Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease

Abstract
:1. Introduction
2. Alcohol Metabolism
3. Alcohol Metabolizing Enzymes
3.1. Alcohol Dehydrogenase (ADH)
3.2. Microsomal Ethanol Oxidizing System
3.3. Catalase
3.4. Aldehyde Dehydrogenase (ALDH)
3.5. Other Minor Pathways for the Generation of Acetaldehyde
4. Genetic Polymorphisms of Alcohol-Metabolizing Enzymes and ALD
5. Ethanol Metabolism and Its Association in ALD Pathogenesis
5.1. Redox State Alterations and ALD
5.2. Role of Oxidative Stress
5.3. Role of Protein Adduct Formation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALD | Alcohol associated liver disease |
| ADH | Alcohol dehydrogenase |
| ALDH | Aldehyde dehydrogenase |
| C/EBP | CCAAT-enhancer-binding proteins |
| COUP-TF | Chicken ovalbumin upstream promoter transcription factor |
| CTF/NF-I | TATAA binding factors and CAAT box-binding transcription factor/nuclear factor-1 |
| CYP | Cytochrome |
| CYP2E1 | Cytochrome P450 2E1 |
| DBP | D-box binding protein |
| HNE | Hydroxynonenal |
| HNF | hepatocyte nuclear factor |
| MDA | Malondialdehyde |
| MEOS | Microsomal ethanol oxidizing system |
| PKC | Protein kinase C |
| NAD | Nicotinamide adenine dinucleotide |
| NADP | Nicotinamide adenine dinucleotide phosphate |
| PKC | protein kinase C |
| PPARs | Peroxisome proliferator activated receptors |
| PPRE | Peroxisome proliferator responsive element |
| PUFA | Polyunsaturated fatty acid |
| ROS | Reactive oxygen species |
| SIRT | Sirtuin |
| SREBP | Sterol regulatory element-binding protein |
| USF | Upstream stimulatory factor |
| VLDL | Very low density lipoprotein |
References
- Liangpunsakul, S.; Haber, P.; McCaughan, G.W. Alcoholic Liver Disease in Asia, Europe, and North America. Gastroenterology 2016, 150, 1786–1797. [Google Scholar] [CrossRef] [Green Version]
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutright, P.; Fernquist, R.M. Predictors of per capita alcohol consumption and gender-specific liver cirrhosis mortality rates: Thirteen European countries, circa 1970–1984 and 1995–2007. Omega (Westport) 2010, 62, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Ramstedt, M. Alcohol consumption and liver cirrhosis mortality with and without mention of alcohol—The case of Canada. Addiction 2003, 98, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Tuyns, A.J.; Pequignot, G. Greater risk of ascitic cirrhosis in females in relation to alcohol consumption. Int. J. Epidemiol. 1984, 13, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Corrao, G.; Bagnardi, V.; Zambon, A.; Arico, S. Exploring the dose-response relationship between alcohol consumption and the risk of several alcohol-related conditions: A meta-analysis. Addiction 1999, 94, 1551–1573. [Google Scholar] [CrossRef]
- Becker, U.; Deis, A.; Sorensen, T.I.; Gronbaek, M.; Borch-Johnsen, K.; Muller, C.F.; Schnohr, P.; Jensen, G. Prediction of risk of liver disease by alcohol intake, sex, and age: A prospective population study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Puri, P.; Shah, V.; Kamath, P.; Sanyal, A.; Urban, T.; Ren, X.; Katz, B.; Radaeva, S.; Chalasani, N.; et al. Effects of Age, Sex, Body Weight, and Quantity of Alcohol Consumption on Occurrence and Severity of Alcoholic Hepatitis. Clin. Gastroenterol. Hepatol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Becker, U.; Gronbaek, M.; Johansen, D.; Sorensen, T.I. Lower risk for alcohol-induced cirrhosis in wine drinkers. Hepatology 2002, 35, 868–875. [Google Scholar] [CrossRef]
- Sozio, M.S.; Liangpunsakul, S.; Crabb, D. The role of lipid metabolism in the pathogenesis of alcoholic and nonalcoholic hepatic steatosis. Semin. Liver Dis. 2010, 30, 378–390. [Google Scholar] [CrossRef]
- Lieber, C.S.; JONES, D.P.; DECARLI, L.M. Effects of prolonged ethanol intake: production of fatty liver despite adequate diets. J. Clin. Investig. 1965, 44, 1009–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.M.; Reinus, J.F. Prevalence and natural history of alcoholic liver disease. Clin. Liver Dis. 2012, 16, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.P.; Lieber, C.S. Ultrastructural alterations in human hepatocytes following ingestion of ethanol with adequate diets. Am. J. Pathol. 1966, 49, 593–603. [Google Scholar] [PubMed]
