Blight, J.; Viveros-Sandoval, M.E.; Janse, C.J.; Khan, S.M.; Hill, A.V.S.; Reyes-Sandoval, A. Tailoring a Plasmodium vivax Vaccine To Enhance Efficacy through a Combination of a CSP Virus-Like Particle and TRAP Viral Vectors. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Mueller, I.; Shakri, A.R.; Chitnis, C.E. Development of vaccines for Plasmodium vivax malaria. Vaccine 2015, 33, 7489–7495. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.W.; Yadava, A.; Tosh, D.; Sattabongkot, J.; Komisar, J.; Ware, L.A.; McCarthy, W.F.; Cowden, J.J.; Regules, J.; Spring, M.D.; et al. Phase 1/2a Trial of Plasmodium vivax Malaria Vaccine Candidate VMP001/AS01B in Malaria-Naive Adults: Safety, Immunogenicity, and Efficacy. PLoS Negl. Trop. Dis. 2016, 10, e0004423. [Google Scholar] [CrossRef]
- Teixeira, L.H.; Tararam, C.A.; Lasaro, M.O.; Camacho, A.G.; Ersching, J.; Leal, M.T.; Herrera, S.; Bruna-Romero, O.; Soares, I.S.; Nussenzweig, R.S.; et al. Immunogenicity of a prime-boost vaccine containing the circumsporozoite proteins of Plasmodium vivax in rodents. Infect. Immun. 2014, 82, 793–807. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, A.M.; Lima, L.C.; Francoso, K.S.; Denapoli, P.M.A.; Panatieri, R.; Bargieri, D.Y.; Thiberge, J.M.; Andolina, C.; Nosten, F.; Renia, L.; et al. Vaccine Containing the Three Allelic Variants of the Plasmodium vivax Circumsporozoite Antigen Induces Protection in Mice after Challenge with a Transgenic Rodent Malaria Parasite. Front. Immunol. 2017, 8, 1275. [Google Scholar] [CrossRef] [Green Version]
- Frietze, K.M.; Peabody, D.S.; Chackerian, B. Engineering virus-like particles as vaccine platforms. Curr. Opin. Virol. 2016, 18, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Fuenmayor, J.; Godia, F.; Cervera, L. Production of virus-like particles for vaccines. N Biotechnol. 2017, 39, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Cimica, V.; Galarza, J.M. Adjuvant formulations for virus-like particle (VLP) based vaccines. Clin. Immunol. 2017, 183, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dai, S.; Wang, M.; Hu, Z.; Wang, H.; Deng, F. Virus like particle-based vaccines against emerging infectious disease viruses. Virol. Sin. 2016, 31, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisci, E.; Barcena, J.; Montoya, M. Virus-like particles: The new frontier of vaccines for animal viral infections. Vet. Immunol. Immunopathol. 2012, 148, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wang, H.; Deng, F. Advances and Challenges in Enveloped Virus-like Particle (VLP)-Based Vaccines. J. Immunol. Sci. 2018, 2, 36–41. [Google Scholar]
- Sekaly, R.P. The failed HIV Merck vaccine study: A step back or a launching point for future vaccine development? J. Exp. Med. 2008, 205, 7–12. [Google Scholar] [CrossRef]
- Slibinskas, R.; Zvirbliene, A.; Gedvilaite, A.; Samuel, D.; Jin, L.; Beard, S.; Staniulis, J.; Sasnauskas, K. Synthesis of mumps virus nucleocapsid protein in yeast Pichia pastoris. J. Biotechnol. 2003, 103, 43–49. [Google Scholar] [CrossRef]
- Allwinn, R.; Zeidler, B.; Steinhagen, K.; Rohwader, E.; Wicker, S.; Rabenau, H.F.; Doerr, H.W. Assessment of mumps virus-specific antibodies by different serological assays: Which test correlates best with mumps immunity? Eur J. Clin. Microbiol Infect. Dis. 2011, 30, 1223–1228. [Google Scholar] [CrossRef]
- Jacob, D.; Ruffie, C.; Dubois, M.; Combredet, C.; Amino, R.; Formaglio, P.; Gorgette, O.; Pehau-Arnaudet, G.; Guery, C.; Puijalon, O.; et al. Whole Pichia pastoris yeast expressing measles virus nucleoprotein as a production and delivery system to multimerize Plasmodium antigens. PLoS ONE 2014, 9, e86658. [Google Scholar] [CrossRef] [Green Version]