- Teli, M.R.; Day, C.P.; Burt, A.D.; Bennett, M.K.; James, O.F. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995, 346, 987–990. [Google Scholar] [CrossRef]
- Lourens, S.; Sunjaya, D.B.; Singal, A.; Liangpunsakul, S.; Puri, P.; Sanyal, A.; Ren, X.; Gores, G.J.; Radaeva, S.; Chalasani, N.; et al. Acute Alcoholic Hepatitis: Natural History and Predictors of Mortality Using a Multicenter Prospective Study. Mayo Clin. Proc. Innov. Qual. Outcomes 2017, 1, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinjuvadia, R.; Liangpunsakul, S. Trends in Alcoholic Hepatitis-related Hospitalizations, Financial Burden, and Mortality in the United States. J. Clin. Gastroenterol. 2015, 49, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Chayanupatkul, M.; Liangpunsakul, S. Alcoholic hepatitis: A comprehensive review of pathogenesis and treatment. World J. Gastroenterol. 2014, 20, 6279–6286. [Google Scholar] [CrossRef]
- Peeraphatdit, T.B.; Kamath, P.S.; Karpyak, V.M.; Davis, B.; Desai, V.; Liangpunsakul, S.; Sanyal, A.; Chalasani, N.; Shah, V.H.; Simonetto, D.A. Alcohol Rehabilitation Within 30 Days of Hospital Discharge is Associated With Reduced Readmission, Relapse, and Death in Patients with Alcoholic Hepatitis. Clin. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Mills, S.J.; Harrison, S.A. Comparison of the natural history of alcoholic and nonalcoholic fatty liver disease. Curr. Gastroenterol. Rep. 2005, 7, 32–36. [Google Scholar] [CrossRef]
- Tapper, E.B.; Parikh, N.D. Mortality due to cirrhosis and liver cancer in the United States, 1999–2016: Observational study. BMJ 2018, 362, k2817. [Google Scholar] [CrossRef] [Green Version]
- Frezza, M.; di, P.C.; Pozzato, G.; Terpin, M.; Baraona, E.; Lieber, C.S. High blood alcohol levels in women. The role of decreased gastric alcohol dehydrogenase activity and first-pass metabolism. N. Engl. J. Med. 1990, 322, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Yamashina, S.; Schemmer, P.; Rivera, C.A.; Bradford, B.U.; Enomoto, A.; Brenner, D.A.; Thurman, R.G. Estriol sensitizes rat Kupffer cells via gut-derived endotoxin. Am. J. Physiol. 1999, 277, G671–G677. [Google Scholar] [CrossRef] [PubMed]
- Crabb, D.W.; Liangpunsakul, S. Acetaldehyde generating enzyme systems: Roles of alcohol dehydrogenase, CYP2E1 and catalase, and speculations on the role of other enzymes and processes. Novartis. Found. Symp. 2007, 285, 4–16. [Google Scholar] [PubMed]
- Paton, A. Alcohol in the body. BMJ 2005, 330, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Beck, I.T.; Paloschi, G.B.; Dinda, P.K.; Beck, M. Effect of intragastric administration of alcohol on the ethanol concentrations and osmolality of pancreatic juice, bile, and portal and peripheral blood. Gastroenterology 1974, 67, 484–489. [Google Scholar] [CrossRef]
- Lieber, C.S. Metabolism of alcohol. Clin. Liver Dis. 2005, 9, 1–35. [Google Scholar] [CrossRef]
- Jones, A.W. Alcohol, its absorption, distribution, metabolism, and excretion in the body and pharmacokinetic calculations. WIREs Forensic Sci. 2019, 1, e1340. [Google Scholar] [CrossRef]
- Ramchandani, V.A.; Bosron, W.F.; Li, T.K. Research advances in ethanol metabolism. Pathol. Biol. (Paris) 2001, 49, 676–682. [Google Scholar] [CrossRef]
- Crabb, D.W.; Matsumoto, M.; Chang, D.; You, M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 2004, 63, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.W.; Plapp, B.V. Progressive sequence alignment and molecular evolution of the Zn-containing alcohol dehydrogenase family. J. Mol. Evol. 1992, 34, 522–535. [Google Scholar] [CrossRef]
- Jornvall, H.; Persson, B.; Jeffery, J. Characteristics of alcohol/polyol dehydrogenases. The zinc-containing long-chain alcohol dehydrogenases. Eur. J. Biochem. 1987, 167, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M.; Osna, N.A.; Clemens, D.L. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int. J. Biochem. Cell Biol. 2006, 38, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Hsu, L.C.; Yasunami, M. Genetics of human alcohol-metabolizing enzymes. Prog. Nucleic Acid Res. Mol. Biol. 1991, 40, 255–287. [Google Scholar] [PubMed]
- Borras, E.; Coutelle, C.; Rosell, A.; Fernandez-Muixi, F.; Broch, M.; Crosas, B.; Hjelmqvist, L.; Lorenzo, A.; Gutierrez, C.; Santos, M.; et al. Genetic polymorphism of alcohol dehydrogenase in europeans: The ADH2*2 allele decreases the risk for alcoholism and is associated with ADH3*1. Hepatology 2000, 31, 984–989. [Google Scholar] [CrossRef]
- Eng, M.Y.; Luczak, S.E.; Wall, T.L. ALDH2, ADH1B, and ADH1C genotypes in Asians: A literature review. Alcohol Res. Health 2007, 30, 22–27. [Google Scholar]
- Thomasson, H.R.; Beard, J.D.; Li, T.K. ADH2 gene polymorphisms are determinants of alcohol pharmacokinetics. Alcohol. Clin. Exp. Res. 1995, 19, 1494–1499. [Google Scholar] [CrossRef]
- Thomasson, H.R.; Crabb, D.W.; Edenberg, H.J.; Li, T.K.; Hwu, H.G.; Chen, C.C.; Yeh, E.K.; Yin, S.J. Low frequency of the ADH2*2 allele among Atayal natives of Taiwan with alcohol use disorders. Alcohol. Clin. Exp. Res. 1994, 18, 640–643. [Google Scholar] [CrossRef]
- Ehrig, T.; Bosron, W.F.; Li, T.K. Alcohol and aldehyde dehydrogenase. Alcohol Alcohol. 1990, 25, 105–116. [Google Scholar] [CrossRef]
- Svensson, S.; Some, M.; Lundsjo, A.; Helander, A.; Cronholm, T.; Hoog, J.O. Activities of human alcohol dehydrogenases in the metabolic pathways of ethanol and serotonin. Eur. J. Biochem. 1999, 262, 324–329. [Google Scholar] [CrossRef] [Green Version]
- Holmes, R.S. Alcohol dehydrogenases: A family of isozymes with differential functions. Alcohol Alcohol. Suppl. 1994, 2, 127–130. [Google Scholar]
- Han, C.L.; Liao, C.S.; Wu, C.W.; Hwong, C.L.; Lee, A.R.; Yin, S.J. Contribution to first-pass metabolism of ethanol and inhibition by ethanol for retinol oxidation in human alcohol dehydrogenase family--implications for etiology of fetal alcohol syndrome and alcohol-related diseases. Eur. J. Biochem. 1998, 254, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.M.; Teixeira, F.M.E.; Sato, M.N. Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases. Mediat. Inflamm. 2018, 2018, 3067126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estonius, M.; Svensson, S.; Hoog, J.O. Alcohol dehydrogenase in human tissues: Localisation of transcripts coding for five classes of the enzyme. FEBS Lett. 1996, 397, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Dohmen, K.; Baraona, E.; Ishibashi, H.; Pozzato, G.; Moretti, M.; Matsunaga, C.; Fujimoto, K.; Lieber, C.S. Ethnic differences in gastric sigma-alcohol dehydrogenase activity and ethanol first-pass metabolism. Alcohol. Clin. Exp. Res. 1996, 20, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688. [Google Scholar] [CrossRef]
- Malik, F.; Wickremesinghe, P.; Saverimuttu, J. Case report and literature review of auto-brewery syndrome: Probably an underdiagnosed medical condition. BMJ Open Gastroenterol. 2019, 6, e000325. [Google Scholar] [CrossRef]
- Salaspuro, M. Microbial metabolism of ethanol and acetaldehyde and clinical consequences. Addict. Biol. 1997, 2, 35–46. [Google Scholar] [CrossRef]
- Tagaino, R.; Washio, J.; Abiko, Y.; Tanda, N.; Sasaki, K.; Takahashi, N. Metabolic property of acetaldehyde production from ethanol and glucose by oral Streptococcus and Neisseria. Sci. Rep. 2019, 9, 10446. [Google Scholar] [CrossRef] [Green Version]
- Potter, J.J.; Cheneval, D.; Dang, C.V.; Resar, L.M.; Mezey, E.; Yang, V.W. The upstream stimulatory factor binds to and activates the promoter of the rat class I alcohol dehydrogenase gene. J. Biol. Chem. 1991, 266, 15457–15463. [Google Scholar]
- Stewart, M.J.; McBride, M.S.; Winter, L.A.; Duester, G. Promoters for the human alcohol dehydrogenase genes ADH1, ADH2, and ADH3: Interaction of CCAAT/enhancer-binding protein with elements flanking the ADH2 TATA box. Gene 1990, 90, 271–279. [Google Scholar] [CrossRef]
- Edenberg, H.J. Regulation of the mammalian alcohol dehydrogenase genes. Prog. Nucleic Acid Res. Mol. Biol. 2000, 64, 295–341. [Google Scholar] [PubMed]