- Leal, M.T.; Camacho, A.G.; Teixeira, L.H.; Bargieri, D.Y.; Soares, I.S.; Tararam, C.A.; Rodrigues, M.M. Immunogenicity of recombinant proteins consisting of Plasmodium vivax circumsporozoite protein allelic variant-derived epitopes fused with Salmonella enterica Serovar Typhimurium flagellin. Clin. Vaccine Immunol. 2013, 20, 1418–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, M.V.; Francoso, K.S.; Lima, L.C.; Camargo, T.M.; Machado, R.L.D.; Costa, F.T.M.; Renia, L.; Nosten, F.; Russell, B.; Rodrigues, M.M.; et al. Generation, characterization and immunogenicity of a novel chimeric recombinant protein based on Plasmodium vivax AMA-1 and MSP119. Vaccine 2017, 35, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Fabris, A.L.; Nunes, A.V.; Schuch, V.; de Paula-Silva, M.; Rocha, G.; Nakaya, H.I.; Ho, P.L.; Silveira, E.L.V.; Farsky, S.H.P. Hydroquinone exposure alters the morphology of lymphoid organs in vaccinated C57Bl/6 mice. Environ. Pollut 2020, 257, 113554. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, D.A.; Yadava, A.; Angov, E.; Maurizio, P.L.; Ockenhouse, C.F.; Zavala, F. Development of a chimeric Plasmodium berghei strain expressing the repeat region of the P. vivax circumsporozoite protein for in vivo evaluation of vaccine efficacy. Infect. Immun. 2013, 81, 2882–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliprandini, E.; Tavares, J.; Panatieri, R.H.; Thiberge, S.; Yamamoto, M.M.; Silvie, O.; Ishino, T.; Yuda, M.; Dartevelle, S.; Traincard, F.; et al. Cytotoxic anti-circumsporozoite antibodies target malaria sporozoites in the host skin. Nat. Microbiol. 2018, 3, 1224–1233. [Google Scholar] [CrossRef]
- Apostolico Jde, S.; Lunardelli, V.A.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, Modus Operandi, and Licensing. J. Immunol. Res. 2016, 2016, 1459394. [Google Scholar] [CrossRef] [Green Version]
- Silveira, E.L.; Kasturi, S.P.; Kovalenkov, Y.; Rasheed, A.U.; Yeiser, P.; Jinnah, Z.S.; Legere, T.H.; Pulendran, B.; Villinger, F.; Wrammert, J. Vaccine-induced plasmablast responses in rhesus macaques: Phenotypic characterization and a source for generating antigen-specific monoclonal antibodies. J. Immunol. Methods 2015, 416, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Kasturi, S.P.; Kozlowski, P.A.; Nakaya, H.I.; Burger, M.C.; Russo, P.; Pham, M.; Kovalenkov, Y.; Silveira, E.L.V.; Havenar-Daughton, C.; Burton, S.L.; et al. Adjuvanting a Simian Immunodeficiency Virus Vaccine with Toll-Like Receptor Ligands Encapsulated in Nanoparticles Induces Persistent Antibody Responses and Enhanced Protection in TRIM5alpha Restrictive Macaques. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- de Camargo, T.M.; de Freitas, E.O.; Gimenez, A.M.; Lima, L.C.; de Almeida Caramico, K.; Francoso, K.S.; Bruna-Romero, O.; Andolina, C.; Nosten, F.; Renia, L.; et al. Prime-boost vaccination with recombinant protein and adenovirus-vector expressing Plasmodium vivax circumsporozoite protein (CSP) partially protects mice against Pb/Pv sporozoite challenge. Sci. Rep. 2018, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Kingston, R.L.; Baase, W.A.; Gay, L.S. Characterization of nucleocapsid binding by the measles virus and mumps virus phosphoproteins. J. Virol. 2004, 78, 8630–8640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Jiang, S.; Wang, Y. Recent advances in the production of recombinant subunit vaccines in Pichia pastoris. Bioengineered 2016, 7, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, R.; Rajpoot, R.K.; Arora, U.; Poddar, A.; Swaminathan, S.; Khanna, N. Pichia pastoris-Expressed Bivalent Virus-Like Particulate Vaccine Induces Domain III-Focused Bivalent Neutralizing Antibodies without Antibody-Dependent Enhancement in Vivo. Front. Microbiol. 2017, 8, 2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraswat, S.; Athmaram, T.N.; Parida, M.; Agarwal, A.; Saha, A.; Dash, P.K. Expression and Characterization of Yeast Derived Chikungunya Virus Like Particles (CHIK-VLPs) and Its Evaluation as a Potential Vaccine Candidate. PLoS Negl. Trop. Dis. 2016, 10, e0004782. [Google Scholar] [CrossRef] [Green Version]