- van Ooij, C.; Snyder, R.C.; Paeper, B.W.; Duester, G. Temporal expression of the human alcohol dehydrogenase gene family during liver development correlates with differential promoter activation by hepatocyte nuclear factor 1, CCAAT/enhancer-binding protein alpha, liver activator protein, and D-element-binding protein. Mol. Cell. Biol. 1992, 12, 3023–3031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.S.; Tsai, T.F.; Chang, H.M.; Chao, K.M.; Su, T.S.; Tsai, S.F. Distant HNF1 site as a master control for the human class I alcohol dehydrogenase gene expression. J. Biol. Chem. 2006, 281, 19809–19821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duester, G. Retinoids and the alcohol dehydrogenase gene family. EXS 1994, 71, 279–290. [Google Scholar] [PubMed]
- Dong, Y.; Poellinger, L.; Okret, S.; Hoog, J.O.; von Bahr-Lindstrom, H.; Jornvall, H.; Gustafsson, J.A. Regulation of gene expression of class I alcohol dehydrogenase by glucocorticoids. Proc. Natl. Acad. Sci. USA 1988, 85, 767–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Lee, C.B.; Park, Y.S.; Ahn, Y.H.; Kim, T.W.; Kee, C.S.; Kang, J.S.; Om, A.S. Effect of thyroid hormone on the alcohol dehydrogenase activities in rat tissues. J. Korean Med. Sci. 2001, 16, 313–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Ronis, M.J.; Badger, T.M. Ethanol induction of class I alcohol dehydrogenase expression in the rat occurs through alterations in CCAAT/enhancer binding proteins beta and gamma. J. Biol. Chem. 2002, 277, 43572–43577. [Google Scholar] [CrossRef] [Green Version]
- Potter, J.J.; Rennie-Tankersley, L.; Mezey, E. Endotoxin enhances liver alcohol dehydrogenase by action through upstream stimulatory factor but not by nuclear factor-kappa B. J. Biol. Chem. 2003, 278, 4353–4357. [Google Scholar] [CrossRef] [Green Version]
- Bosron, W.F.; Crabb, D.W.; Li, T.K. Relationship between kinetics of liver alcohol dehydrogenase and alcohol metabolism. Pharmacol. Biochem. Behav. 1983, 18 (Suppl. S1), 223–227. [Google Scholar] [CrossRef]
- Crabb, D.W.; Bosron, W.F.; Li, T.K. Steady-state kinetic properties of purified rat liver alcohol dehydrogenase: Application to predicting alcohol elimination rates in vivo. Arch. Biochem. Biophys. 1983, 224, 299–309. [Google Scholar] [CrossRef]
- Lieber, C.S.; DeCarli, L.M. Ethanol oxidation by hepatic microsomes: Adaptive increase after ethanol feeding. Science 1968, 162, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Niemela, O.; Parkkila, S.; Juvonen, R.O.; Viitala, K.; Gelboin, H.V.; Pasanen, M. Cytochromes P450 2A6, 2E1, and 3A and production of protein-aldehyde adducts in the liver of patients with alcoholic and non-alcoholic liver diseases. J. Hepatol. 2000, 33, 893–901. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Kolwankar, D.; Pinto, A.; Gorski, J.C.; Hall, S.D.; Chalasani, N. Activity of CYP2E1 and CYP3A enzymes in adults with moderate alcohol consumption: A comparison with nonalcoholics. Hepatology 2005, 41, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. The discovery of the microsomal ethanol oxidizing system and its physiologic and pathologic role. Drug Metab. Rev. 2004, 36, 511–529. [Google Scholar] [CrossRef] [PubMed]
- Quertemont, E. Genetic polymorphism in ethanol metabolism: Acetaldehyde contribution to alcohol abuse and alcoholism. Mol. Psychiatry 2004, 9, 570–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Hall, S.D.; Maya, J.F.; Li, L.; Asghar, A.; Gorski, J.C. Diabetes mellitus increases the in vivo activity of cytochrome P450 2E1 in humans. Br. J. Clin. Pharmacol. 2003, 55, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.Y.; Pan, J.M.; Gonzalez, F.J.; Gelboin, H.V.; Yang, C.S. The induction of a specific form of cytochrome P-450 (P-450j) by fasting. Biochem. Biophys. Res. Commun. 1987, 142, 1077–1083. [Google Scholar] [CrossRef]