- Vanloubbeeck, Y.; Pichyangkul, S.; Bayat, B.; Yongvanitchit, K.; Bennett, J.W.; Sattabongkot, J.; Schaecher, K.; Ockenhouse, C.F.; Cohen, J.; Yadava, A.; et al. Comparison of the immune responses induced by soluble and particulate Plasmodium vivax circumsporozoite vaccine candidates formulated in AS01 in rhesus macaques. Vaccine 2013, 31, 6216–6224. [Google Scholar] [CrossRef]
- Yu, M.; Levine, S.J. Toll-like receptor, RIG-I-like receptors and the NLRP3 inflammasome: Key modulators of innate immune responses to double-stranded RNA viruses. Cytokine Growth Factor Rev. 2011, 22, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Perez-Mazliah, D.; Langhorne, J. CD4 T-cell subsets in malaria: TH1/TH2 revisited. Front. Immunol. 2014, 5, 671. [Google Scholar] [CrossRef]
- Schwenk, R.; DeBot, M.; Porter, M.; Nikki, J.; Rein, L.; Spaccapelo, R.; Crisanti, A.; Wightman, P.D.; Ockenhouse, C.F.; Dutta, S. IgG2 antibodies against a clinical grade Plasmodium falciparum CSP vaccine antigen associate with protection against transgenic sporozoite challenge in mice. PLoS ONE 2014, 9, e111020. [Google Scholar] [CrossRef]
- Angeletti, D.; Gibbs, J.S.; Angel, M.; Kosik, I.; Hickman, H.D.; Frank, G.M.; Das, S.R.; Wheatley, A.K.; Prabhakaran, M.; Leggat, D.J.; et al. Defining B cell immunodominance to viruses. Nat. Immunol. 2017, 18, 456–463. [Google Scholar] [CrossRef]
- Angeletti, D.; Kosik, I.; Santos, J.J.S.; Yewdell, W.T.; Boudreau, C.M.; Mallajosyula, V.V.A.; Mankowski, M.C.; Chambers, M.; Prabhakaran, M.; Hickman, H.D.; et al. Outflanking immunodominance to target subdominant broadly neutralizing epitopes. Proc. Natl. Acad. Sci. USA 2019, 116, 13474–13479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadava, A.; Waters, N.C. Rationale for Further Development of a Vaccine Based on the Circumsporozoite Protein of Plasmodium vivax. PLoS Negl. Trop. Dis. 2017, 11, e0005164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisalu, N.K.; Idris, A.H.; Weidle, C.; Flores-Garcia, Y.; Flynn, B.J.; Sack, B.K.; Murphy, S.; Schon, A.; Freire, E.; Francica, J.R.; et al. A human monoclonal antibody prevents malaria infection by targeting a new site of vulnerability on the parasite. Nat. Med. 2018, 24, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Paparoditis, P.; Horton, A.P.; Fruhwirth, A.; McDaniel, J.R.; Jung, J.; Boutz, D.R.; Hussein, D.A.; Tanno, Y.; Pappas, L.; et al. Persistent Antibody Clonotypes Dominate the Serum Response to Influenza over Multiple Years and Repeated Vaccinations. Cell Host Microbe 2019, 25, 367–376 e365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.; Amino, R.; Mueller, I. Theoretical Implications of a Pre-Erythrocytic Plasmodium vivax Vaccine for Preventing Relapses. Trends Parasitol. 2017, 33, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, R.; Wirtz, R.A.; Schneider, I.; Burge, R. An estimation of the number of malaria sporozoites ejected by a feeding mosquito. Trans. R Soc. Trop. Med. Hyg. 1990, 84, 209–212. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, R.F.; Gimenez, A.M.; Aliprandini, E.; Novais, J.T.; Cury, D.P.; Watanabe, I.-S.; Dominguez, M.R.; Silveira, E.L.V.; Amino, R.; Soares, I.S. Protective Malaria Vaccine in Mice Based on the Plasmodium vivax Circumsporozoite Protein Fused with the Mumps Nucleocapsid Protein. Vaccines 2020, 8, 190. https://doi.org/10.3390/vaccines8020190
Marques RF, Gimenez AM, Aliprandini E, Novais JT, Cury DP, Watanabe I-S, Dominguez MR, Silveira ELV, Amino R, Soares IS. Protective Malaria Vaccine in Mice Based on the Plasmodium vivax Circumsporozoite Protein Fused with the Mumps Nucleocapsid Protein. Vaccines. 2020; 8(2):190. https://doi.org/10.3390/vaccines8020190
Chicago/Turabian StyleMarques, Rodolfo F., Alba Marina Gimenez, Eduardo Aliprandini, Janaina T. Novais, Diego P. Cury, Ii-Sei Watanabe, Mariana R. Dominguez, Eduardo L. V. Silveira, Rogerio Amino, and Irene S. Soares. 2020. "Protective Malaria Vaccine in Mice Based on the Plasmodium vivax Circumsporozoite Protein Fused with the Mumps Nucleocapsid Protein" Vaccines 8, no. 2: 190. https://doi.org/10.3390/vaccines8020190