- Johansson, I.; Lindros, K.O.; Eriksson, H.; Ingelman-Sundberg, M. Transcriptional control of CYP2E1 in the perivenous liver region and during starvation. Biochem. Biophys. Res. Commun. 1990, 173, 331–338. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Takahashi, T.; Lasker, J.M.; Rosman, A.S.; Lieber, C.S. Induction of cytochrome P-4502E1 in the human liver by ethanol is caused by a corresponding increase in encoding messenger RNA. Hepatology 1993, 17, 236–245. [Google Scholar] [PubMed]
- Lagadic-Gossmann, D.; Lerche, C.; Rissel, M.; Joannard, F.; Galisteo, M.; Guillouzo, A.; Corcos, L. The induction of the human hepatic CYP2E1 gene by interleukin 4 is transcriptional and regulated by protein kinase C. Cell Biol. Toxicol. 2000, 16, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, D.; Tolleson, W.H.; Yu, L.R.; Green, B.; Zeng, L.; Chen, Y.; Chen, S.; Ren, Z.; Guo, L.; et al. A systematic evaluation of microRNAs in regulating human hepatic CYP2E1. Biochem. Pharmacol. 2017, 138, 174–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsumi, M.; Lasker, J.M.; Takahashi, T.; Lieber, C.S. In vivo induction of hepatic P4502E1 by ethanol: Role of increased enzyme synthesis. Arch. Biochem. Biophys. 1993, 304, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Weathermon, R.; Crabb, D.W. Alcohol and medication interactions. Alcohol Res. Health 1999, 23, 40–54. [Google Scholar]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Oshino, N.; Oshino, R.; Chance, B. The characteristics of the “peroxidatic” reaction of catalase in ethanol oxidation. Biochem. J. 1973, 131, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, E.; Peiroten, A.; Landete, J.M.; Medina, M.; Arques, J.L. Gut Catalase-Positive Bacteria Cross-Protect Adjacent Bifidobacteria from Oxidative Stress. Microbes Environ. 2015, 30, 270–272. [Google Scholar] [CrossRef] [Green Version]
- Tillonen, J.; Kaihovaara, P.; Jousimies-Somer, H.; Heine, R.; Salaspuro, M. Role of catalase in in vitro acetaldehyde formation by human colonic contents. Alcohol. Clin. Exp. Res. 1998, 22, 1113–1119. [Google Scholar] [CrossRef]
- Orellana, M.; Rodrigo, R.; Valdes, E. Peroxisomal and microsomal fatty acid oxidation in liver of rats after chronic ethanol consumption. Gen. Pharmacol. 1998, 31, 817–820. [Google Scholar] [CrossRef]
- Girnun, G.D.; Domann, F.E.; Moore, S.A.; Robbins, M.E. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol. Endocrinol. 2002, 16, 2793–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Thomasson, H.R.; Crabb, D.W.; Edenberg, H.J.; Li, T.K. Alcohol and aldehyde dehydrogenase polymorphisms and alcoholism. Behav. Genet. 1993, 23, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Nene, A.; Chen, C.H.; Disatnik, M.H.; Cruz, L.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 activation and coevolution of its epsilonPKC-mediated phosphorylation sites. J. Biomed. Sci. 2017, 24, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, M.; Fischer, M.; Cho, W.K.; Crabb, D. Transcriptional control of the human aldehyde dehydrogenase 2 promoter by hepatocyte nuclear factor 4: Inhibition by cyclic AMP and COUP transcription factors. Arch. Biochem. Biophys. 2002, 398, 79–86. [Google Scholar] [CrossRef]
- Stewart, M.J.; Dipple, K.M.; Estonius, M.; Nakshatri, H.; Everett, L.M.; Crabb, D.W. Binding and activation of the human aldehyde dehydrogenase 2 promoter by hepatocyte nuclear factor 4. Biochim. Biophys. Acta 1998, 1399, 181–186. [Google Scholar] [CrossRef]
- Pinaire, J.; Hasanadka, R.; Fang, M.; Chou, W.Y.; Stewart, M.J.; Kruijer, W.; Crabb, D. The retinoid X receptor response element in the human aldehyde dehydrogenase 2 promoter is antagonized by the chicken ovalbumin upstream promoter family of orphan receptors. Arch. Biochem. Biophys. 2000, 380, 192–200. [Google Scholar] [CrossRef]
- Xue, L.; Xu, F.; Meng, L.; Wei, S.; Wang, J.; Hao, P.; Bian, Y.; Zhang, Y.; Chen, Y. Acetylation-dependent regulation of mitochondrial ALDH2 activation by SIRT3 mediates acute ethanol-induced eNOS activation. FEBS Lett. 2012, 586, 137–142. [Google Scholar] [CrossRef]
- Castro, G.D.; Delgado de Layno, A.M.; Costantini, M.H.; Castro, J.A. Cytosolic xanthine oxidoreductase mediated bioactivation of ethanol to acetaldehyde and free radicals in rat breast tissue. Its potential role in alcohol-promoted mammary cancer. Toxicology 2001, 160, 11–18. [Google Scholar] [CrossRef]
- Diaz Gomez, M.I.; Castro, G.D.; de Layno, A.M.; Costantini, M.H.; Castro, J.A. Cytochrome P450 reductase-mediated anaerobic biotransformation of ethanol to 1-hydroxyethyl-free radicals and acetaldehyde. Toxicology 2000, 154, 113–122. [Google Scholar] [CrossRef]
- Couzigou, P.; Coutelle, C.; Fleury, B.; Iron, A. Alcohol and aldehyde dehydrogenase genotypes, alcoholism and alcohol related disease. Alcohol Alcohol. Suppl. 1994, 2, 21–27. [Google Scholar] [PubMed]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728–1744. [Google Scholar] [CrossRef] [PubMed]
- Chambers, G.K.; Marshall, S.J.; Robinson, G.M.; Maguire, S.; Newton-Howes, J.; Chong, N.L. The genetics of alcoholism in Polynesians: Alcohol and aldehyde dehydrogenase genotypes in young men. Alcohol. Clin. Exp. Res. 2002, 26, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Loh, E.W.; Hsu, Y.P.; Chen, C.C.; Yu, J.M.; Cheng, A.T. Alcohol-metabolising genes and alcoholism among Taiwanese Han men: Independent effect of ADH2, ADH3 and ALDH2. Br. J. Psychiatry 1996, 168, 762–767. [Google Scholar] [CrossRef]
- Higuchi, S. Polymorphisms of ethanol metabolizing enzyme genes and alcoholism. Alcohol Alcohol. Suppl. 1994, 2, 29–34. [Google Scholar]
- Yin, S.J. Alcohol dehydrogenase: Enzymology and metabolism. Alcohol Alcohol. Suppl. 1994, 2, 113–119. [Google Scholar]
- Mizoi, Y.; Yamamoto, K.; Ueno, Y.; Fukunaga, T.; Harada, S. Involvement of genetic polymorphism of alcohol and aldehyde dehydrogenases in individual variation of alcohol metabolism. Alcohol Alcohol. 1994, 29, 707–710. [Google Scholar]
- Neumark, Y.D.; Friedlander, Y.; Durst, R.; Leitersdorf, E.; Jaffe, D.; Ramchandani, V.A.; O’Connor, S.; Carr, L.G.; Li, T.K. Alcohol dehydrogenase polymorphisms influence alcohol-elimination rates in a male Jewish population. Alcohol. Clin. Exp. Res. 2004, 28, 10–14. [Google Scholar] [CrossRef]
- Yokoyama, A.; Taniki, N.; Hara, S.; Haysashi, E.; Nakamoto, N.; Mizukami, T.; Maruyama, K.; Yokoyama, T. Slow-metabolizing ADH1B and inactive heterozygous ALDH2 increase vulnerability to fatty liver in Japanese men with alcohol dependence. J. Gastroenterol. 2018, 53, 660–669. [Google Scholar] [CrossRef]
- Grove, J.; Brown, A.S.; Daly, A.K.; Bassendine, M.F.; James, O.F.; Day, C.P. The RsaI polymorphism of CYP2E1 and susceptibility to alcoholic liver disease in Caucasians: Effect on age of presentation and dependence on alcohol dehydrogenase genotype. Pharmacogenetics 1998, 8, 335–342. [Google Scholar] [CrossRef]
- Hayashi, S.; Watanabe, J.; Kawajiri, K. Genetic polymorphisms in the 5’-flanking region change transcriptional regulation of the human cytochrome P450IIE1 gene. J. Biochem. 1991, 110, 559–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; He, Y.; Niu, F.; Yan, M.; Li, J.; Yuan, D.; Jin, T. Polymorphisms of drug-metabolizing enzyme CYP2E1 in Chinese Uygur population. Medicine 2018, 97, e9970. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Hayashi, S.; Kawajiri, K. Different regulation and expression of the human CYP2E1 gene due to the RsaI polymorphism in the 5’-flanking region. J. Biochem. 1994, 116, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Kodydkova, J.; Vavrova, L.; Kocik, M.; Zak, A. Human catalase, its polymorphisms, regulation and changes of its activity in different diseases. Folia Biol. (Praha) 2014, 60, 153–167. [Google Scholar]
- Plemenitas, A.; Kastelic, M.; Porcelli, S.; Serretti, A.; Rus Makovec, M.; Kores Plesnicar, B.; Dolzan, V. Genetic variability in CYP2E1 and catalase gene among currently and formerly alcohol-dependent male subjects. Alcohol Alcohol. 2015, 50, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zhao, H.; Gelernter, J. Strong protective effect of the aldehyde dehydrogenase gene (ALDH2) 504lys (*2) allele against alcoholism and alcohol-induced medical diseases in Asians. Hum. Genet. 2012, 131, 725–737. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Huang, I.Y.; Ikawa, M. Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals. Proc. Natl. Acad. Sci. USA 1984, 81, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.B.; Martin, N.G. Aversive reactions and alcohol use in Europeans. Alcohol. Clin. Exp. Res. 1993, 17, 131–134. [Google Scholar] [CrossRef]
- Petersen, E.N. The pharmacology and toxicology of disulfiram and its metabolites. Acta Psychiatr. Scand. Suppl. 1992, 369, 7–13. [Google Scholar] [CrossRef]
- Chang, B.; Hao, S.; Zhang, L.; Gao, M.; Sun, Y.; Huang, A.; Teng, G.; Li, B.; Crabb, D.W.; Kusumanchi, P.; et al. Association Between Aldehyde Dehydrogenase 2 Glu504Lys Polymorphism and Alcoholic Liver Disease. Am. J. Med. Sci. 2018, 356, 10–14. [Google Scholar] [CrossRef]
- Li, H.; Borinskaya, S.; Yoshimura, K.; Kal’ina, N.; Marusin, A.; Stepanov, V.A.; Qin, Z.; Khaliq, S.; Lee, M.Y.; Yang, Y.; et al. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann. Hum. Genet. 2009, 73, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunnet, N.; Kondrup, J. The effect of ethanol on the beta-oxidation of fatty acids. Alcohol. Clin. Exp. Res. 1986, 10, 64S–68S. [Google Scholar] [CrossRef] [PubMed]
- Liangpunsakul, S.; Bennett, R.; Westerhold, C.; Ross, R.A.; Crabb, D.W.; Lai, X.; Witzmann, F.A. Increasing serum pre-adipocyte factor-1 (Pref-1) correlates with decreased body fat, increased free fatty acids, and level of recent alcohol consumption in excessive alcohol drinkers. Alcohol 2014, 48, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, A.; Price, D.; Crabb, D. High-level expression of rat class I alcohol dehydrogenase is sufficient for ethanol-induced fat accumulation in transduced HeLa cells. Hepatology 1999, 29, 1164–1170. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- You, M.; Jogasuria, A.; Taylor, C.; Wu, J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 88–100. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [Green Version]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Shahidi, F.; Zhong, Y. Lipid oxidation and improving the oxidative stability. Chem. Soc. Rev. 2010, 39, 4067–4079. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, S.; Gaal, K.; Britton, R.S.; Bacon, B.R.; Triadafilopoulos, G.; Tsukamoto, H. Increased 4-hydroxynonenal levels in experimental alcoholic liver disease: Association of lipid peroxidation with liver fibrogenesis. Hepatology 1992, 16, 448–453. [Google Scholar] [CrossRef]
- Shearn, C.T.; Backos, D.S.; Orlicky, D.J.; Smathers-McCullough, R.L.; Petersen, D.R. Identification of 5’ AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J. Biol. Chem. 2014, 289, 15449–15462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Smathers, R.L.; Galligan, J.J.; Stewart, B.J.; Petersen, D.R. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem. Biol. Interact. 2011, 192, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanji, A.A.; Zhao, S.; Sadrzadeh, S.M.; Dannenberg, A.J.; Tahan, S.R.; Waxman, D.J. Markedly enhanced cytochrome P450 2E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol. Clin. Exp. Res. 1994, 18, 1280–1285. [Google Scholar] [CrossRef]
- Assiri, M.A.; Roy, S.R.; Harris, P.S.; Ali, H.; Liang, Y.; Shearn, C.T.; Orlicky, D.J.; Roede, J.R.; Hirschey, M.D.; Backos, D.S.; et al. Chronic Ethanol Metabolism Inhibits Hepatic Mitochondrial Superoxide Dismutase via Lysine Acetylation. Alcohol. Clin. Exp. Res. 2017, 41, 1705–1714. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Miller, M.E.; Cave, M.C.; Joshi-Barve, S.; McClain, C.J. Alcoholic Liver Disease: Update on the Role of Dietary Fat. Biomolecules 2016, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Nanji, A.A.; Jokelainen, K.; Tipoe, G.L.; Rahemtulla, A.; Dannenberg, A.J. Dietary saturated fatty acids reverse inflammatory and fibrotic changes in rat liver despite continued ethanol administration. J. Pharmacol. Exp. Ther. 2001, 299, 638–644. [Google Scholar]
- Chiang, D.J.; McCullough, A.J. The impact of obesity and metabolic syndrome on alcoholic liver disease. Clin. Liver Dis. 2014, 18, 157–163. [Google Scholar] [CrossRef]
- Ruhl, C.E.; Everhart, J.E. Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin. Gastroenterol. Hepatol. 2005, 3, 1260–1268. [Google Scholar] [CrossRef]
- Mahli, A.; Hellerbrand, C. Alcohol and Obesity: A Dangerous Association for Fatty Liver Disease. Dig Dis 2016, 34 (Suppl. S1), 32–39. [Google Scholar] [CrossRef] [Green Version]
- Emery, M.G.; Fisher, J.M.; Chien, J.Y.; Kharasch, E.D.; Dellinger, E.P.; Kowdley, K.V.; Thummel, K.E. CYP2E1 activity before and after weight loss in morbidly obese subjects with nonalcoholic fatty liver disease. Hepatology 2003, 38, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. CYP2E1 potentiates toxicity in obesity and after chronic ethanol treatment. Drug Metab. Drug Interact. 2012, 27, 125–144. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Akbar, M.; Jo, I.; Hardwick, J.P.; Abdelmegeed, M.A. Translational Implications of the Alcohol-Metabolizing Enzymes, Including Cytochrome P450-2E1, in Alcoholic and Nonalcoholic Liver Disease. Adv. Pharmacol. 2015, 74, 303–372. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Kim, S.J.; Im, G.Y.; Nahas, J.; Dhesi, B.; Vergis, N.; Sinha, A.; Ghezzi, A.; Rink, M.R.; McCune, A.; et al. Obesity in acute alcoholic hepatitis increases morbidity and mortality. EBioMedicine 2019, 45, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, Y.; Hurwitz, E.; Niemela, O.; Arnon, R. Monoclonal and polyclonal antibodies against acetaldehyde-containing epitopes in acetaldehyde-protein adducts. Proc. Natl. Acad. Sci. USA 1986, 83, 7923–7927. [Google Scholar] [CrossRef] [Green Version]
- Tuma, D.J.; Casey, C.A. Dangerous byproducts of alcohol breakdown--focus on adducts. Alcohol Res. Health 2003, 27, 285–290. [Google Scholar]
- Niemela, O. Distribution of ethanol-induced protein adducts in vivo: Relationship to tissue injury. Free Radic. Biol. Med. 2001, 31, 1533–1538. [Google Scholar] [CrossRef]
- Niemela, O. Aldehyde-protein adducts in the liver as a result of ethanol-induced oxidative stress. Front. Biosci. 1999, 4, D506–D513. [Google Scholar] [CrossRef] [Green Version]
- Tuma, D.J.; Hoffman, T.; Sorrell, M.F. The chemistry of acetaldehyde-protein adducts. Alcohol Alcohol. Suppl. 1991, 1, 271–276. [Google Scholar]
- Tuma, D.J.; Smith, S.L.; Sorrell, M.F. Acetaldehyde and microtubules. Ann. N. Y. Acad. Sci. 1991, 625, 786–792. [Google Scholar] [CrossRef]
- Guillot, A.; Ren, T.; Jourdan, T.; Pawlosky, R.J.; Han, E.; Kim, S.J.; Zhang, L.; Koob, G.F.; Gao, B. Targeting liver aldehyde dehydrogenase-2 prevents heavy but not moderate alcohol drinking. Proc. Natl. Acad. Sci. USA 2019, 116, 25974–25981. [Google Scholar] [CrossRef] [PubMed]
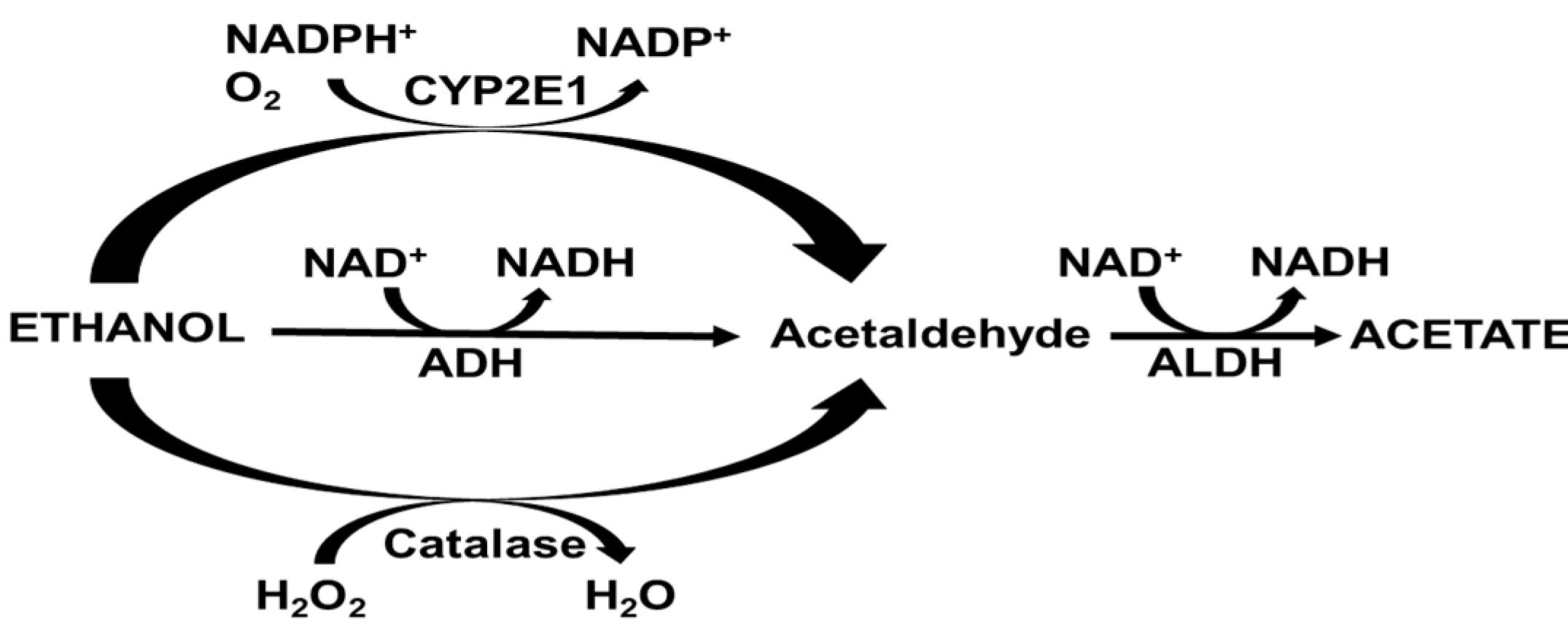

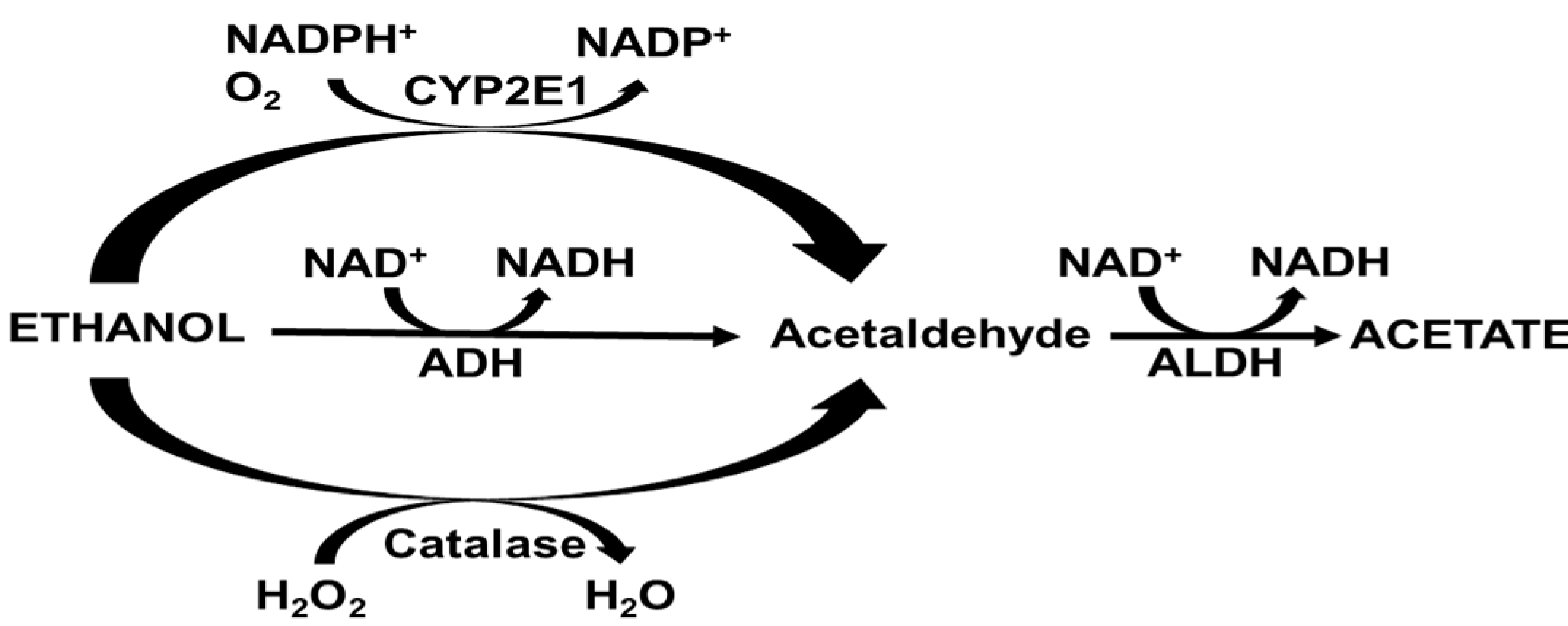{kind=link}
| Gene Locus | Subunit | Km for Ethanol (mM) | Vmax (min−1) | Major Ethnicity | Tissue Distribution |
|---|---|---|---|---|---|
| Class I | |||||
| ADH1B*1 | β1 | 0.05 | 54 | Caucasians, African-Americans | Liver, lung |
| ADH1B*2 | β2 | 0.9 | - | Asians | |
| ADH1B*3 | β2 | 34 | - | African-Americans | |
| ADH1C*1 | γ1 | 1 | - | All groups | Liver, stomach |
| ADH1C*2 | γ2 | 0.63 | - | Caucasians | |
| Class II: ADH2 | π | 34 | 40 | Liver | |
| Class III: ADH3 | χ | 1000 | - | Ubiquitous | |
| Class IV: ADH4 | σ, µ | 20 | 1510 | Stomach and esophagus |
| Gene Locus | Km for Acetaldehyde | Tissue Distribution |
|---|---|---|
| Class I ALDH1 | 30 µM | Liver and many other tissues |
| Class II ALDH2 | <1 µM | Low levels in most tissues with the expression highest in liver compared to kidney and muscle |
| Class III ALDH3 | 11 mM | Stomach, liver, cornea |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease. Biomedicines 2020, 8, 50. https://doi.org/10.3390/biomedicines8030050
Jiang Y, Zhang T, Kusumanchi P, Han S, Yang Z, Liangpunsakul S. Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease. Biomedicines. 2020; 8(3):50. https://doi.org/10.3390/biomedicines8030050
Chicago/Turabian StyleJiang, Yanchao, Ting Zhang, Praveen Kusumanchi, Sen Han, Zhihong Yang, and Suthat Liangpunsakul. 2020. "Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease" Biomedicines 8, no. 3: 50. https://doi.org/10.3390/biomedicines8030050